Mesoporous silica nanoparticles for the design of smart delivery nanodevices
Montserrat
Colilla
ab,
Blanca
González
ab and
María
Vallet-Regí
*ab
aDepartamento de Química Inorgánica y Bioinorgánica, Facultad de Farmacia, Universidad Complutense de Madrid, Plaza Ramón y Cajal s/n, 28040 Madrid, Spain. E-mail: vallet@farm.ucm.es; Fax: +34913941786; Tel: +34913941861
bNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain. Fax: +34913941786; Tel: +34913941861
First published on 21st September 2012
Abstract
Mesoporous silica nanoparticles (MSNPs) are receiving growing attention by the scientific community for their groundbreaking potential in nanomedicine. It is possible to load huge amounts of cargo into the mesopore voids and capping the pore entrances with different nanogates. Different internal or external stimuli can provoke the nanocap removal and trigger the departure of the cargo, which permits the design of stimuli-responsive drug delivery nanodevices. It is also feasible to combine the multifunctionality of MSNPs with the wide range of applications of magnetic nanoparticles (mNPs), giving rise to advanced smart nanosystems whose features and functionality can be tailored attending to specific clinical needs. This review describes the possible combinations of MSNPs, stimuli-responsive nanocaps and mNPs and the current scientific challenges aimed at accelerating the progression from bench to bedside.
 Montserrat Colilla | Montserrat Colilla was born in Madrid, Spain, in 1975. She studied chemistry at Universidad Autónoma de Madrid and received her PhD degree there in 2004, after a predoctoral fellowship at Instituto de Ciencia de Materiales de Madrid of the Spanish Council for Scientific Research. In 2005 she moved to the Department of Inorganic and Bioinorganic Chemistry at Universidad Complutense de Madrid, where she is Associate Professor since 2011. Her research is focused on bioceramics for bone tissue regeneration and drug delivery applications. She has many publications in international scientific journals and book chapters on organic–inorganic hybrid materials and ordered mesoporous materials for biomedical applications. |
 Blanca González | Blanca González was born in Madrid, Spain, in 1974. She graduated in Chemistry at Universidad Autónoma de Madrid (1998) and received her PhD degree from the same university (2003) on the field of electroactive organometallic macromolecules. In 2006 she moved to the Inorganic and Bioinorganic Chemistry Department of the Faculty of Pharmacy at Universidad Complutense de Madrid, where she currently holds an Associate Professor position. Her research interests are focused on organic–inorganic hybrid materials, including dendritic macromolecules and bioceramics, for biomedical applications and nanomedicine. |
 María Vallet-Regí | María Vallet-Regí was born in Las Palmas, Spain. She is Professor of Inorganic Chemistry and head of the research group Smart Biomaterials at Universidad Complutense de Madrid. She has written over 600 articles and several books, being the most cited Spanish scientist (ISI Web of Knowledge) in Materials Science. A Fellow of the International College of Fellows of Biomaterials Science and Engineering, and Numbered Fellow of the Spanish Royal Academy of Engineering and the Royal National Academy of Pharmacy, she has received many awards such as the Spanish National Research award in Engineering 2008 and the RSEQ Research award and Gold Medal 2011. |
1. Introduction
This review illustrates how the combination of three pivotal elements, mesoporous silica nanoparticles (MSNPs) as multifunctional nanoplatforms, suitable moieties acting as gatekeepers that can be opened and closed by different triggering stimuli, and magnetic nanoparticles (mNPs), permits the design and fabrication of smart nanosystems for clinical nanomedicine. The main effort carried out by authors during the writing of this article has been focused on showing how to combine these three elements to be able to design multicomponent systems with appropriate properties to fulfil concrete clinical needs.MSNPs with typical particle diameters in the 50–300 nm range and narrow pore size distributions of 3–6 nm are receiving growing interest by the scientific community for their use as drug delivery systems both in vitro and in vivo.1–17 MSNPs exhibit attractive characteristics, such as the possibility of controlling their particle size, chance of functionalization of the silica surface, biocompatibility, degradability under physiological conditions and large surface areas and pore volumes, which allow entrapping large amounts of cargo molecules.2,3,5,7 It is also possible to provide MSNPs of smart drug delivery capability by capping the pore entrances with suitable gatekeepers that prevent the premature release of the cargo before reaching the target. Upon exposure to internal or external stimuli the nanogates are opened and the departure of the entrapped cargo takes place. The different gatekeepers are designed depending on the stimulus that is going to be used as release trigger.1,3–5,12,13,18–21 Among them, the used stimuli can be either internal (pH, redox potential, temperature, biomolecules) or external (light, magnetic field), or even the combination of more than one stimulus (dual-stimuli or multi-responsive release systems). It is worth noting that these “zero-release” systems are especially important for cancer treatment. Currently, the cancer treatment by chemotherapy requires the administration of high dosages of cytotoxic drugs, due to their lack of specificity, which provokes severe cytotoxic effects on healthy tissues and organs and, therefore, severe adverse side effects in the patient. For this purpose, one of the current scientific goals is the design of targeted smart drug delivery nanosystems that can transport an effective drug dosage to tumour cells. To reach this goal MSNPs should exhibit multifunctional properties7,8,22–25 and, furthermore, the external surface of MSNPs can be modified by using targeting agents such as certain peptides, antibodies or folic acid. Biocompatible polymers such as polyethylene glycol (PEG) can be also attached to the MSNPs surface to provide the system of “stealth” properties by minimizing proteins adsorption (opsonisation), which would lead to a rapid clearance of nanoparticles.26–29 Moreover, these nanosystems can be also used for bioimaging by using fluorescent dyes or magnetic resonance imaging (MRI) active complexes.30–34
On the other hand mNPs with high values of saturation magnetization and magnetic susceptibility offer interesting features that make them suitable for nanomedicine.35–39 First of all, they can be synthesized with controlled size from a few to tens of nanometres and therefore they can interact with biological material. In addition, the nanoparticle magnetism offers the possibility of manipulation by an external magnetic field gradient. This property permits the transportation or immobilization of mNPs for the release of drugs, genes or proteins. Finally, mNPs can respond to the action of alternating magnetic fields and an energy transference from the field to the particle takes place. Thus, the mNP could be used to transmit certain amounts of thermal energy into tumour cells, which is the basis of the antitumor therapy by hyperthermia.
Among the wide variety of mNPs, colloidal superparamagnetic iron oxide nanoparticles (IONPs), such as magnetite (Fe3O4) or its oxidized form maghemite (γ-Fe2O3), offer great potential in nanomedicine.
The use of mNPs constitutes a promising alternative for drug targeting, as mNPs can carry a drug either attached on their surface or entrapped in a surface coating and the drug could be magnetically guided to the target organ for specific release. The combination of mNPs with gene vectors would permit to apply the same principles of magnetically targeted drug delivery to the gene transfection methods.40,41
It is possible to combine MSNPs and mNPs to design stimuli-responsive targeted drug delivery systems with the advantage of incorporating iron oxide magnetic nuclei that could allow the magnetic guidance of the system, synergistic hyperthermia and chemotherapy treatments and even MRI.
In this manuscript we review the different approaches developed so far to design and improve the behaviour of these smart nanodevices. We describe the recent advances achieved by the scientific community derived from in vitro experiments and some preliminary in vivo tests. Such investigations must be improved and the experiments must be carefully designed to acquire the necessary knowledge that permits the transit from bench to bedside.
2. Mesoporous silica nanoparticles for the design of smart delivery nanodevices: possibilities
There is an increasing interest in the nanomedicine field in the design of nanoparticles capable of acting as drug delivery devices.42,43 Among the different types of nanoparticles with applications in nanomedicine, ceramic (inorganic) nanoparticles are receiving huge attention due to their increased mechanical strength, chemical stability, biocompatibility and resistance to microbial attack as compared to their organic (polymeric) equivalents.44–46 In addition, the ceramic matrix efficiently protects entrapped guest molecules against enzymatic degradation or denaturation induced by external pH and temperature as no swelling or porosity changes take place as a response to environmental changes such as pH.47–49After introduction of mesoporous materials in the drug delivery landscape in the early 2000s,50 MSNPs have been also shown to be excellent candidates for cell-specific delivery. There are two main synthetic approaches to carry out the synthesis of MSNPs. The first one, the so-called “modified Stöber method”, consists in the condensation of silica under basic medium in the presence of a cationic structure directing agent,51 which yields monodisperse spherical MSNPs with sizes in the 50–200 nm range and containing pores of ca. 2 nm in diameter. The second strategy is the aerosol-assisted synthesis, which allows use of not only cationic, but also anionic and non-ionic surfactants.52–60 The last step of the synthesis, independent of the used approach, is the surfactant removal, which can be carried out using different methods61–65 and normally leads to materials with cylindrical mesopores organised in a two-dimensional hexagonal way typical of MCM-41-type materials.62 MSNPs are receiving growing attention due to their unique features: (i) tuneable particle size in the 50–300 nm range; (ii) stable and rigid framework compared to other polymer-based drug carriers; (iii) uniform and tuneable pore sizes that can be tailored between 2–6 nm; (iv) high surface areas (700–1000 m2 g−1) and large pore volumes (0.6–1 cm3 g−1); (v) two functional surfaces: an internal surface into the pore channels and an external surface corresponding to the exterior particle surface. This feature allows the selective functionalization of the internal and/or external surface of MSNPs; (vi) unique porous structure. The unique hexagonally ordered pore structures of MSNPs offer the possibility of obtaining a “perfect” capping of mesopore channels. This is essential for certain applications, for instance the delivery of toxic antitumor drugs, which requires “zero release” before reaching the targeted cells or tissues. Then, drug release would take place at specific sites after application of a suitable stimulus, in the so-called stimuli-responsive delivery systems, that will be described in more depth below.
Another foremost advantage of MSNPs is that they can be endocytosed by a great number of mammalian cells (tumour and non-tumour cells) and even plant cells in short times, usually within 30 min after they are incorporated into the culture medium. This phenomenon is widely dependent on surface functionalization,66 size67 and morphology of the particles.68 The in vitro toxicology of MSNPs has been recently investigated, indicating that these materials are well-tolerated for different cells lines at dosages <100 μg mL−1,69 and that only transitory effects related to alterations in cellular metabolism have been reported.70,71 Tamanoi and co-workers have investigated in depth the in vivo biocompatibility of these materials, finding that dosages lower than 200 mg kg−1 are well-tolerated.72 Lin and co-workers73 demonstrated the good hemocompatibility of MSNPs, which has a great significance for intravenous administration of these drug nanocarriers. In addition, Asefa and co-workers reported that these particles significantly enhance the cytotoxicity of Pt-based anticancer drugs loaded inside their pores, even using the less toxic trans-platinum.74 This effect could be attributed to the formation of hydroxyl or oxygen radicals in the mesoporous channels,75 which can catalyze the hydrolytic reactions of the Pt drugs, therefore enhancing the cytotoxic effect and showing that MSNPs improve the therapeutic effect of their loads and could act as more than simple carriers.
Traditional methods of pharmaceuticals administration, such as oral delivery or injection, are usually associated with a rapid release and removal of the drug from the blood (Fig. 1). Therefore, a high initial drug dosage is needed to keep therapeutic concentrations over a prolonged time period. In some cases, such as chemotherapy for cancer treatment, the administration of high dosages of cytotoxic drugs has severe adverse side effects and, what is more, only limited effectiveness due to the lack of target specificity of the current antitumor drugs. For this reason, in oncology the ratio of risk–benefit associated with chemotherapeutic agents is frequently unmanageable. To overcome this drawback, targeted drug delivery systems that can transport an effective dosage of drug to the unhealthy cells and tissues is a good alternative (Fig. 1). Premature release of guest molecules present a challenging problem and it is necessary to design “zero-release” nanocarriers able to reach the target tissue before releasing their loads.
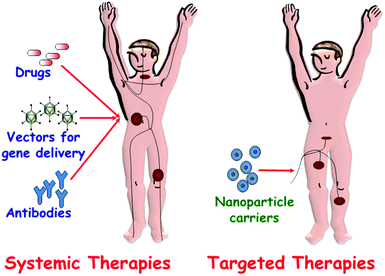 | ||
| Fig. 1 A schematic depiction of drug administration through systemic therapies versus targeted therapies. | ||
A ground-breaking approach is the possibility of employing the available channels of MSNPs as drug reservoirs using different nanocaps that can be opened and closed by different triggers, in the so-called stimuli-responsive release systems (Fig. 2).1,3–5,12,13,18–21 Stimuli used as release triggers can be found in the interior of biological systems (pH, temperature, redox potential and biomolecules) or can be applied externally from biological systems (light, magnetic field). Stimuli-responsive controlled delivery systems are especially valuable when the triggering stimuli are unique to the targeted pathology. The different gatekeepers and the stimuli used as triggers for uncapping of mesopores and subsequently controlled release of their cargos are here summarized.
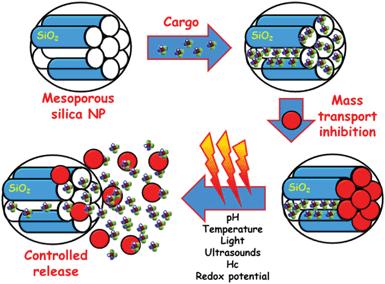 | ||
| Fig. 2 A schematic representation of the different steps involved in the performance of MSNPs as stimuli-responsive drug delivery devices. Firstly, drug is loaded into the mesopore channels. The pore openings are then closed using nanocaps to prevent premature drug departure. Finally, the application of a stimulus provokes the removing of the gatekeepers allowing the release of the entrapped drug. | ||
2.1. pH-Responsive gatekeepers
The use of pH as a release trigger in stimuli-responsive release systems is based on the fact that certain tissues of the body, such as tumours and inflammatory tissues (pH ≈ 6.8), as well as endosomal and lysosomal cell compartments (pH ≈ 5.5), have a more acidic pH than blood or healthy tissues (pH ≈ 7.4).76 To achieve successful pH-responsive delivery systems the carrier must be stable at physiological (pH ≈ 7.4), but release its loaded cargo in acidic environments. Considering these concepts, Park et al. reported the pH-controlled release of guest molecules entrapped into the pores of MSNPs that were blocked by the surface-grafted pH-responsive polyethyleneimine/cyclodextrin (PEI/CD) polypseudorotaxane.77 The mesopores of PEI-modified MSNPs were filled with a guest molecule (calcein) and then blocked with CD at pH 11. At pH 5.5, the calcein molecules were released from the pores by the reversible dethreading of CDs from the PEI chain.The group of Prof. Zink has deeply investigated the potential applications of CDs in the development of pH-sensitive nanovalves.2,13 Thus, in 2009 the group reported a new category of mechanized NPs consisting of hollow MSNPs capped by supramolecular machines based on CDs.78 These authors prepared pH-responsive nanovalves controlled by a supramolecular system containing α-CD rings interacting by hydrogen bonds with anilinoalkane stalks that were tethered to the silica surface. When the α-CD rings were complexed with the stalk at neutral pH, the α-CD rings were located near the nanopore openings blocking departure of cargo molecules that were loaded in the nanopores and hollow interior of the particle. When the nitrogen atoms on the aniline residues were protonated at lower pH, the binding affinities between the α-CD rings and the stalks decreased, releasing the α-CD and allowing the cargo molecules to escape. The same research group also reported a system based on the function of β-CD nanovalves that were also responsive to the endosomal acidification.79 In some recent works, this group investigated the opposite recognition, i.e. the β-CD rings were immobilized and the stalks were removable. In this case, the removable rhodamine B/benzidine stalks acted as nanopistons and moved in and out of the cylindrical cavities provided by the β-CD rings in response to changes in pH.80
A different approach consisted of the introduction of nanocaps tethered to the mesopore entrances of MSNPs through acid-cleavable chemical bonds. Thus, Au81 and Fe3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 82 nanoparticles have been used as blocking caps to control the transport of cargo from mesoporous silica through a reversible pH-dependent boronate ester bond, which is hydrolysed under acid pH. An acid-labile acetal linker has been also reported as pH-responsive nanogated ensemble by capping the pores of MSNPs with Au nanoparticles through the formation of these pH-sensitive bonds.83
82 nanoparticles have been used as blocking caps to control the transport of cargo from mesoporous silica through a reversible pH-dependent boronate ester bond, which is hydrolysed under acid pH. An acid-labile acetal linker has been also reported as pH-responsive nanogated ensemble by capping the pores of MSNPs with Au nanoparticles through the formation of these pH-sensitive bonds.83
Recently, Chen et al. developed an imaginative pH-responsive release system based on DNA nanoswitch-controlled organization of Au NPs attached to MSNPs.84 In this system, the hybridization and dehybridization of DNA between strands 1 and 2 were controlled by adjusting the pH of aqueous media. This structural transformation allows opening and closing the mesopore openings, leading to the controlled release of loaded molecules from pore voids at acid conditions.
Another strategy consisted of using polymers as pH-responsive gatekeepers. For instance, the grafting poly(4-vinyl pyridine) on the mesoporous silica surface creates a nanoshell that acts as a pH-sensitive barrier controlling the release of the molecules trapped inside the pores.85 Poly(acrylic acid) (PAA)-grafted MSNPs have also been prepared, demonstrating that the drug release rate was pH-dependent and increased with the decrease of pH.86,87 Sun et al. have recently developed pH-responsive nanodevices consisting of MSNPs poly(2-(diethylamino)ethyl methacrylate) (PDEAEMA) grafted to MSNPs.88
2.2. Temperature-responsive gatekeepers
Temperature is another internal stimulus that can be used to trigger the delivery of molecules from MSNPs. For instance, it has been revealed that the local temperature in many tumours is slightly higher than normal body temperature. Therefore, a temperature-responsive delivery system able to release its payload only at temperatures higher than 37 °C, but preserving the drugs entrapped while in circulation is required. The attachment of thermo-sensitive polymers, such as poly(N-isopropylacrylamide) (PNIPAM) and its derivatives, onto the surface of MSNPs is used in the design of thermo-responsive release systems and it has been reported in the recent years89–92 that PNIPAM changes its molecular chain conformation in response to temperature in aqueous environments.93 Thus, the molecular chains of PNIPAM are hydrated below the lower critical solution temperature (LCST) of 32 °C, resulting in an extended chain conformation that prevents the departure of the drugs loaded inside the mesopore channels. Increasing the temperature above the LCST dehydrates the polymer chains, resulting in a collapsed conformation, pore opening and subsequent release of the cargo. An increase in the LCST under physiological conditions would be desirable for biomedical applications. This can be achieved by performing modifications of the polymer composition by copolymerization with other monomers, such as acrylamide94,95 or N-isopropylmethacrylamide.96,97Very recently, Baeza et al. developed a novel nanodevice able to perform remotely controlled delivery of small molecules and proteins in response to an alternating magnetic field.98 This device is based on MSNPs with IONPs encapsulated inside the silica matrix and decorated on the outer surface with a thermo-responsive copolymer of poly(ethylenimine)-β-poly(N-isopropylacrylamide) (PEI/NIPAM). The polymer structure was designed with a double aim, to act as temperature-responsive gatekeeper for the drugs loaded inside the mesopores and to retain proteins into the polymer shell by electrostatic or hydrogen bond interactions. The nanodevice avoids premature cargo departure at low temperature (20 °C) and delivers the entrapped molecules when the temperature exceeds 35–40 °C.
Another strategy reported by Schlossbauer and co-workers consisted of developing a molecular valve that releases entrapped fluorescein upon heating to the specific melting temperature of double-stranded DNA sequences that were attached to the pore openings of MSNPs.99
Martínez-Mañez and co-workers also reported temperature-responsive drug delivery nanosystems.100 In this case, MSNPs were loaded with a fluorescent cargo and functionalized with octadecyltrimethoxysilane. The alkyl chains interact with paraffin, which builds a hydrophobic layer around the particle. Upon melting of the paraffin, the guest molecule is released. Following this strategy, the release temperature can be modulated by selecting appropriate paraffin.
2.3. Redox potential-responsive gatekeepers
The intracellular glutathione (GSH) levels in most tumour cells are 100–1000-fold higher than the extracellular levels, therefore the naturally occurring redox potentials between the mildly oxidizing extracellular space and the reducing intracellular space can be utilized as an internal stimulus to trigger the cargo release from MSNPs.101 Relying on this knowledge, different redox potential-responsive release systems were developed, which consisted of using different gatekeepers, such as CdS102 Fe3O4103 or Au104 nanoparticles, covalently linked to the MSNPs through chemically labile disulphide linkages. These nanocaps were removed by cleaving such linkages using disulphide-reducing agents, such as dithiothreitol (DTT) or mercaptoethanol (ME), leading to the release of the entrapped cargo. Liu et al. described the use of cross-linked poly(N-acryloxysuccinimide) attached at the pore entrance of MSNPs.105 After loading the dye molecules into MSNPs, the openings were blocked by the addition of cystamine, a disulphide-based bifunctional primary amine that allows polymer chains to be cross-linked through the reaction with N-oxysuccimide groups along the polymer chain. The presence of DTT cleaved the disulphide bond of the cystamine causing a disruption in the polymeric network and leading to the redox potential-controlled delivery. Other authors have proposed collagen immobilized on the exterior surfaces of MSNPs as redox potential-responsive gatekeepers for targeted drug delivery of cancer cells.106 Zink and co-workers have reported the fabrication of snap-top systems using MSNPs functionalized with rotaxanes incorporating disulphide bonds in their stalks, which are encircled by curcurbit[6]uril or α-CD rings.107 Upon exposition to DTT, there is a reductive cleavage of disulphide bonds in the stalks, resulting in the snapping of the stalks of the rotaxanes, leading to cargo release from the inside of the MSNPs. A similar approach carried out by Kim et al. consisted in using glutathione-induced intracellular release of cargos from MSNPs with CD gatekeepers covalently connected onto the particle surface via a disulphide unit.108
However, different research studies have evidenced the low reductive or even oxidative medium in endosomes.109 Therefore, the contact with the cytosol is essential to cleavage the disulphide linkages and release the cargo. The use of photoactive compounds to open the endosome is one possibility to overcome this challenge.110,111 Being and co-workers demonstrated that there was no release of disulphide-bound dye from MSNPs endocytosed by cells.112 Inefficient endosomal escape is generally a bottleneck for molecular delivery into the cytoplasm. However, after photochemical rupture of the endosomes by means of a photosensitizer, MSNPs successfully released the disulphide-bound dye into the cytoplasm, showing that the reducing milieu of the cytoplasm is enough to cleave the disulphide linkages.
2.4. Biomolecules-responsive gatekeepers
Biomolecules are receiving increasing interest as internal stimulus to trigger molecules release due to their biocompatibility and interesting biological activities. Among biomolecules, we will focus on enzymes, glucose, antigens and “aptamer–targets” as release triggers.Very recently, multi-enzyme-responsive capped MSNPs have been developed using bulky organic moieties containing amide and urea linkages to block the mesopores.118 Departure of the loaded cargo takes place upon addition of amidase and urease. Amidase induced an immediate, yet not complete, release of the cargo. On the other hand, urease allowed a near total cargo release that was delayed in time. This research demonstrates that the incorporation of different enzyme-hydrolysable groups in well-defined locations of the capping molecule permits control of the drug delivery profiles.
Enzyme-responsive polymers have also been attached to MSNPs to give them bioresponsive drug delivery capability. Thus, Singh et al.119 reported an innovative strategy to prepare MSNPs able to release the antitumor drug doxorubicin in response to proteases present at a tumour site in vivo, resulting in cellular apoptosis.
One of the first smart mesoporous nanosystems based on aptamer–target interactions consisted of MSNPs whose pores were capped with Au NPs modified with adenosine triphosphate (ATP) aptamer.124 The presence of the aptamer–target, i.e. ATP molecules, provokes the removal of Au NPs by a competitive displacement reaction, allowing the cargo release.
More recently, Özalp and Schäfer developed switchable controlled drug delivery systems using aptamers as nanovalves.125 For the proof of principle, MSNPs were modified with a hairpin form of ATP-binding DNA, which allows encapsulation and release of guest molecules controlled by ATP. This reversible aptamer-based nanogate function could provide novel trigger systems for any desired biological stimulus in drug-delivery applications.
2.5. Light-responsive gatekeepers
Since processes involving external light activation are rapid and directional, light-responsive controlled release systems are receiving increasing interest. In 2003, Fujiwara and co-workers demonstrated for the first time that the uptake, storage and release of organic molecules in MCM-41 could be regulated through the photo-controlled and reversible intermolecular dimerization of coumarin derivatives attached to the pore outlets.126,127 The same research group developed a multifunctional, fully controlled storage and release system by attaching azobenzene groups on the mesopore outlets.128 The release of guest molecules included in the pores of mesoporous silica was promoted by simultaneous irradiation with UV and visible light, which made the azobenzene molecules act as both impellers and gatekeepers. The reversible cis–trans photoisomerization of azobenzene substituents on the pore surface by a rotation–inversion mechanism caused a stirring action that accelerates the diffusion of the guest from the mesopores.The research group headed by Zink reported the use of light-operated mechanized MSNPs whose performance was based on the high binding affinity in aqueous solution between β-CD and trans-azobenzene derivatives and low, if any, binding between β-CD and cis-azobenzene derivatives.129 Irradiating with 351 nm light causes the isomerization of azobenzene to the cis conformation and therefore pore uncapping. An alternative strategy using CDs was proposed by Park et al., which consisted of covalently linking β-CD on the surface of MSNP through a photocleavable o-nitrobenzyl ester moiety.130 Upon exposure to UV light, the guest molecules were released from the pore by removal of the CD gatekeeper.
Lin and co-workers reported on the use of Au nanoparticles as light sensitive gatekeepers by the surface attachment through the photoresponsive linker thioundecyl-tetraethyleneglycoestero-nitrobenzylethyldimethylammonium bromide (TUNA).131 Upon UV irradiation, TUNA would lead to the negatively charged thioundecyltetraethyleneglycolcarboxylate (TUEC), leading to the dissociation of the Au NPs from the MSNP surface due to charge repulsion. Thus, there was an uncapping of the mesopores and the release of guest molecules took place.
An innovative strategy consisted of grafting a light-responsive polymer on the pore outlets of MSNPs.132 This approach relied upon the fact that the incorporation of hydrophobic or hydrophilic monomers into the PNIPAM backbone can lead to a decrease or increase in the LCST, respectively. Therefore, when monomers bearing a light-responsive moiety, such as azobenzene and 2-nitrobenzyl groups, were incorporated in the PNIPAM backbone, the resultant polymers were light-responsive. Their LCST could be easily modulated by applying UV irradiation since the polarity of the light-responsive moieties changes under irradiation. Thus, upon UV irradiation, the polymer changed its conformation from collapsed (insoluble) to coil (soluble) state so that the gate was opened and subsequently the entrapped molecules escaped out of the pores.
In a recent study, sulforhodamine 101 was loaded inside the mesopores of mercaptopropyl-functionalized MSNPs and the cargo molecules were entrapped by the presence of Ru(bpy)2(PPh3)-moieties, coordinated to mercaptopropyl functional groups.133 Upon irradiation with visible light (λ = 455 nm), Ru–S coordination bond was cleaved, triggering the release of capping species and loaded molecules.
2.6. Magnetic-responsive gatekeepers
Magnetic fields can also serve as external stimulus to trigger the release of molecules from MSNPs. Chen et al. have recently reported the capping of MSNPs with Fe3O4 mNPs.134 For this purpose, MSNPs were first functionalized with 3-aminopropyltrimethoxysilane (APTS) and then loaded with the antitumor drug camptothecin (CPT). The mesopore entrances with the CPT-loaded amine-MSNPs were covalently capped through amidation of the APTES bound at the pore surface with meso-2,3-dimercaptosuccinic acid functionalized superparamagnetic IONPs with an average diameter of 5.6 nm. Under the application of a magnetic trigger, the Fe3O4 nanocaps were removed due to the cleavage of chemical bonds and this subsequently led to a fast-responsive drug release.Vallet-Regí and co-workers have reported the use of alternative magnetic fields as an external release trigger to develop “on–off” stimuli-responsive drug delivery systems.135 In such a system, oligonucleotide-modified MSNPs, encapsulating superparamagnetic IONPs, are loaded with fluorescein and subsequently capped with IONPs functionalized with the complementary strand, which act as gatekeepers (Fig. 3). DNA duplex was selected to display a melting temperature of 47 °C, which corresponds to the upper limit of therapeutic magnetic hyperthermia. The magnetic-responsive release of this system was tested by exposition of this fluorescein-loaded MSNPs to an alternating magnetic field. Once the temperature reaches 47 °C the pore channels were uncapped and the cargo molecule was released (Fig. 3). It should be highlighted that the nanocapping system here acts as a reversible gatekeeper, i.e. fluorescein release is triggered when the temperature is raised, whereas cargo release is hindered when the temperature is stabilized. This “on–off” behaviour makes this system a potential on-demand drug delivery device, with potential applications in nanomedicine, such as the synergistic combination of drug delivery and hyperthermia treatments for cancer treatment.
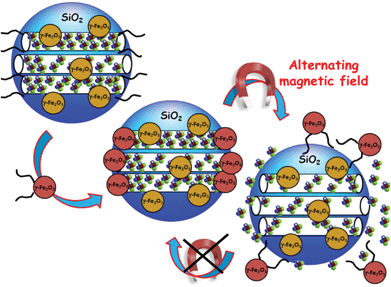 | ||
| Fig. 3 A schematic representation of reversible smart drug delivery nanodevices consisting of magnetic MSNPs modified with single-stranded DNA and γ-Fe2O3 nanoparticles modified with the complementary DNA strand acting as nanogates. The hybridization of both DNA strands provokes the mesopore capping. The progressive double-stranded DNA dehybridization upon application of an alternating magnetic field provokes the nanocap removal and the subsequent cargo release.135 | ||
2.7. Dual stimuli-responsive gatekeepers
Dual-controlled or multi-responsive controlled delivery systems are those able to respond to two or more stimuli, either in an independent or in a synergistic fashion. Martínez-Mañez and co-workers anchored suitable polyamines on the MSNP surface to obtain pH sensitive and anion-controllable gate-like ensembles capable of controlling the release of a ruthenium dye trapped inside the mesoporous matrix. To achieve this goal, they varied the pH value and content of certain anions into the release medium.136 The same group also modified the pore outlets of MSNPs with boronic acid-functionalized Au nanoparticles acting as nanocaps. These NPs were linked to the surface of saccharide-functionalized MSNPs through the formation of boronate ester bonds, which are hydrolysed under acid conditions (pH = 3). The nanosystem exhibited an “on–off” response because of the reversibility of the boronate bond formation. Moreover, the metallic nanoparticles could be heated by laser irradiation at 1064 nm (266 mJ) causing a plasmon-resonance light-induction release due to the thermal boronate bond cleavage. Dual-stimuli controlled release MSNPs could also be used as AND logic gates, as have been recently demonstrated by Angelos et al.137 These molecular machines were designed to operate in tandem with one another in such a way that the dual-controlled nanoparticle systems function as AND logic gates. In this case, two different molecular machines have been mounted on the mesoporous silica surface, azobenzene as light-activated nanoimpellers and [2]pseudorotaxanes as pH-responsive nanovalves. The two systems can act separately, but only the simultaneous activation of both molecular machines conducts the load release, showing that these kind of devices could not only be used in drug delivery applications, but could also perform simple logic operations.Liu et al. reported a multi-responsive supramolecular capped mesoporous silica system by grafting β-CD-bearing polymer on the surface of mesoporous silica and cross-linking by the addition of disulphide groups to form a polymeric network that blocked the pore entrances.138 The nanodevice was able to release their cargo in response to three different stimuli: UV light (causing the isomeric transformation of azobenzene groups), presence of α-CD (as competitive ligands to displace β-CD), and the addition of disulphide reductive agents, such as DTT (to cleave the disulphide bond between β-CD and polymer main chains).
Very recently, Chang et al. reported the synthesis of core–shell MSNPs with thermo/pH-coupling sensitivity.139 Magnetic MSNPs were used as the core and cross-linked poly(N-isopropylacrylamide-co-methacrylic acid) (P(NIPAM-co-MAA)) polymer was used for the outer shell. The thermo-sensitive volume phase transition (VPTT) could be precisely regulated by the molar ratios of MAA to NIPAM and the concentration of NaCl. The amount of drug released was small below the VPTT and increased above such a value, exhibiting an apparent thermo/pH-response controlled drug release. In a very recent work, Chen et al. reported the synthesis of dual stimuli-responsive vehicles by attaching self-complementary duplex DNA to the openings of MSNPs, which resulted in a cap for trapping guest molecules.140 The duplex DNA cap could be either denatured by heating or hydrolysed by endonucleases, thus opening the nanopores and releasing the cargo.
3. Multifunctionality of mesoporous silica particles
MSNPs possess well-defined structure and high density of surface silanol groups, which can be modified with a wide range of organic functional groups.141–143 The surface functional groups can play several roles in MSNPs in nanomedicine: (i) to increase the host–guest (matrix–drug) interactions when drugs are adsorbed into the mesoporous cavities with the aim of enhancing drug adsorption and/or slow-down drug release rates; (ii) to control the surface charge of MSNPs; (iii) to chemically graft functional molecules (prodrugs, fluorescent molecules, MRI active agents, etc.) inside or outside the pores; and (iv) to link nanogates at the mesopore entrances to prevent premature release of entrapped cargo. The unique topology of MSNPs gives them three well-defined domains that can be distinctively functionalized: the silica framework, the nanopores and the most external surface. These paramount attributes make MSNPs ideal nanoplatforms to incorporate different multifunctionalities for therapy and diagnosis of several diseases (Fig. 4).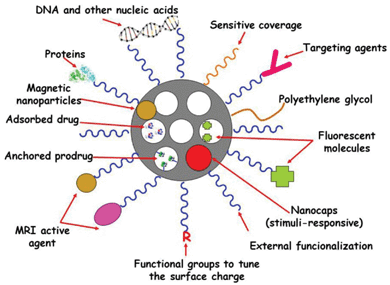 | ||
| Fig. 4 Multifunctionality and cargo loading possibilities of MSNPs. Small drug molecules can be loaded into the mesopores by adsorption from solution. It is also possible to attach some prodrugs to the functional groups present in the inner part of mesoporous silica walls. Biomolecules, such as proteins, targeting agents or nucleic acids can be attached to the external surface of MSNPs. Biocompatible polymers, such as polyethylene glycol (PEG), can be linked to the external surface of MSNPs to give them “stealth” properties. The surface charge of MSNPs can be modulated by using different functional groups to increase their dispensability in biological fluids. Different nanocaps can be used to design stimuli-responsive drug delivery devices. Magnetic nanoparticles can be incorporated into the silica matrix to provide the resulting MSNPs with magnetic properties suitable for magnetic guidance, hyperthermia and magnetic resonance imaging (MRI). The systems can be made bio-imageable by attachment of fluorescent moieties and MRI-active agents. | ||
Usually, the functionalization of the mesopore walls of MSNPs is carried out by using co-condensation or a one-step method, which allows incorporation of diverse functional groups homogeneously distributed during the synthesis stage. The co-condensation method of a tetraalkoxysilane ((RO)4Si) and one or more organoalkoxysilanes ((RO)3SiR′) has been widely employed to synthesize mesoporous organic–inorganic hybrid materials.141,142 Thus, a wide range of functional groups have been introduced to MSNPs to adsorb drug molecules or covalently attach fluorophores.18,144 Usually, the fluorophore is pre-reacted with an alkoxysilane that is subsequently used in the co-condensation synthesis, yielding inherently fluorescent MSNPs for cell imaging.5,22,66,145–148
The high intrinsic specific surface areas and pore volumes of MSNPs permit us to host huge amounts of drug, sometimes exceeding 30 wt%.18 Drug loading can be modulated, which allows use of appropriate nanoparticle concentrations to reach therapeutic drug levels in vitro and in vivo, while minimizing particle-induced toxicity. Drug loading into MSNPs is usually carried out as the final step, i.e. once the MSNPs have been synthesized. This fact permits the independent optimization of the structural and physicochemical properties of the carrier (host) and the conditions for the drug (guest) loading.7 The incorporation of drugs into the MSNPs can be achieved using three main different approaches: physical adsorption from solution into the mesopores, physical adsorption from solution onto the outermost surface of MSNPs, and covalent linking. The chosen loading method depends on the molecular structure and the size of the drug regarding the mesopore diameter, its water solubility and cytotoxicity and the chemical stability of the drug.
One of the most significant advantages of MSNPs is their capability to host hydrophobic drugs, which may be difficult when using other nanocarriers. In this case, the driving forces for drug adsorption are mainly hydrogen-bonding or polar interactions. It is also possible to promote the host–guest interactions by incorporating functional groups onto a mesoporous matrix that shows high affinity for the drug, such as amino groups in the case of adsorption of drugs bearing carboxylic acids. For more hydrophilic drugs, the electrostatic adsorption can be increased by incorporating functional groups, such as weak acids and bases like carboxylic acids or amines, onto the silica surface.18,149 It is also possible to covalently link a given drug to diverse functional groups present on the mesopore walls,150–152 which is especially attractive to prevent premature drug release. However, the drug activity should be preserved after decoupling from the pore wall, which involves the original functional group of the drug molecule used for the covalent grafting being re-formed upon detachment. Recent advances have been achieved to covalently link drugs or prodrugs by stimuli-sensitive functional groups, such as the pH-hydrolysable hydrazone bond.153,154
However, when aiming at targeted delivery applications, additional moieties have to be introduced to the outer particle surface by post-synthesis functionalization.7 The different multifunctionalization of MSNPs and the possibilities of loading and release of drugs and other biologically active molecules are schematically illustrated in Fig. 4.
Thus, the most external surface of MSNPs can be modified by targeting moieties, such as peptides, antibodies or simple molecules, such as folic acid. At this point, it is also important to ensure that extensive protein adsorption to the particles, which would lead to the MSNPs clearance by the mononuclear phagocytic system, does not occur. This can be avoided by providing MSNPs with “stealth” properties by functionalizing their external surface with biocompatible polymers, such as PEG.
As commented in previous sections, it can be necessary to introduce pore capping agents onto the outer particle surface to create gate-keeping properties, i.e. capping the pore openings to prevent physically adsorbed cargo from desorbing from the carrier before reaching the target cells. Then, the nanocaps would be opened upon an appropriated stimulus, in the so-called stimuli-responsive release systems. Moreover, the MSNP system can be made bio-imageable, for instance by the post-synthesis attachment of fluorescent dyes144 or MRI-active complexes to the particle surface.155 In the next sections, we will tackle some of the main aspects concerning biologically relevant functionalities that can be given to MSNPs.
3.1. “Stealth” properties
Extensive protein adsorption to the MSNPs under physiological conditions is an extremely important issue that must be avoided because it would decrease the targetability of the particles, as well as increase the recognition of the particles as foreign by the body defense mechanisms (the reticuloendothelial system, RES), which would lead to rapid removal from the blood circulation. In addition, protein adsorption onto the MSNPs may also influence their toxicity. The PEGylation or PEG-functionalization of NPs has been frequently used as a general and effective approach to reduce the nonspecific binding of NPs to blood proteins and macrophages providing them with the so-called “stealth” properties. Thus, their enhanced permeability and retention (EPR) effect at the tumour site can be greatly enhanced and their blood circulation half-lives can be prolonged.156,157 The “stealth” features of PEGylated particles are generally ascribed to the steric hindrance and repulsion effects of PEG chains against blood proteins and macrophages, which are closely correlated to the PEG molecular weight, surface chain density and conformation. Surface chain density and conformation of the PEG chain are two interrelated parameters. The “mushroom” and “brush” conformations could form at low and high surface PEG chain densities, respectively. The different conformations and molecular weights of PEG chains would directly affect their flexibility and hydrophilicity, respectively, and consequently their steric repulsion against blood proteins and macrophages.26,27 One challenge is to find the optimal PEG molecular weight and chain density on MSNPs for both the minimum nonspecific binding to blood proteins and the minimum uptake by human macrophages. Tsai et al. reported the synthesis of PEG-functionalized MSNPs that could be targeted to breast cancer cells overexpressing the HER2/neu receptor under in vitro conditions.28,29 In such work, the targeting monoclonal antibody was first linked to PEG (molecular weight 5000) before covalent attachment to thiol-functionalized MSNPs. The non-selective uptake of MSNPs was noticeably reduced when the particles were covered by PEG.Recently, He et al. studied the influence of PEG molecular weight and packing density on the adsorption of serum proteins to MSNPs, whose PEG chains were linked to the MSNPs using silane coupling chemistry.158 From the results derived from this work it was concluded that PEG molecular weights of 10000–20000 were optimum for minimizing non-specific protein adsorption. In view of these results, it could be postulated that the optimum PEG molecular weight may be a function of the chemistry used to covalently link PEG to the MSNP surface.
3.2. Targeting agents
The abnormally large fenestrations of tumour vasculature and the inefficient lymphatic drainage allow MSNPs exit the blood vessels and accumulate at the tumour site by passive targeting via the EPR effect.159–161 The diffusion rate in the extracellular spaces of the tumour is governed by the size and the surface charge of the particles. Although different surface modifications, such as PEGylation, can reduce unspecific interactions with proteins and thus increase the circulation times, active cellular uptake at the tumour site and enhanced retention times can be further facilitated by active targeting. As mentioned above, targeting is especially relevant in the context of cancer therapies, as most of the commonly used anticancer drugs have serious side-effects due to unspecific action on healthy cells. The selectivity is a function of the ability of the NPs to be internalized by the targeted cell population. Active targeting requires the knowledge of which receptors are overexpressed on the outer cell membrane for the given cancer type.Therefore, different moieties, such as peptides, antibodies or more simple molecules, such as folic acid can be used as targeting agents. The folate receptor (FR), for instance, has been found to be overexpressed on the surface of several cancer cell types, such as ovarian, endometrial, colorectal, breast, lung, renal cell carcinoma, brain metastases derived from epithelial cancer, and neuroendocrine carcinoma.162,163 Because of this distinctive characteristic between normal and cancer cells, folic acid (FA) has emerged as an attractive targeting ligand for selective delivery.164 To date, MSNPs have been functionalized with different ligands, such as sugar moieties,106,165–168 folic acid,22,72,144,148,150,165,169,170 monoclonal antibodies28 and DNA aptamers171 for active targeting purposes. The effectiveness of the active targeting depends on the capability of the ligand linked to the MSNP to bind to the cell-surface receptors to trigger receptor-mediated endocytosis. Such effectiveness can be assessed by carrying out appropriate in vitro experiments, which should include the following stages:8 (i) quantification of uptake of non-targeted and targeted MSNP systems; (ii) selective uptake in receptor-expressing cells versus cells not expressing the targeted receptor; (iii) verify the internalization of the MSNP; (iv) verify the uptake mechanisms, where the interaction between the ligand linked to the MSNP and the targeted receptor is perturbed by the presence of increasing concentrations of free ligand. More in-depth studies regarding the uptake mechanism should be performed by blocking the selective endocytosis processes; (v) the functionality of the developed system should be tested by model drugs on cells expressing different levels of the targeted receptor; (vi) further proof for specificity of the targeted carrier should be obtained by carrying out the analysis in co-culture conditions with different cell types expressing different levels of the targeted receptor in an attempt to mimic the complexity of cellular organization and composition in vivo. Critical evaluation of targetability of MSNPs in properly designed biological settings in vitro will provide relevant information and speed up the transit from bench to bedside, while keeping in vivo testing at reasonable levels.
To date, there are only two reported in vivo studies on the biodistribution72 and tumour suppression efficacy172 of targeted vs. non-targeted MSNPs, both reported by the group of Tamanoi and Zink. In the former study they were the first to demonstrate that MSPNs are effective for antitumor drug delivery and that the tumour suppression was significant with a subcutaneous human breast cancer xenograft in mice.72 For this purpose, fluorescent MSNPs (FSNPs) with sizes in the 100–130 nm range were synthesized. Both folic-acid-conjugated (FA-FSNPs) and non-folic-acid-conjugated (FSNPs) MSNPs were used. The authors investigated the effect of FA conjugation on the cellular uptake of NPs using two breast cancer cell lines, with (SK-BR-3 cells) and without (MCF10F) enhanced expression of folate receptor. The results indicated that although the cellular internalization of FMSNs was observed in both cell-lines, the highest amount of internalized FA-FMSNs was found in cells overexpressing folate receptor (SK-BR-3 cells). The up-regulated folate receptor expression on SK-BR-3 cells, which may facilitate the recognition of the FA-conjugated MSNPs and enhance the uptake through folate-receptor-mediated endocytosis was also confirmed after loading particles with the antitumor drug camptothecin (CPT). So, the next step was to investigate the effect of CPT-loaded FMSNs and CPT-loaded FA-FMSNs in human tumour xenografts in mice. Surprisingly, conjugation with FA on the surface showed a slight, but not significant increase in the tumour-suppressing effect. The authors suggested two reasons to explain these unpredicted results: first, the high dosages and large number of injections carried out may suppress the tumours as quickly so that could be masking the enhanced effect of using FA-FMSNs; second, experiments were carried out in xenografts of human breast cancer cells MCF-7, whereas in vitro tests were carried out in SK-BR-3 cells. Therefore, the specific binding between FA-FMSNs and MCF-7 cells might not be strong enough to promote a significant difference in tumour-suppressing ability in animals.
To further examine the effects of targeting MSNPs, these authors have reported in a second article an animal tumor study of MSNPs on pancreatic cancer xenografts, using two different mice species.172 In this study it is shown that although CPT-loaded MSNs are effective in tumor growth inhibition, CPT-loaded FMSN is much more effective, pointing to the enhancement of tumor inhibition efficacy by adding a targeting moiety to MSNs. The authors also examined different dosages to obtain dose-dependent inhibition of tumor growth and, moreover, their preliminary observation that MSNs are excreted in the urine was addressed.
3.3. Imaging
Non-invasive imaging techniques are of paramount importance in clinical diagnostics. Imaging technologies benefit greatly from using nanoparticle-based contrast agents with the following properties: (i) forming stable colloidal solutions in diverse in vitro and in vivo environments; (ii) chemical stability under diverse physiological conditions; (iii) limited nonspecific binding to avoid macrophagocytic system uptake; (iv) owing programmed clearance mechanisms; (v) high sensitivity and selectivity for the target (i.e. antigen, cell or tissue); (vi) exhibiting good image contrast, with high signal-to-noise ratio; (vii) long circulation time in the blood if administered intravenously.23,33,173–175 MSNPs-based contrast agents are promising platforms to fulfil the above-mentioned prerequisites. Moreover, MSNPs are effectively “transparent”, i.e. they do not absorb light in the near-infrared (NIR), visible, and ultraviolet regions or interfere with magnetic fields. MSNPs are mainly used for optical imaging (OI), magnetic resonance imaging (MRI), or a combination of both modalities. OI is a relative low cost technology that allows for rapid screening, while MRI can offer high resolution and the capability to simultaneously obtain physiological and anatomical information.MSNPs have arisen as powerful nanoplatforms to design novel OI contrast agents. Thus, the incorporation of dye molecules into MSNPs has been carried out to investigate cellular internalization and cell tracking.5,9,66,145,147,176 For instance, Rosenholm et al. developed MSNPs incorporating both fluorescent and targeting moieties.144 In a first step MSNPs were first functionalized by surface hyperbranching polymerization of poly(ethylene imine) (PEI). Then, a fluorescent dye, in this case fluorescein isothiocyanate (FITC), was covalently linked to PEI, making MSNPs visible by fluorescence microscopy and flow cytometry. Folic acid (FA), which was also covalently linked to the hyperbranched PEI layer, was used as the targeting ligand and the cancer-specific internalization of these particles was tested on tumour and healthy cells. The presence of FITC allowed use of flow cytometry to quantify the mean number of MSNPs internalized per cell. The biospecifically tagged MSNPs–PEI–FITC–FA system was shown to be non-cytotoxic and able to specifically target folate receptor-expressing cancer cells also under co-culture conditions. Later on, the same research group evaluated the intracellular drug delivery ability of this system. For this purpose they used two hydrophobic fluorophores, DiI (1,1′-dioactadecyl-3,3,3′,3′-tetramethindocarbocyanine perchlorate) and DiO (3,3-dioactadecyoloxacarbocyanine perchlorate), as model drugs, which made it possible to monitor the intracellular release by confocal fluorescence microscopy. Likewise, the use of fluorophores as model cargo allowed quantification of the intracellular delivery using flow cytometry. In vitro assays demonstrated that the nanoparticles were taken up by receptor-mediated endocytosis followed by accumulation in the endosomal compartment and subsequent release of cargo into the interior of the cell. Besides the selectivity of the developed nanoparticles for cancer cells, the incorporated agent was able to escape from the endosomes into the cytoplasm, which is essential for successful intracellular delivery.
However, it is well known that OI usually suffers from the attenuation of photon propagation in living tissue and poor signal-to-noise ratio due to tissue autofluorescence. To overcome these limitations, Lee and co-workers developed NIR MSNPs-based probes.177 The authors entrapped indocyanine green (ICG), an optical contrast agent exhibiting characteristic fluorescent excitation and emission wavelengths in the NIR window, into MSNPs by electrostatic interactions with trimethylammonium groups incorporated into the silica matrix, affording MSNP–TA–ICG. MSNPs prevented aggregation of ICG molecules and protected them from degradation. The authors evaluated the biodistribution of this OI probe in rat and mouse models. Optical images revealed that after intravenous injection, the MSNPs rapidly accumulated in liver, followed by the kidneys lungs, spleen and heart. This was the first report of MSNPs functionalized with NIR-ICG for in vivo OI. Later on, the same research group evaluated the role of the surface charge of ICG-loaded MSNPs on their biodistribution, clearance from circulation, and excretion.178 MSNPs were incorporated with ICG via covalent or ionic bonding, to derive comparable constructs of different net surface charge. The results showed that tailoring the surface charge of MSNs permits to control excretion rates and biodistribution of MSNPs, a functionality that could lead their widespread clinical use as targetable contrast agents and traceable drug delivery platforms.
Very recently, Lin and co-workers have reported a potential T1 positive contrast agent based on luminescent rattle-type mesoporous silica microspheres.181 The system is a multifunctional composite microsphere that integrates several advantages of mesoporous, luminescence and temperature responses into one single entity. Firstly, the hollow mesoporous silica capsules were fabricated via a sacrificial template route. Then, Gd2O3:Eu3+ luminescent nanoparticles are incorporated into the internal cavities to form rattle-type mesoporous silica nanocapsules by an incipient wetness impregnation method. Finally, the rattle-type capsules serve as a nanoreactor for successfully filling a temperature-responsive hydrogel via photoinduced polymerization of PNIPAM-based hydrogel into the inner cavity of the rattle-type mesoporous silica spheres. For in vitro magnetic resonance imaging (MRI), the sample showed the promising spin-lattice relaxation time (T1) weighted effect. Moreover, the multifunctional carriers exhibited a remarkable positive temperature sensitive on–off modulation for indomethacin (IMC) release.
4. Magnetic mesoporous silica nanoparticles
4.1. Magnetic nanoparticles in nanomedicine
Magnetic nanoparticles (mNPs) are an important class of nanomaterials that consist of typical magnetic elements, such as iron, nickel, cobalt, manganese, chromium and gadolinium, as well as their chemical compounds. Furthermore, magnetic particles with controllable size in the nanometre range also possessing high values of saturation magnetization and magnetic susceptibility are applicable in biotechnology. Recently, these materials have attracted significant interest in biomedical research because they provide advanced therapeutic and diagnostic capabilities with dual-mode manipulation controlled either by using a magnetic field or through surface ligand engineering.182Colloidal nanoparticles that form a ferrofluid have superparamagnetic properties. They are mono-domain particles whose magnetic moments are randomly oriented. Under the action of an external magnetic field, these moments will rapidly rotate to be placed in the direction of the field, so as to increase the magnetic flux. Once the field is ceased, a return to the random initial arrangement is produced without keeping any magnetic remanence.183 In addition, Brownian motion of the nanoparticles is sufficient to cause thermal energy. The aim is to design colloidal fluids stable at physiological pH and salinity, in which mNPs do not form aggregates able to impair circulation in the bloodstream. To accomplish this requirement, the dimension of the NPs should be sufficiently small to avoid precipitation due to gravitation forces, and the charge and surface chemistry must provide both steric and coulombic repulsions, which can be achieved by convenient functionalization or coating of the mNPs. These particles may achieve high values of magnetization under the action of body-tolerated magnetic field intensities and frequencies, eventually losing the magnetization on suspending the magnetic field.
From a therapeutic point of view, the suitability of magnetic nanomaterials is largely determined by three factors: toxicity, magnetic performance and biocompatibility.35,184 The inorganic nanocrystals based on non-toxic elements that offer a high potential for several uses in nanomedicine are colloidal superparamagnetic IONPs, such as magnetite (Fe3O4) or its oxidized form maghemite (γ-Fe2O3). The characteristics and properties of IONPs in terms of magnetic susceptibility, narrow size distribution, superparamagnetic behaviour, surface chemistry and toxicity are designed in their synthesis.35,37,38 Size, charge and surface chemistry of the particles are very important factors that will strongly influence the properties of the mNPs to be used in bioapplications.185–187
As aforementioned, therapeutic and diagnostic applications are the main biomedical uses of mNPs.188 Remarkably, the superparamagnetic behaviour provides the final biomaterial with multifunctionality through magnetic targeting in several biotechnological applications, such as bioseparation,189 magnetic guided drug delivery, hyperthermia treatment, magnetic resonance imaging and magnetofection.
Magnetic nanoparticles can be synthesized with controlled size from a few to tens of nanometres, therefore being smaller than or in a similar range as a cell (10–100 μm), a virus (20–450 nm), a protein (5–50 nm) or a gene (2 nm wide and 10–100 nm long). This fact means that they can interact with biological material and can even be attached to biological molecules that may direct the mNPs to the site of interest in the body.190 Secondly, the particle magnetism provides the chance for manipulation by an external magnetic field gradient. This feature allows either transportation or immobilization of nanoparticles and nanoparticle/biological entity composites, for the release of drugs, genes or proteins.191 Finally, mNPs can respond to the action of alternating magnetic fields, so there is a transfer of energy from the field to the particle. For example, the nanoparticle could be used to transmit certain amounts of thermal energy into tumour cells, which forms the basis of antitumour therapy by hyperthermia.192,193
Regarding magnetic resonance imaging (MRI), as explained in Section 3.1.3,179 superparamagnetic nanoparticles represent a class of nuclear magnetic resonance (NMR) contrast agents that are referred to as T2 (transversal relaxation time) contrast agents as opposed to T1 (longitudinal relaxation time) agents, such as paramagnetic gadolinium(III) chelates. A current trend regarding the use of magnetic NPs is to integrate in a multifunctional nanosystem simultaneous imaging and therapy,36,194 or to generate multimodal imaging probes that integrate into a single system a mNP, radionuclide, an optical tag and a targeting moiety.155
When mNPs are combined in gene vectors it is possible to apply the same principles of magnetically targeted drug delivery to the gene transfection methods. The viral and non-viral alternatives of this technology were demonstrated by Scherer, Plank and co-workers, who coined the term magnetofection.40,195 The magnetofection technique consists of the association of superparamagnetic NPs with molecules able to interact with DNA fragments facilitating DNA binding and protection during extra- and intracellular trafficking. Then, the coupling of genetic material, DNA fragments, plasmids (pDNA) or small interfering RNA (siRNA), to mNPs produces the magnetic vector, which is focused on the target site or cells via a magnetic field gradient. This technique improves the uptake and expression of DNA since the transfection into cells can be assisted by the application of an external magnetic field, which targets and reduces the duration of the gene delivery, enhancing the efficiency of the DNA vector. In a recent example developed by our group,196 poly(propyleneimine) dendrimers were covalently bonded to maghemite nanoparticles, thus obtaining nanosystems that act as non-viral vectors for in vitro gene magnetofection.
Methods of surface modification to obtain magnetic hybrid composites are usually employed to provide the nanoparticles with functionality and biocompatibility, and also stabilizing the colloid at the same time. Encapsulation of the iron oxide nanoparticles in either organic, such as polymeric surfactants, or inorganic matrices, mainly silica, may prevent the flocculation or the aggregation of the nanoscale particulate. Furthermore, the modified surface can impart non toxicity and bear functional groups to allow the covalent grafting of biomolecules, therapeutic agents or specific ligands for targeting.
Inorganic coatings present some advantages over polymeric matrices, such as chemical and mechanical stability and the lack of swelling or porosity changes with the pH. They also protect doped molecules (enzymes, drugs, etc.) against denaturalisation induced by extreme pH and temperature. Many of the systems proposed in the literature are based on core–shell settings, in which species such as silica or gold197 coat the magnetic nanocrystals.
The silica coating on a magnetic core leads to stable and biocompatible ferrofluids in a wide range of pH and at high electrolyte concentrations. This coating also prevents oxidation and degradation of the mNPs during and after synthesis. An advantage of having a surface enriched in silica is the presence of surface silanol groups that can easily react with silane coupling agents and provide an ideal anchorage for covalent binding of specific ligands. The preparation of core–shell magnetic silica nanoparticles involves the synthesis of the mNPs and a series of reactions to grow the coating silica layer based on the Stöber method.198 In the synthetic procedure known as the Stöber method the silica species grow in alkaline mixtures of ethanol and water.
In addition to core–shell structures, magnetic nanocrystals can be encapsulated into the mesostructured network of silica nanospheres.5 Hence, the MSNPs with superparamagnetic IONPs embedded in the matrix can be externally manipulated by using magnetic fields, used for both optical and MRI, and used to store and deliver chemotherapeutic drugs into cancer cells.
Currently, different possibilities for the combination of mesoporous silica nanoparticles and magnetic nanoparticles that further extend their functionalities are gaining great attraction in the field of nanomedicine.199 The different possibilities of combination between mesoporous silica and mNPs are addressed in the following subsections.
4.2. Magnetic nanoparticles@mesoporous silica: core–shell structures
Some examples in which a mesoporous silica coating acts as an inorganic shell of mNPs are detailed to illustrate the multifunctionality of these systems.The group of Lin has reported core–shell multifunctional drug carrier systems with mesoporous, magnetic and luminescent properties. These composites can act as a multifunctional drug carrier system, which can perform the targeting and monitoring of drugs simultaneously. In a first study,200 hydrothermally synthesized Fe3O4 microspheres were encapsulated with nonporous silica, giving rise to a monodisperse spherical silica-coated magnetite core. This core was successfully encapsulated by a further layer of ordered mesoporous silica as the shell, using CTAB as a template. The surface of the outer silica shell was further functionalized by the deposition of YVO4:Eu3+ phosphors, giving rise to the sandwich structured material Fe3O4@nSiO2@mSiO2@YVO4:Eu3+. A drug release test using ibuprofen as the model drug indicated that the multifunctional system shows drug-sustained properties. This composite is an ideal system for targeting drug delivery via its superparamagnetic feature and the luminescence property can be used to track the position and release efficiency of the drugs. In this former study, relatively larger Fe3O4 submicrospheres (300 nm) were selected instead of smaller ones (<50 nm) in order to obtain larger magnetization for drug targeting. A well-known efficient phosphor (YVO4:Eu3+) was also used, instead of up converting (near infrared-excitable) lanthanide ion (Er3+), to achieve suitable luminescence intensity to correlate it with the extent of drug release. Enlightened by this study, further research was extended to the effects of the magnetite size on the drug storage/release property, and the combination of up-conversion nanophosphors with the magnetic nanoparticles (<50 nm) that would be very promising for in vivo biomedical labels and controlled drug release systems using 980 nm near infrared as the excitation source (the optical window of human tissues).
Then, monodisperse core–shell-structured Fe3O4@nSiO2@mSiO2@NaYF4:Yb3+,Er3+/Tm3+ nanocomposites were synthesized as previously described in the former study through a simple two-step sol–gel process.201 The nanocomposites showed typical ordered mesoporous characteristics and a monodisperse spherical morphology with narrow size distribution (around 80 nm). In addition, they exhibited high magnetization (38.0 emu g−1, thus it is possible for drug targeting under a foreign magnetic field) and unique up-conversion emission (green for Yb3+/Er3+ and blue for Yb3+/Tm3+) under 980 nm laser excitation, even after loading with drug molecules. Drug release tests suggested that the multifunctional nanocomposites have a controlled drug release property. Interestingly, the up conversion emission intensity of the multifunctional carrier increases with the released amount of ibuprofen model drug, thus allowing the release process to be monitored and tracked by the change of photoluminescence intensity.
Among the core–shell structures a promising T2 contrast agent for magnetic resonance imaging has been developed by Yeh and co-workers.202 The system consists of a magnetic Fe3O4 nanoparticle core covered by a mesoporous silica shell, where chelating ligands for Gd3+ ions have been anchored. The Gd3+ ions within the silica shell enhance the transversal relaxation of the magnetite NPs having an effective magnetic resonance imaging effect. In short-term cytotoxicity studies this T2 contrast agent showed no adverse effect on organ tissues or Kupffer cells, indicating that the nanosystem is biocompatible.
A multifunctional core–shell structured MSNPs for simultaneous magnetic resonance and fluorescence imaging, cell targeting and photodynamic therapy has been synthesized as a promising material for cancer diagnosis and therapy.203 Superparamagnetic magnetite NPs and fluorescent dyes are co-encapsulated inside nonporous silica NPs as the core to provide dual-imaging capabilities (MR and optical). Then, the photosensitizer molecules, tetra-substituted carboxyl aluminium phthalocyanine (AlC4Pc), are covalently linked to the mesoporous silica shell and exhibit excellent photo-oxidation efficiency. Finally, the surface modification of the core–shell silica nanoparticles with folic acid enhances the delivery of photosensitizers to the targeting cancer cells that overexpress the folate receptor, thereby decreasing their toxicity to the surrounding normal tissues.
4.3. Rattle-type magnetic nanoparticles@hollow mesoporous silica spheres
This kind of magnetic mesoporous silica nanocomposite is based on core–shell structures where iron oxide nanoparticles are encapsulated within a mesoporous silica shell possessing a hollow interlayer in the middle. The inner Fe3O4 movable core endows the spheres with magnetic property and the outer mesoporous silica shell provides the pathway for guest molecules to diffuse into or out from the hollow core. Therefore, this kind of magnetic hollow mesoporous sphere is a very promising candidate material for application in a targeted drug delivery system.The group of Shi reported a novel kind of rattle-type hollow magnetic mesoporous sphere with Fe3O4 particles encapsulated in the cores of mesoporous silica microspheres.204 Such composite spheres were fabricated by sol–gel reactions on hematite particles followed by cavity generation with hydrothermal treatment and H2 reduction of the hematite cores to magnetite. The ring-shaped cavity between the magnetic core and the mesoporous silica shell was generated by selectively etching away the middle silica interlayer in a simple hydrothermal treatment process. The capability as drug carriers was explored using ibuprofen as the model drug. The prepared rattle-type hollow magnetic mesoporous spheres exhibited a significantly higher storage capacity of drug because of the rattle-type structure than the corresponding magnetic/mesoporous spheres without the cavities. Moreover, the release experiments showed a sustained-release behavior of IBU from the drug storage system, following Fick's law.
Then, following this previous work on synthesis, Shi and co-workers synthesized a multifunctional rattle-type magnetic mesoporous silica nanosphere system.205 The surface properties of the former nanospheres were modified with poly(ethylene glycol) (PEG) as a biocompatible polymer and folic acid as a cancer-cell-specific ligand, with the aim of specifically targeting cancer cells. The final system combined magnetic and receptor-specific targeting effects, magnetic resonance imaging and drug delivery into one. The nanocomposites specifically targeted cancer cells overexpressing FA receptors and displayed very low in vitro toxicity and negligible hemolytic activity against MCF-7 and HeLa cells, which is favorable for further biological applications. Furthermore, docetaxel (DOC), a highly hydrophobic anticancer drug, was loaded into the nanospheres and the cytotoxic effect of the DOC-loaded spheres on cells that overexpress the folate receptor was demonstrated. In addition, the in vivo transport of RMMSN-PEG/FA to a designated organ under an external magnetic field was also preliminarily investigated. In summary, such a multifunctional system may be used for the diagnosis and targeted therapy of various types of cancer.
Kaskel and co-workers have developed an efficient colloidal carbon sphere templating route to prepare rattle-type Fe3O4@SiO2 hollow mesoporous spheres with large cavities.206 The spheres were well monodisperse and nearly uniform in dimensions with a particle size of ca. 900 nm. The thickness of the mesoporous silica shell was about 100 nm and only one Fe3O4 particle of ca. 100 nm in diameter was encapsulated in each hollow mesoporous silica sphere. In this synthetic route the first step involved the preparation of the colloidal carbon spheres adsorbed with iron precursor by a one-pot hydrothermal treatment. In the next step, the organosilicate-incorporated silica shells were deposited on the colloidal carbon spheres through the simultaneous sol–gel polymerization of tetraethoxysilane (TEOS) and n-octadecyltrimethoxysilane (C18TMS). Finally, the rattle-type Fe3O4@SiO2 hollow mesoporous spheres were obtained after the calcination to remove the carbon templates and the organic groups of C18TMS, followed by reduction under hydrogen atmosphere. Using aspirin as a model drug, the Fe3O4@SiO2 hollow mesoporous spheres showed high drug loading capacity and sustained release property governed by Fickian diffusion.
In a more recent paper, these researchers reported the preparation of rattle-type Fe3O4@SiO2 hollow mesoporous spheres with different particle sizes, different mesoporous shell thicknesses, and different levels of Fe3O4 content using colloidal carbon spheres as templates.207 The effects of particle size and concentration of Fe3O4@SiO2 hollow mesoporous spheres on cell uptake and their in vitro cytotoxicity to HeLa cells were evaluated. The spheres exhibited relatively fast cell uptake and concentrations of up to 150 μg mL−1 showed no cytotoxicity, whereas a concentration of 200 μg mL−1 showed a small amount of cytotoxicity after 48 h of incubation. Doxorubicin hydrochloride (DOX), an anticancer drug, was loaded into the Fe3O4@SiO2 hollow mesoporous spheres, and the DOX-loaded spheres exhibited a somewhat higher cytotoxicity than free DOX. These results indicated the potential of Fe3O4@SiO2 hollow mesoporous spheres for drug loading and delivery into cancer cells to induce cell death.
4.4. Magnetic nanoparticles encapsulated within mesoporous silica nanospheres
As aforementioned, systems in which silica nanospheres encapsulate a significant amount of mNPs have been investigated. A large amount of magnetic cores can be introduced during the synthesis of MSNPs (Fig. 5), so the final material can reach temperatures in the range of hyperthermia under the action of alternating magnetic fields.208 Therefore, it is also feasible to use hyperthermia combined with antitumor drugs to treat certain cancerous tissues without affecting healthy tissues.54,209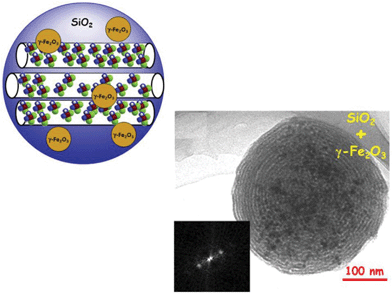 | ||
| Fig. 5 A schematic representation of MSNPs encapsulating γ-Fe2O3 mNPs and a Transmission Electronic Microscopy (TEM) image of this system and its corresponding Fourier Transform (FT).54 | ||
Both the mesoporous ordering and the magnetic properties of the biomaterial can be adjusted by varying the ratio of surfactant and silica, the kind of surfactant and the quantity of encapsulated mNPs. Different synthetic approaches have been followed for the preparation of these systems, such as aerosol-assisted methods and the modified Stöber method. The latter offers some advantages, such as the possibility of reaching homogeneous nanoparticle size distribution and tailor-made pore ordering.210
In the temperature-controlled pyrolysis of an aerosol including silica precursors and magnetic nanoparticle ferrofluid at a suitable pH, the pore size distribution in the carriers can be tuned by the use of different surfactant molecules in the self-assembly process. When the non-ionic Pluronic P123 was employed as structure directing agent, 5.6 nm pore size matrices were produced by an aerosol-assisted method (Fig. 5). In this case, encapsulated mNPs kept crystallinity and superparamagnetic behaviour after the silica coating, so the original properties of mNPs are retained after the pyrolysis process. The silica NPs presented an ordered majority hexagonal array of pores, with high values of specific surface area and pore volume and the ability to load and subsequently release a model drug was evidenced.209
 | ||
| Fig. 6 TEM images of pure silica and silica incorporating γ-Fe2O3 MSNPs. For many of the applications in nanomedicine it is possible to decorate the outermost surface of the MSNPs with targeting ligands able to specifically interact with the cell receptors present in the target cell. | ||
The biocompatibility of highly magnetic MSNPs and their ability to conduct magnetic hyperthermia upon exposure to a low-frequency alternating magnetic field have been evaluated in a research work by our group.211 The assays performed in vitro using human cells of a cancerous nature showed that the particles are efficiently internalized by human A549, Saos-2 and HepG2 cells, and that are excluded from the nuclear compartment. Moreover, the particles do not interfere with morphological features, metabolic activity or endogenous heat-shock response, suggesting a good biocompatibility of the material. Magnetic hyperthermia experiments showed the ability to control the temperature rise in the cell culture environment upon the treatment with magnetic MSNPs and alternating magnetic field exposure, thus generating heat treatments that severely compromise cell survival. The possibility of fine-tuning the heating power output, together with efficient uptake by tumour cells in vitro, makes the magnetic MSNPs a promising agent for hyperthermia treatments combined with intracellular delivery of chemotherapeutic drugs, aimed toward remission of solid tumours. It is also feasible to graft targeting agents on the surface of MSNPs that could specifically interact with cell receptors to be then internalised in the cell to release their cargo, as schematically depicted in Fig. 6.
Applications in drug delivery and optical imaging have been proposed for magnetic MSNPs covalently bonded with near-infrared (NIR) luminescent lanthanide complexes. The composites have been prepared via incorporation of an alkoxysilane chelate ligand for Nb3+ and Yb3+ in the magnetic mesoporous framework so finally exhibit magnetic separation ability and NIR luminescent properties.212
4.5. Mesoporous silica nanoparticles decorated with magnetic nanoparticles
The functionalities associated to the mNPs can also be introduced in the MSNPs by assembling the superparamagnetic IONPs onto the outer surface of MSNPs. In this sense, multifunctional nanosystems for simultaneous T2 MR and optical imaging and drug delivery have been developed. The superparamagnetic property of magnetite nanocrystals enables the NPs to be used as a contrast agent in magnetic resonance imaging, and a fluorescent molecule in the silica framework imparts optical imaging modality. In addition, an anticancer drug can be introduced in the porous system to induce cell death.Hyeon and co-workers have used a method in which dye-doped MSNPs are first prepared by incorporating an organic dye into the silica walls of MNSPs by co-condensation of 3-aminopropyltriethoxysilane-modified fluorescein isothiocyanate or Rhodamine B isothiocyanate during the sol–gel method, i.e., via a one-pot synthesis. Subsequently, the surface of the MSNPs is functionalized with amine groups. The ligands of superparamagnetic magnetite nanocrystals, having 2-bromo-2-methylpropionic acid, are assembled by condensation reaction with the amine groups on the surface of the MSNPs. Finally, to make the system more biocompatible and with higher colloidal stability, the outer surface is coated with PEG. Although characterization studies revealed that pores seemed to be partially blocked by Fe3O4 nanocrystals, the surface area of Fe3O4-MSNPs was sufficiently large for use as a drug delivery vehicle.
In their first approximation213 amine functionalization of the MSNPs was carried out by treating the dye-doped MSNPs with 3-aminopropylsilane. For the drug delivery assays the chemotherapeutic agent doxorubicin was loaded into the pores of the magnetic MSNPs.
An improvement of the initial system has been achieved by immobilizing pH-responsive hydrazone bonds on the surface of the MSNPs, therefore a pH-sensitive drug release has been achieved.214 The pH-sensitive bond was introduced by first functionalizing the MSNPs with 3-bromopropylsilane and subsequently treating them with an excess of hydrazine. The resulting hydrazine-functionalized MSNPs were conjugated via the pH-sensitive hydrazone bonding with doxorubicin as the anticancer drug. A time and pH-dependent doxorubicin profile of the system is verified in the drug delivery assays, and in vitro cell culture experiments, followed by confocal laser scanning microscopy, demonstrate internalization of the magnetic MSNPs in the cytoplasm and doxorubicin release to the nucleus. The cytotoxic effect of the system was tested on breast cancer cells showing a cytotoxic efficacy that increases with the system concentration in the cell culture, whereas no cytotoxicity was found for the system without doxorubicin.
As previously detailed in Section 2 of this review, mNPs attached on the outer surface of the MSNPs can act as caps for retaining a therapeutic drug inside the mesopores. These nanogated systems are usually stimuli-responsive so the mNPs can be removed via magnetic,134,135 pH82 or redox activation,103 therefore releasing the drugs on demand.
5. Future prospects
Mesoporous silica nanoparticles (MSNPs) have emerged as excellent multifunctional nanoplatforms to design smart delivery nanodevices. However, their application in clinical nanomedicine is still in its infancy. Multifunctional stimuli-responsive drug and gene delivery nanodevices for nanomedicine must be designed attending to the following issues: biocompatibility and absence of toxicity of all of its components, biocompatible trigger stimuli and efficiency to target the suitable cell or tissue, considering the huge complexity of the body. Significant achievements derived from in vitro experiments have been made during the last decade. In addition, in vivo findings are encouraging from the perspective of moving the MSNPs platforms into clinical trials.Nanomedicine is currently evolving to encompass integrated, biocompatible multifunctional nanoplatforms able to play dual roles as diagnostic and therapeutic agents. This trend has experienced so burgeoning expansion that the term “theranostic”, derived from thera(py) + (diag)nostic has been coined. Within this milieu much research endeavor is being dedicated to design new multifunctional MSNPs that combine imaging and therapeutic agents for both diagnostics and therapy. The mesoporous cavities of MSNPs can host and protect a wide variety of organic molecules, such as drugs, proteins, nucleic acids or photosensitizers for photodynamic therapy (PDT). These moieties can be just adsorbed or covalently linked to the inner mesopore walls. In addition, the silica framework can incorporate organic molecules as optical imaging (OI) or magnetic resonance imaging (MRI) contrast agents. Molecular nanogates can be covalently linked to the pore outlets blocking the mesopores and preventing the premature departure of entrapped molecules. The aperture of the nanogates in response to internal or external stimuli triggers cargo release in the place where needed. Moreover, the outermost surface of MSNPs can be decorated with targeting ligands for specific cellular uptake, functional groups to tune surface charge, polymeric coatings such as polyethylene glycol (PEG) or stimuli-responsive polymers.
Of foremost interest is the possibility of incorporating magnetic nanoparticles (mNPs) into MSNPs, since the synergistic combination of both components would permit design of theranostic nanosystems. mNPs can play several roles when combined with MSNPs, including: (i) acting as contrast agents for MRI; (ii) acting as thermoseeds for hyperthermia treatment of cancer under the action of alternating magnetic fields; (iii) acting as magnetic-responsive gatekeepers to avoid premature release of chemotherapeutic agents entrapped within the mesoporous cavities; (iv) permitting magnetic guidance of the nanosystem to the target tissue.
Significant advances derived from in vitro experiments and scarcely in vivo tests have been attained. However, we are just at the beginning of an exciting scientific journey and much research endeavour is still needed to find answers to several open questions that permit the transit from bench to bedside.
Acknowledgements
We thank financial support by the Spanish CICYT through project MAT2008-00736, Spanish National CAM project S2009MAT-1472 and the Network of Excellence CSO2010-11384-E. We also thank J. M. Moreno for his technical help.Notes and references
- S. Saha, K. C. F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS.
- B. G. Trewyn, S. Giri, I. I. Slowing and V. S. Y. Lin, Chem. Commun., 2007, 3236–3245 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS.
- K. K. Coti, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoddart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251–6257 RSC.
- Y. Zhao, J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, Expert Opin. Drug Delivery, 2010, 7, 1013–1029 CrossRef CAS.
- J. M. Rosenholm, C. Sahlgren and M. Linden, Nanoscale, 2010, 2, 1870–1883 RSC.
- J. Rosenholm, C. Sahlgren and M. Linden, J. Mater. Chem., 2010, 20, 2707–2713 RSC.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS.
- M. Vallet-Regí and E. Ruiz-Hernández, Adv. Mater., 2011, 23, 5177–5218 CrossRef.
- M. Vallet-Regí, E. Ruiz-Hernández, B. González and A. Baeza, J. Biomater. Tissue Eng., 2011, 1, 6–29 Search PubMed.
- M. W. Ambrogio, C. R. Thomas, Y.-L. Zhao, J. I. Zink and J. F. Stoddart, Acc. Chem. Res., 2011, 44, 903–913 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS.
- S. Dufort, L. Sancey and J.-L. Coll, Adv. Drug Delivery Rev., 2012, 64, 179–189 CrossRef CAS.
- H. Yamada, C. Urata, Y. Aoyama, S. Osada, Y. Yamauchi and K. Kuroda, Chem. Mater., 2012, 24, 1462–1471 CrossRef CAS.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef.
- M. Manzano, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Delivery, 2009, 6, 1383–1400 CrossRef CAS.
- M. Manzano and M. Vallet-Regi, J. Mater. Chem., 2010, 20, 5593–5604 RSC.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673–2685 RSC.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS.
- M. Vallet-Regí, ISRN Mater. Sci., 2012, 608548, DOI:10.5402/2012/608548.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- P. M. Claesson, E. Blomberg, J. C. Fröberg, T. Nylander and T. Arnebrant, Adv. Colloid Interface Sci., 1995, 57, 161–227 CrossRef CAS.
- C.-P. Tsai, C.-Y. Chen, Y. Hung, F.-H. Chang and C.-Y. Mou, J. Mater. Chem., 2009, 19, 5737–5743 RSC.
- M. M. J. Kamphuis, A. P. R. Johnston, G. K. Such, H. H. Dam, R. A. Evans, A. M. Scott, E. C. Nice, J. K. Heath and F. Caruso, J. Am. Chem. Soc., 2010, 132, 15881–15883 CrossRef CAS.
- Y.-S. Lin, Y. Hung, J.-K. Su, R. Lee, C. Chang, M.-L. Lin and C.-Y. Mou, J. Phys. Chem. B, 2004, 108, 15608–15611 CrossRef CAS.
- K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154–2155 CrossRef CAS.
- J.-K. Hsiao, C.-P. Tsai, T.-H. Chung, Y. Hung, M. Yao, H.-M. Liu, C.-Y. Mou, C.-S. Yang, Y.-C. Chen and D.-M. Huang, Small, 2008, 4, 1445–1452 CrossRef CAS.
- K. M. L. Taylor-Pashow, J. Della Rocca, R. C. Huxford and W. Lin, Chem. Commun., 2010, 46, 5832–5849 RSC.
- S. A. Li, H. A. Liu, L. Li, N. Q. Luo, R. H. Cao, D. H. Chen and Y. Z. Shao, Appl. Phys. Lett., 2011, 98 Search PubMed.
- A. J. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- J. R. McCarthy and R. Weissleder, Adv. Drug Delivery Rev., 2008, 60, 1241–1251 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- A. G. Roca, R. Costo, A. F. Rebolledo, S. Veintemillas-Verdaguer, P. Tartaj, T. González-Carreño, M. P. Morales and C. J. Serna, J. Phys. D: Appl. Phys., 2009, 42, 224002 CrossRef.
- T. D. Schladt, K. Schneider, H. Schild and W. Tremel, Dalton Trans., 2011, 40, 6315–6343 RSC.
- F. Scherer, M. Anton, U. Schillinger, J. Henke, C. Bergemann, A. Kruger, B. Gansbacher and C. Plank, Gene Ther., 2002, 9, 102–109 CrossRef CAS.
- D. Kami, S. Takeda, Y. Itakura, S. Gojo, M. Watanabe and M. Toyoda, Int. J. Mol. Sci., 2011, 12, 3705–3722 CrossRef CAS.
- P. R. Gil and W. J. Parak, ACS Nano, 2008, 2, 2200–2205 CrossRef CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- C. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, Adv. Mater., 2004, 16, 1959–1966 CrossRef.
- W. Tan, K. Wang, X. He, X. J. Zhao, T. Drake, L. Wang and R. P. Bagwe, Med. Res. Rev., 2004, 24, 621–638 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- I. Gill, Chem. Mater., 2001, 13, 3404–3421 CrossRef CAS.
- C.-H. Lee, T.-S. Lin and C.-Y. Mou, Nano Today, 2009, 4, 165–179 CrossRef CAS.
- A. Bouamrani, Y. Hu, E. Tasciotti, L. Li, C. Chiappini, X. Liu and M. Ferrari, Proteomics, 2010, 10, 496–505 CrossRef CAS.
- M. Vallet-Regi, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- M. Grün, I. Lauer and K. K. Unger, Adv. Mater., 1997, 9, 254–257 CrossRef.
- Y. Lu, H. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- E. Ruiz-Hernández, A. López-Noriega, D. Arcos, I. Izquierdo-Barba, O. Terasaki and M. Vallet-Regí, Chem. Mater., 2007, 19, 3455–3463 CrossRef.
- M. Colilla, M. Manzano, I. Izquierdo-Barba, M. Vallet-Regi, C. Boissiere and C. Sanchez, Chem. Mater., 2010, 22, 1821–1830 CrossRef CAS.
- C. Boissiere, D. Grosso, A. Chaumonnot, L. Nicole and C. Sanchez, Adv. Mater., 2011, 23, 599–623 CrossRef CAS.
- F. Qu, G. Zhu, S. Huang, S. Li, J. Sun, D. Zhang and S. Qiu, Microporous Mesoporous Mater., 2006, 92, 1–9 CrossRef CAS.
- X.-H. Li, R.-Z. Zhang, X.-D. Yang and H. Zhang, J. Molec. Struct. (THEOCHEM), 2007, 815, 151–156 CrossRef CAS.
- D. Arcos, A. López-Noriega, E. Ruiz-Hernández, O. Terasaki and M. Vallet-Regí, Chem. Mater., 2009, 21, 1000–1009 CrossRef CAS.
- Y. Zhu, J. Shi, H. Chen, W. Shen and X. Dong, Microporous Mesoporous Mater., 2005, 84, 218–222 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- C. Urata, Y. Aoyama, A. Tonegawa, Y. Yamauchi and K. Kuroda, Chem. Commun., 2009, 5094–5096 RSC.
- N. Lang and A. Tuel, Chem. Mater., 2004, 16, 1961–1966 CrossRef CAS.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- I. Slowing, B. G. Trewyn and V. S. Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792–14793 CrossRef CAS.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Small, 2009, 5, 1408–1413 CrossRef CAS.
- B. G. Trewyn, J. A. Nieweg, Y. Zhao and V. S. Y. Lin, Chem. Eng. J., 2008, 137, 23–29 CrossRef CAS.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045–4055 CrossRef CAS.
- Z. Tao, M. P. Morrow, T. Asefa, K. K. Sharma, C. Duncan, A. Anan, H. S. Penefsky, J. Goodisman and A.-K. Souid, Nano Lett., 2008, 8, 1517–1526 CrossRef CAS.
- D.-M. Huang, T.-H. Chung, Y. Hung, F. Lu, S.-H. Wu, C.-Y. Mou, M. Yao and Y.-C. Chen, Toxicol. Appl. Pharmacol., 2008, 231, 208–215 CrossRef CAS.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS.
- Y. Zhao, X. Sun, G. Zhang, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, ACS Nano, 2011, 5, 1366–1375 CrossRef CAS.
- Z. Tao, B. Toms, J. Goodisman and T. Asefa, ACS Nano, 2010, 4, 789–794 CrossRef CAS.
- Z. Tao, G. Wang, J. Goodisman and T. Asefa, Langmuir, 2009, 25, 10183–10188 CrossRef CAS.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 CAS.
- C. Park, K. Oh, S. C. Lee and C. Kim, Angew. Chem., Int. Ed., 2007, 46, 1455–1457 CrossRef CAS.
- L. Du, S. Liao, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 15136–15142 CrossRef CAS.
- H. Meng, M. Xue, T. Xia, Y.-L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690–12697 CrossRef CAS.
- Y.-L. Zhao, Z. Li, S. Kabehie, Y. Y. Botros, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 13016–13025 CrossRef CAS.
- E. Aznar, C. Coll, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, P. Amorós, J. Cano and E. Ruiz, Chem.–Eur. J., 2009, 15, 6877–6888 CrossRef CAS.
- Q. Gan, X. Lu, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and C. Liu, Biomaterials, 2011, 32, 1932–1942 CrossRef CAS.
- R. Liu, Y. Zhang, X. Zhao, A. Agarwal, L. J. Mueller and P. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS.
- L. Chen, J. Di, C. Cao, Y. Zhao, Y. Ma, J. Luo, Y. Wen, W. Song, Y. Song and L. Jiang, Chem. Commun., 2011, 47, 2850–2852 RSC.
- R. Liu, P. Liao, J. Liu and P. Feng, Langmuir, 2011, 27, 3095–3099 CrossRef CAS.
- C.-Y. Hong, X. Li and C.-Y. Pan, J. Mater. Chem., 2009, 19, 5155–5160 RSC.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CAS.
- Y.-Z. You, K. K. Kalebaila, S. L. Brock and D. Oupický, Chem. Mater., 2008, 20, 3354–3359 CrossRef CAS.
- J.-H. Park, Y.-H. Lee and S.-G. Oh, Macromol. Chem. Phys., 2007, 208, 2419–2427 CrossRef CAS.
- S. Zhu, Z. Zhou, D. Zhang, C. Jin and Z. Li, Microporous Mesoporous Mater., 2007, 106, 56–61 CrossRef CAS.
- Y. Zhu, S. Kaskel, T. Ikoma and N. Hanagata, Microporous Mesoporous Mater., 2009, 123, 107–112 CrossRef CAS.
- Q. Fu, G. V. Rama Rao, T. L. Ward, Y. Lu and G. P. Lopez, Langmuir, 2007, 23, 170–174 CrossRef CAS.
- A. Zintchenko, M. Ogris and E. Wagner, Bioconjugate Chem., 2006, 17, 766–772 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2007, 23, 9409–9415 CrossRef CAS.
- M. Keerl, V. Smirnovas, R. Winter and W. Richtering, Macromolecules, 2008, 41, 6830–6836 CrossRef CAS.
- T. Hoare, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 9, 3651–3657 CrossRef CAS.
- A. Baeza, E. Guisasola, E. Ruiz-Hernández and M. Vallet-Regí, Chem. Mater., 2012, 24, 517–524 CrossRef CAS.
- A. Schlossbauer, S. Warncke, P. M. E. Gramlich, J. Kecht, A. Manetto, T. Carell and T. Bein, Angew. Chem., Int. Ed., 2010, 49, 4734–4737 CrossRef CAS.
- E. Aznar, L. Mondragón, J. V. Ros-Lis, F. Sancenón, M. D. Marcos, R. Martínez-Máñez, J. Soto, E. Pérez-Payá and P. Amorós, Angew. Chem., Int. Ed., 2011, 50, 11172–11175 CrossRef CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS.
- F. Torney, B. G. Trewyn, V. S. Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295–300 CrossRef CAS.
- R. Liu, X. Zhao, T. Wu and P. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS.
- Z. Luo, K. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS.
- M. W. Ambrogio, T. A. Pecorelli, K. Patel, N. M. Khashab, A. Trabolsi, H. A. Khatib, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Org. Lett., 2010, 12, 3304–3307 CrossRef CAS.
- H. Kim, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4283 CrossRef CAS.
- C. D. Austin, X. Wen, L. Gazzard, C. Nelson, R. H. Scheller and S. J. Scales, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17987–17992 CrossRef CAS.
- K. G. de Bruin, C. Fella, M. Ogris, E. Wagner, N. Ruthardt and C. Bräuchle, J. Controlled Release, 2008, 130, 175–182 CrossRef CAS.
- S. b. Febvay, D. M. Marini, A. M. Belcher and D. E. Clapham, Nano Lett., 2010, 10, 2211–2219 CrossRef CAS.
- A. M. Sauer, A. Schlossbauer, N. Ruthardt, V. Cauda, T. Bein and C. Bräuchle, Nano Lett., 2010, 10, 3684–3691 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS.
- A. Bernardos, E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, J. M. Barat and P. Amorós, Angew. Chem., Int. Ed., 2009, 48, 5884–5887 CrossRef CAS.
- A. Bernardos, L. Mondragón, E. Aznar, M. D. Marcos, R. n. Martínez-Máñez, F. l. Sancenón, J. Soto, J. M. Barat, E. Pérez-Payá, C. Guillem and P. Amorós, ACS Nano, 2010, 4, 6353–6368 CrossRef CAS.
- A. Agostini, L. Mondragón, C. Coll, E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, E. Pérez-Payá and P. Amorós, ChemistryOpen, 2012, 1, 17–20 CrossRef CAS.
- N. Singh, A. Karambelkar, L. Gu, K. Lin, J. S. Miller, C. S. Chen, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2011, 133, 19582–19585 CrossRef CAS.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS.
- E. Climent, A. Bernardos, R. Martínez-Máñez, Á. Maquieira, M. D. Marcos, N. Pastor-Navarro, R. Puchades, F. Sancenón, J. Soto and P. Amorós, J. Am. Chem. Soc., 2009, 131, 14075–14080 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- V. C. Özalp, F. Eyidogan and H. A. Oktem, Pharmaceuticals, 2011, 4, 1137–1157 CrossRef.
- C.-L. Zhu, C.-H. Lu, X.-Y. Song, H.-H. Yang and X.-R. Wang, J. Am. Chem. Soc., 2011, 133, 1278–1281 CrossRef CAS.
- V. C. Özalp and T. Schäfer, Chem.–Eur. J., 2011, 17, 9893–9896 CrossRef.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS.
- N. K. Mal, M. Fujiwara, Y. Tanaka, T. Taguchi and M. Matsukata, Chem. Mater., 2003, 15, 3385–3394 CrossRef CAS.
- S. Angelos, E. Choi, F. Vogtle, L. De Cola and J. I. Zink, J. Phys. Chem. C, 2007, 111, 6589–6592 CAS.
- D. P. Ferris, Y.-L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS.
- J. Lai, X. Mu, Y. Xu, X. Wu, C. Wu, C. Li, J. Chen and Y. Zhao, Chem. Commun., 2010, 46, 7370–7372 RSC.
- N. Z. Knezevic, B. G. Trewyn and V. S. Y. Lin, Chem. Commun., 2011, 47, 2817–2819 RSC.
- P.-J. Chen, S.-H. Hu, C.-S. Hsiao, Y.-Y. Chen, D.-M. Liu and S.-Y. Chen, J. Mater. Chem., 2011, 21, 2535–2543 RSC.
- E. Ruiz-Hernández, A. Baeza and M. a. Vallet-Regí, ACS Nano, 2011, 5, 1259–1266 CrossRef.
- R. Casasús, E. Climent, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, P. Amorós, J. Cano and E. Ruiz, J. Am. Chem. Soc., 2008, 130, 1903–1917 CrossRef.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS.
- R. Liu, Y. Zhang and P. Feng, J. Am. Chem. Soc., 2009, 131, 15128–15129 CrossRef CAS.
- B. Chang, X. Sha, J. Guo, Y. Jiao, C. Wang and W. Yang, J. Mater. Chem., 2011, 21, 9239–9247 RSC.
- C. Chen, J. Geng, F. Pu, X. Yang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2011, 50, 882–886 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS.
- F. Hoffmann and M. Froba, Chem. Soc. Rev., 2011, 40, 608–620 RSC.
- D. Bruhwiler, Nanoscale, 2010, 2, 887–892 RSC.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197–206 CrossRef CAS.
- Y.-S. Lin, C.-P. Tsai, H.-Y. Huang, C.-T. Kuo, Y. Hung, D.-M. Huang, Y.-C. Chen and C.-Y. Mou, Chem. Mater., 2005, 17, 4570–4573 CrossRef CAS.
- Y.-S. Lin, S.-H. Wu, Y. Hung, Y.-H. Chou, C. Chang, M.-L. Lin, C.-P. Tsai and C.-Y. Mou, Chem. Mater., 2006, 18, 5170–5172 CrossRef CAS.
- J. Lu, M. Liong, S. Sherman, T. Xia, M. Kovochich, A. Nel, J. Zink and F. Tamanoi, NanoBiotechnology, 2007, 3, 89–95 CrossRef CAS.
- J. M. Rosenholm, E. Peuhu, J. E. Eriksson, C. Sahlgren and M. Lindén, Nano Lett., 2009, 9, 3308–3311 CrossRef CAS.
- F. Balas, M. Manzano, P. Horcajada and M. Vallet-Regí, J. Am. Chem. Soc., 2006, 128, 8116–8117 CrossRef CAS.
- J. M. Rosenholm, E. Peuhu, L. T. Bate-Eya, J. E. Eriksson, C. Sahlgren and M. Lindén, Small, 2010, 6, 1234–1241 CrossRef CAS.
- R. Mortera, J. Vivero-Escoto, I. I. Slowing, E. Garrone, B. Onida and V. S. Y. Lin, Chem. Commun., 2009, 3219–3221 RSC.
- C. Tourne-Peteilh, D. Brunel, S. Begu, B. Chiche, F. Fajula, D. A. Lerner and J.-M. Devoisselle, New J. Chem., 2003, 27, 1415–1418 RSC.
- I.-P. Huang, S.-P. Sun, S.-H. Cheng, C.-H. Lee, C.-Y. Wu, C.-S. Yang, L.-W. Lo and Y.-K. Lai, Mol. Cancer Ther., 2011, 10, 761–769 CrossRef CAS.
- F. Jianquan, F. Gang, W. Xiaodan, Z. Fang, X. Yufei and W. Shuizhu, Nanotechnology, 2011, 22, 455102 CrossRef.
- J. Cheon and J.-H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS.
- G. R. Harper, M. C. Davies, S. S. Davis, T. F. Tadros, D. C. Taylor, M. P. Irving and J. A. Waters, Biomaterials, 1991, 12, 695–700 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS.
- F. M. Muggia, Clin. Cancer Res., 1999, 5, 7–8 CAS.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS.
- H. Elnakat and M. Ratnam, Adv. Drug Delivery Rev., 2004, 56, 1067–1084 CrossRef CAS.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS.
- J. Gu, W. Fan, A. Shimojima and T. Okubo, Small, 2007, 3, 1740–1744 CrossRef CAS.
- I. Y. Park, I. Y. Kim, M. K. Yoo, Y. J. Choi, M.-H. Cho and C. S. Cho, Int. J. Pharm., 2008, 359, 280–287 CrossRef CAS.
- D. Brevet, M. Gary-Bobo, L. Raehm, S. Richeter, O. Hocine, K. Amro, B. Loock, P. Couleaud, C. Frochot, A. Morere, P. Maillard, M. Garcia and J.-O. Durand, Chem. Commun., 2009, 1475–1477 RSC.
- O. Hocine, M. Gary-Bobo, D. Brevet, M. Maynadier, S. Fontanel, L. Raehm, S. Richeter, B. Loock, P. Couleaud, C. Frochot, C. Charnay, G. Derrien, M. Smaïhi, A. Sahmoune, A. Morère, P. Maillard, M. Garcia and J.-O. Durand, Int. J. Pharm., 2010, 402, 221–230 CrossRef CAS.
- V. Lebret, L. Raehm, J.-O. Durand, M. Smaïhi, M. Werts, M. Blanchard-Desce, D. Méthy-Gonnod and C. Dubernet, J. Sol-Gel Sci. Technol., 2008, 48, 32–39 CrossRef CAS.
- L.-S. Wang, L.-C. Wu, S.-Y. Lu, L.-L. Chang, I. T. Teng, C.-M. Yang and J. A. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS.
- C.-L. Zhu, X.-Y. Song, W.-H. Zhou, H.-H. Yang, Y.-H. Wen and X.-R. Wang, J. Mater. Chem., 2009, 19, 7765–7770 RSC.
- J. Lu, Z. Li, J. I. Zink and F. Tamanoi, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 212–220 CrossRef CAS.
- S.-H. Wu, Y. Hung and C.-Y. Mou, Chem. Commun., 2011, 47, 9972–9985 RSC.
- H. S. Choi and J. V. Frangioni, Mol. Imaging, 2010, 9, 291–310 CAS.
- E. C. Cho, C. Glaus, J. Chen, M. J. Welch and Y. Xia, Trends Mol. Med., 2010, 16, 561–573 CrossRef CAS.
- J. Lu, M. Liong, J. I. Zink and F. Tamanoi, Small, 2007, 3, 1341–1346 CrossRef CAS.
- C.-H. Lee, S.-H. Cheng, Y.-J. Wang, Y.-C. Chen, N.-T. Chen, J. Souris, C.-T. Chen, C.-Y. Mou, C.-S. Yang and L.-W. Lo, Adv. Funct. Mater., 2009, 19, 215–222 CrossRef CAS.
- J. S. Souris, C.-H. Lee, S.-H. Cheng, C.-T. Chen, C.-S. Yang, J.-a. A. Ho, C.-Y. Mou and L.-W. Lo, Biomaterials, 2010, 31, 5564–5574 CrossRef CAS.
- C. Khemtong, C. W. Kessinger and J. Gao, Chem. Commun., 2009, 3497–3510 RSC.
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach and E. Toth, John Wiley & Sons Ltd, 2001 Search PubMed.
- X. Kang, Z. Cheng, D. Yang, P. Ma, M. Shang, C. Peng, Y. Dai and J. Lin, Adv. Funct. Mater., 2012, 22, 1539–1539 CrossRef.
- X. Xue, F. Wang and X. Liu, J. Mater. Chem., 2011, 21, 13107–13127 RSC.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42(15pp), 224001 CrossRef.
- C. Fang and M. Zhang, J. Mater. Chem., 2009, 19, 6258–6266 RSC.
- B. Polyak and G. Friedman, Expert Opin. Drug Delivery, 2009, 6, 53–70 CrossRef CAS.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Jon, Angew. Chem., Int. Ed., 2008, 47, 5362–5365 CrossRef CAS.
- J. Xie, J. Huang, X. Li, S. Sun and X. Chen, Curr. Med. Chem., 2009, 16, 1278–1294 CrossRef CAS.
- E.-Q. Song, J. Hu, C.-Y. Wen, Z.-Q. Tian, X. Yu, Z.-L. Zhang, Y.-B. Shi and D.-W. Pang, ACS Nano, 2011, 5, 761–770 CrossRef CAS.
- R. Bardhan, W. Chen, M. Bartels, C. Perez-Torres, M. F. Botero, R. W. McAninch, A. Contreras, R. Schiff, R. G. Pautler, N. J. Halas and A. Joshi, Nano Lett., 2010, 10, 4920–4928 CrossRef CAS.
- S. P. Foy, R. L. Manthe, S. T. Foy, S. Dimitrijevic, N. Krishnamurthy and V. Labhasetwar, ACS Nano, 2010, 4, 5217–5224 CrossRef CAS.
- F. K. H. van Landeghem, K. Maier-Hauff, A. Jordan, K.-T. Hoffmann, U. Gneveckow, R. Scholz, B. Thiesen, W. Brück and A. von Deimling, Biomaterials, 2009, 30, 52–57 CrossRef CAS.
- C. Vauthier, N. Tsapis and P. Couvreur, Nanomedicine, 2010, 6, 99–109 CrossRef.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- J. Dobson, Gene Ther., 2006, 13, 283–287 CrossRef CAS.
- B. González, E. Ruiz-Hernandez, M. J. Feito, C. López de Laorden, D. Arcos, C. Ramírez-Santillán, C. Matesanz, M. T. Portolés and M. Vallet-Regí, J. Mater. Chem., 2011, 21, 4598–4604 RSC.
- M. P. Melancon, A. Elliott, X. Ji, A. Shetty, Z. Yang, M. Tian, B. Taylor, R. J. Stafford and C. Li, Invest. Radiol., 2011, 46, 132–140 CrossRef.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Acc. Chem. Res., 2011, 44, 893–902 CrossRef CAS.
- S. Gai, P. Yang, C. Li, W. Wang, Y. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166–1172 CrossRef CAS.
- P. Yang, Z. Quan, Z. Hou, C. Li, X. Kang, Z. Cheng and J. Lin, Biomaterials, 2009, 30, 4786–4795 CrossRef CAS.
- C.-C. Huang, C.-Y. Tsai, H.-S. Sheu, K.-Y. Chuang, C.-H. Su, U. S. Jeng, F.-Y. Cheng, C.-H. Su, H.-Y. Lei and C.-S. Yeh, ACS Nano, 2011, 5, 3905–3916 CrossRef CAS.
- F. Wang, X. Chen, Z. Zhao, S. Tang, X. Huang, C. Lin, C. Cai and N. Zheng, J. Mater. Chem., 2011, 21, 11244–11252 RSC.
- W. Zhao, H. Chen, Y. Li, L. Li, M. Lang and J. Shi, Adv. Funct. Mater., 2008, 18, 2780–2788 CrossRef CAS.
- H. Wu, G. Liu, S. Zhang, J. Shi, L. Zhang, Y. Chen, F. Chen and H. Chen, J. Mater. Chem., 2011, 21, 3037–3045 RSC.
- Y. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- Y. Zhu, T. Ikoma, N. Hanagata and S. Kaskel, Small, 2010, 6, 471–478 CrossRef CAS.
- B. Julián-Lopez, C. Boissiére, C. Chaneac, D. Grosso, S. Vasseur, S. Miraux, E. Duguet and C. Sanchez, J. Mater. Chem., 2007, 17, 1563–1569 RSC.
- E. Ruiz-Hernández, A. López-Noriega, D. Arcos and M. Vallet-Regí, Solid State Sci., 2008, 10, 421–426 CrossRef.
- T.-W. Kim, P.-W. Chung and V. S. Y. Lin, Chem. Mater., 2010, 22, 5093–5104 CrossRef CAS.
- F. M. Martín-Saavedra, E. Ruíz-Hernández, A. Boré, D. Arcos, M. Vallet-Regí and N. Vilaboa, Acta Biomater., 2010, 6, 4522–4531 CrossRef.
- J. Feng, S.-Y. Song, R.-P. Deng, W.-Q. Fan and H.-J. Zhang, Langmuir, 2010, 26, 3596–3600 CrossRef CAS.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552–557 CrossRef CAS.
- J. E. Lee, D. J. Lee, N. Lee, B. H. Kim, S. H. Choi and T. Hyeon, J. Mater. Chem., 2011, 21, 16869–16872 RSC.
| This journal is © The Royal Society of Chemistry 2013 |