Enzyme responsive materials: design strategies and future developments
Mischa
Zelzer
*ab,
Simon J.
Todd
c,
Andrew R.
Hirst
d,
Tom O.
McDonald
e and
Rein V.
Ulijn
*a
aWestCHEM, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, U.K.. E-mail: rein.ulijn@strath.ac.uk; Fax: +44 (0) 141548 4822; Tel: +44 (0) 141 548 2110
bEindhoven University of Technology, Department of Chemical Engineering and Chemistry, Den Dolech 2, 5612 AZ Eindhoven, The Netherlands. E-mail: m.zelzer@tue.nl; Tel: +31 (0) 40 247 3598
cRenephra Ltd., Core Technology Facility, 46 Grafton Street, Manchester, M13 9NT, U.K.. E-mail: simon@renephra.com
dPolymers and Complex Fluids Group, School of Physics and Astronomy, University of Leeds, Leeds, LS2 9JT, U.K.. E-mail: A.hirst@leeds.ac.uk
eDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZD, U.K.. E-mail: thomas.mcdonald@liverpool.ac.uk
First published on 21st September 2012
Abstract
Enzyme responsive materials (ERMs) are a class of stimuli responsive materials with broad application potential in biological settings. This review highlights current and potential future design strategies for ERMs and provides an overview of the present state of the art in the area.
 Mischa Zelzer | Mischa Zelzer completed his first degree in Chemistry at the Technical University Graz (AT) in 2005. He then moved to the UK where he obtained his PhD from the University of Nottingham in 2009. Subsequently, he joined the University of Strathclyde (UK) in 2010 as a postdoctoral researcher. In 2012, Mischa was awarded a Marie Curie Fellowship and moved to the Technical University of Eindhoven (NL). Mischa's research interests are in stimuli responsive materials and interfaces with a particular emphasis on enzyme responsiveness and biological applications. |
 Simon J. Todd | Simon J. Todd after obtaining a BSc (Hons) degree in Biomedical Materials Science from the University of Nottingham, including a final year project under Prof. D. Grant on the functionalisation of diamond-like carbon with albumin, took a year out to travel in North America. He completed a PhD in the construction of enzyme responsive surfaces at the University of Manchester supervised by Dr Ulijn and Dr J. Gough. Simon then went on to work for the award-winning start up company Renephra. |
 Andrew R. Hirst | Andrew R. Hirst graduated in Colour and Polymer Chemistry in 1997 and received his PhD in polymer solution chemistry (supervisors Prof. J. T. Guthrie and Dr R. J. English) in 2001, both from the University of Leeds. Between 2002 and 2009, he was a postdoctoral researcher at the University of York, UK (working with Prof. D. K. Smith) and at the Department of Materials at Manchester Interdisciplinary Biocentre (working with Prof. R. V. Ulijn). He now currently works in the International Centre, University of Leeds facilitating international strategic partnerships and the development of international teaching and research activities. |
 Tom O. McDonald | Tom O. McDonald received his BSc (2004) and MSc (2005) from the University of Manchester. He then undertook a PhD in Materials Science with Rein Ulijn studying enzyme responsive polymer hydrogels, graduating in 2009 also from the University of Manchester. He then joined the Department of Chemistry at the University of Liverpool as a postdoctoral research associate, initially as part of the Rannard group but has recently become a member of the Cooper group. His research interests are focused on the application of materials chemistry to medical applications, specifically nanomaterials, polymer chemistry and drug delivery. |
 Rein V. Ulijn | Rein V. Ulijn is currently a WestCHEM Research Professor at The University of Strathclyde in Glasgow. He is the holder of an ERC Starting Grant and Leverhulme Trust Leadership Award and was awarded the 2007 Macro Group UK Young Researchers Medal. He previously held an EPSRC Advanced Research Fellowship. He has authored over 90 peer reviewed research articles and presented over 80 invited and keynote lectures at international conferences. Since 2004 he has generated a grant portfolio worth in excess of £4.5 M with funding from ERC, EPSRC, BBSRC, the Leverhulme Trust, Human Frontiers Science Program (HFSP), Airforce Office of Scientific Research and Industry (Smith & Nephew, Johnson & Johnson, Roslin Cellab). He was co-founder of the Manchester based spin out company Renephra Ltd. He gained his MSc degree in Biotechnology at the University of Wageningen (NL) (thesis 1998), PhD in Physical Chemistry at the University of Strathclyde (thesis 2001) and postdoctoral training at the University of Edinburgh. From 2003–2008 he was in the School of Materials, University of Manchester (promoted to Senior Lecturer in 2006 and Reader in 2007). |
1. Introduction
Smart or responsive materials have emerged over the last few decades to meet the demand for more versatile and dynamic material properties. This has led to the development of a plethora of materials able to change form and/or function in response to cues such as pH, temperature, light, electric fields etc.1–4 Devices based on such ‘smart’ technologies have found widespread application in science as well as in every-day life, facilitating advances in areas such as electronics,5 healthcare6–9 and energy creation and storage.10The stimulus or trigger applied to induce the change in the material's properties is an essential part of the responsive material technology. In living organisms all processes are ultimately controlled by enzymes and hence biology has a vast repertoire of materials at its disposal that respond to enzymatic stimuli. Inspired by this, in addition to the above mentioned stimuli, enzymes are increasingly used as triggers to change the properties of artificial materials to create biomimetic materials.11–14 We define such enzyme responsive materials (ERMs) as materials that change their functionality as a result of the action of an enzyme on the material. Compared to our initial description of ERMs in 2006,15 the present definition includes not only materials that undergo structural changes on a macroscopic level upon enzymatic action (e.g. hydrogels, particles), it also includes materials with microscopic structural changes (mesoporous silica particles, self-immolative materials) and materials that display different interactions with their environments after exposure to the enzymatic stimulus (e.g. surfaces, particles).
The definition we use for ERMs in this review only encompasses materials whose structure or functionality changes after direct action of the enzyme. This excludes materials where enzymes are simply immobilised as well as systems where enzymes are merely employed to create a material (i.e. give the material its final form and function but otherwise leave the material inert to further enzymatic action) unless the enzymatic formation of the material in itself performs a function such as the formation of a hydrogel. The line between enzymatically formed materials and ERMs is sometimes not very clear. A strict differentiation between the two may not always be possible and depends on how these materials are being used in the final application. Many examples exist of materials that are indirectly enzyme responsive.16–23 These are usually materials whose properties are changed upon exposure to the product of an enzymatic reaction that occurs nearby. While these materials can be cleverly designed and present attractive solutions to the challenges in sensing,16–19 drug delivery20–23 and other applications, we do not include them in this review because the material does not respond directly to the action of the enzyme.
Work on enzyme responsive materials has been growing substantially over the last few years. Since the first review on ERMs in 2006, a number of manuscripts have been published that summarise the advances in enzyme responsive technology in particular areas such as peptide hydrogels,13,24,25 polymers and polymer conjugates11,26 and particle technology.12,24,27,28 The strategies employed to incorporate enzyme sensitive functionalities in these diverse materials is often based on common principles, albeit some unique approaches have been developed that merit closer attention.
The goal of this manuscript is to review the design strategies for ERMs and highlight notable advances made within the last six years and strategies with promising application potential. After identifying key features for ERMs, we will discuss strategies for the incorporation of enzyme sensitive functionalities into different types of materials, before introducing strategies to design ERMs for specific functions.
2. Key features of ERMs
2.1. Enzymes as stimuli
When employed in a biological setting—as indeed most ERMs are—the natural occurrence of enzymes provides additional advantages over stimuli such as pH or temperature. Because a variety of enzymes is already present in the body, the stimulus does not need to be added externally but can be supplied by the biological environment itself, provided that the naturally present enzyme matches the triggering enzyme of the responsive material.15 In nature, the presence and activity of enzymes is finely tuned to regulate biological processes. Imbalances in the expression or activity of enzymes often occur in disease states and are attractive cues that can be picked up by ERMs and translated into a suitable material response. Finally, enzymatic action is often localised and their expression—and hence their activity—can be temporarily controlled by the biology (i.e. cells), making ERMs highly attractive for dynamic and targeted changes in material properties.
| Enzyme | Catalysed reaction/substrate | Natural function | Occurrence/relevance | Ref. |
|---|---|---|---|---|
| α-Amylase | Hydrolysis of the 1→4 glycosidic bond between β-D-glucoses | Degradation of starch | Present in mammalian saliva and pancreas, produced by numerous bacteria | 212, 241, 242 |
| α-Chymotrypsin | Hydrolysis of peptide bonds C-terminal to hydrophobic amino acids | Protein digestion in the small intestine | Mammalian pancreas | 98, 206, 221 |
| Acetylcholine-esterase | Hydrolysis of acetylcholine | Regulates the neurotransmitter acetylcholine | Present at the vertebrate neuromuscular junction; implicated in Alzheimer's disease | 243, 244 |
| Azoreductase | Reduction of azo compounds | Implicated in electron transport during redox processes inside cells | Expressed by bacteria; present in the colon of the human intestine | 147, 245–247 |
| β-D-Galactosidase | Hydrolysis of 1→4 glycosidic bonds between β-D-galactose and β-D-glucose in lactose | Hydrolysis of saccharides | Present in humans in the enterocytes lining the villi of the small intestine | 43, 212 |
| β-Glucuronidase | Hydrolysis of β-D-glucuronic acid | Degradation of complex carbohydrates | High concentrations present in necrotic tissue and several cancer types | 248, 249 |
| β-Lactamase | Hydrolysis of β-lactams | Degradation of β-lactam antibiotics | Produced by bacteria | 121, 180, 217 |
| BamH I | 5′-GGATCC-3′ | Restriction enzyme | Produced in Bacillus amyloliquefaciens H | 250, 251 |
| 3′-CCTAGG-5′ | ||||
| Caspases | Peptide bond hydrolysis after aspartic acid, very specific activity modulated by the four amino acids preceding aspartic acid and the tertiary protein structure | Cysteine proteases that regulate inflammation and apoptosis | Present in vertebrates and bacteria; down-regulated in several cancer types | 252–257 |
| Catalytic antibody 38C2 | Catalyses retro-aldol/retro-Michael reactions | — | Engineered protein | 258, 259 |
| Cathepsins | Peptide bond hydrolysis with broad specificity | Family of cysteine protease involved in extracellular matrix degradation | Present in mammalian cell lysosomes; over-expressed in various cancer types | 202, 260–262 |
| Chitosanase | Endohydrolysis of β-1→4-bonds between D-glucosamine residues | Degradation of chitosan | Produced by various plants and bacteria | 263 |
| Cutinase | Ester hydrolysis | Degradation of cutin on plant cuticles | Excreted by fungi | 264 |
| D-Aminopeptidase | Hydrolysis of peptide bonds in oligopeptides containing N-terminal D-alanine or D-serine | Hydrolysis of D-amino acid based peptides | Present in bacteria | 265 |
| Dextranase | Hydrolysis of 1→6-α-glucosidic bonds | Degradation of dextran | Present in colon, produced by various bacteria | 37, 266 |
| Dipeptidyl peptidase IV | Peptide bond hydrolysis after proline or alanine at the penultimate position from the amino terminus | Regulation of cellular processes, inactivation of insulin-sensing hormones | Involved in type II diabetes, implicated in various cancer types | 267–270 |
| Dpn II endonuclease | 5′-↓GATC-3′ | Restriction enzyme | Produced by Diplococcus pneumoniae | 176 |
| 3′-CTAG↓-5′ | ||||
| EcoR I | 5′-G↓AATTC-3′ | Restriction enzyme | Produced by Escherichia coli | 175, 189 |
| 3′-CTTAA↓G-5′ | ||||
| EcoR V | 5′-GAT↓ATC-3′ | Restriction enzyme | Produced by Escherichia coli | 183, 198 |
| 3′-CTA↓TAG-5′ | ||||
| Elastase | Peptide bond hydrolysis between two small amino acids (glycine or alanine) | Degradation of elastin in connective tissue | Present in mammals and bacteria, implicated in chronic wounds and inflammation processes | 101, 102, 201 |
| Furin | Peptide bond hydrolysis after RXKR or RXRR | Proprotein convertase | Intracellular protease expressed in eukaryotic and several mammalian cells; implicated in HIV and various cancer types | 210 |
| Glutathione-S-transferase | Coupling of reduced glutathione to electrophilic compounds | Detoxification and intracellular binding of proteins | Present in the cytosol of most mammalian organs and various other species | 271 |
| Glycosyltransferases | Catalysis of glycosidic bond formation of carbohydrate donors with nucleotide or lipid phosphates | Break-down of glucan | Present in animals and plants | 272 |
| HaeIII | 5′-GGCC-3′ | Restriction enzyme | Produced by Haemophilus aegyptius | 198 |
| 3′-CCGG-5′ | ||||
| Horseradish peroxidase (HRP) | Catalysis of the oxidation of a broad number of substrates in the presence of H2O2 | Catalyses cross-linking reactions, implicated in infection resistance and metabolic processes | Present in horseradish roots | 273 |
| Kinases | Phosphorylation of hydroxyl groups in peptide sequences | Activation of signal transducers and activators of transcription factors | Located in the cell membrane; aberrantly activated in several cancer cells | 274–280 |
| Lethal factor | Hydrolysis of a peptide bond in YBYBX↓ZHXZH (X—any amino acid; YB —any basic amino acid; ZH—any hydrophobic amino acid) | Inhibition of signalling pathways by cleavage of mitogen-activated protein kinases | Present in Bacillus anthracis | 281, 282 |
| Lipase | Formation/hydrolysis of esters | Break down of lipids | Present in many mammals, fungi and bacteria | 283, 284 |
| Matrix metalloproteinases (MMPs) | Hydrolysis of peptide bonds with various specificities in the presence of metals | Involved in tissue remodelling and anti-inflammatory processes; interact with biomolecules such as cell receptors and affect cell behaviour | Present in most multicellular organisms including animals and plants, implicated in several diseases including arthritis and cancer | 285, 286 |
| Papain | Peptide bond hydrolysis with broad specificity | Cysteine protease | Present in papaya | 95, 261 |
| Penicillin G acylase | Cleaves acyl chains of penicillins | Implicated in the metabolism of aromatic compounds | Present in bacteria, yeast and fungi | 287 |
| Phosphatases | Hydrolysis of orthophosphoric monoesters in peptide sequences | Regulation of protein activity and signal transduction | Present in plants and mammals (both cell membrane-bound and secreted); activity levels affected by various diseases (cancer, diabetes, multiple sclerosis) | 44, 50, 288–291 |
| Plasmin | Peptide bond hydrolysis with a preference for cleavage after arginine or lysine | Involved in fibrinolysis and wound healing | Present in animals; increased concentrations present in cancer cells | 93, 94 |
| Porcine liver esterase | Ester hydrolysis with wide specificity | Enantioselective serine hydrolase | Isolated from the liver of pigs | 292 |
| Proteinase K | Peptide bond hydrolysis with broad specificity | Degradation of keratin | Isolated from Tritirachium album Limber | 293, 294 |
| Pyroglutamate aminopeptidase | Hydrolysis of pyroglutamate/peptide bonds | Removal of N-terminal pyroglutamyl groups | Present in mammals, bacteria and plants | 295 |
| Rennin | Peptide bond hydrolysis | Aspartyl protease involved in blood pressure regulation | Secreted by juxtaglomerular cells in the kidney | 296 |
| Subtilisin | Peptide bond hydrolysis C-terminal of hydrophobic amino acids | Scavenging of nutrients | Produced by bacteria, excreted into the extracellular environment | 297, 298 |
| Thermolysin | Preferential hydrolysis of peptide bonds N-terminal of hydrophobic or bulky amino acids | Degradation of extracellular proteins and peptides for bacterial nutrition | Present in bacteria, fungi and archaea; involved in various diseases and infections | 299 |
| Thrombin | Selective peptide bond hydrolysis between R and G | Converts fibrinogen into fibrin and causes blood coagulation | Can mediate the formation of acute thrombus and promote malignancy in cancer | 174, 195, 221 |
| Transglutaminase | Catalysis of peptide bond formation between the α-carboxamide group of glutaminyl and primary amines | Cross-linking of proteins, involved in fibrin clot formation | Present in fluids and extracellular matrix throughout the body | 49, 65, 79 |
| Tyrosinase | Oxidation of a wide range of phenols into o-quinones | Insect sclerotization, setting of muscle glue and melanin production | Widely present in plants and animals | 86, 87 |
| Trypsin | Peptide bond hydrolysis C-terminal of lysine and arginine | Involved in digestive processes | Present in the pancreas; increased levels are associated with some pancreatic diseases | 177 |
| Urokinase plasminogen activator | Conversion of plasminogen into plasmin | Degradation of extracellular matrix | Present in urine and the blood stream; implicated in cancer invasion and metastasis | 225, 300 |
It should be noted that the interaction between enzymes and many ERMs is very different from enzymatic reactions that involve solubilised substrates. The kinetic models used to describe the enzymatic conversion of solubilised substrates (e.g. Michaelis Menten kinetics) cannot be applied to ERMs if the material is considerably larger—and hence less mobile—than the enzyme. This is the case for most ERMs, including surfaces, particles and hydrogels, where the enzyme is required to move towards its substrate. In addition to these aspects on diffusion kinetics, substrates bound to other materials may also differ from solubilised substrates in terms of enzyme specificity and steric effects. Thus, factors such as the chemical and physical properties of the material, the concentration of the enzyme substrate on/in the material, and the manner in which the substrate is anchored to the material are important considerations in the design of ERMs.30,31 Several recent studies have presented models to describe the kinetic parameters associated with enzyme action at surfaces32–35 but much yet remains to be elucidated before the interaction of enzymes with ERMs can be better understood.
2.2. Design of ERMs
 | ||
| Fig. 1 The design requirements for ERMs illustrated with the example of a self-assembling switch peptide. (TEM micrographs reprinted with permission from ref. 47, © 2005 American Chemical Society.) The abbreviations for the three components (ESF, Transl and MResp) will be used in subsequent figures to indicate if these components are separate parts within the ERM or if these components are (partly) combined in a single functional unit of the ERM. | ||
 | ||
| Fig. 2 Examples of enzyme-catalysed reactions used in ERMs. | ||
| Inorganic | Organic | |||
|---|---|---|---|---|
| Metals | Silicates | Polymers | Peptides | |
| Naturally derived | Artificial | |||
| CdSe/ZnS218,221,223,228 | Mesoporous silica41–43,171,213,215 | Alginate85 | Poly(acrylamide)104 | Polypeptides48,126,127 |
| CdTe64 | Glass/quartz70,167 | Amylose39 | Poly(acrylic acid) (PAAc)96 | Oligopeptides50,122,124,132 |
| Ironoxides183–186,192 | Cellulose84,234 | Poly(allylamine)99 | Aliphatic peptide amphiphiles (Ac: acyl)61,118,137 | |
| Gold40,63,67,168,172,176,179,182,191,192,199 | Chitosan38,86,87,205 | Poly(butyl methacrylate) (PBMA)91,92 | Aromatic peptide amphiphiles (Fmoc: fluorenylmethoxycarbonyl; Nap: naphthyl)52,62,114,120 | |
| Cyclodextrin213 | Poly(4-((dihydroxyphosphoryl)oxy) butyl acrylate) (POPBA)125 | |||
| Dextran37,60,104 | Poly(ethoxydi(ethylene glycol) acrylate) (PDEGEA)125 | |||
| DNA129,135,175,182,198 | Poly(ethylene glycol) (PEG)/poly(ethylene oxide) (PEO)36,48,50,55,79,94,97,125,167,185,196 | |||
| Gelatin78,86,87,191 | Poly(ethylene glycol acrylamide) (PEGA)57,68,100–102 | |||
| Poly(4-hydroxybutyl acrylate) (POHBA)125 | ||||
| Poly(2-hydroxyethyl methacrylate) (PHEMA)97 | ||||
| Poly(N-isopropyl acrylamide) (PNIPAM)37 | ||||
| Poly(2-isopropyl-2-oxazoline) (PiPrOx)56 | ||||
| Poly(N-(2-(2-pyridyldithio))ethyl methacrylamide (PDTEMA)98 | ||||
| Poly(N,N-dimethylacrylamide) (PDMA)37 | ||||
The enzymatic formation of bonds in ERMs has been accomplished via the condensation of amino acids or peptide fragments45—including the formation of cross-links between the side groups of amino acids49—and the phosphorylation of amino acids44 or polymer side chains.50 The bond cleavage of a large variety of peptides51 and amino acid esters52 as well as ester bonds between polymers and small molecules53 has been employed as ESFs in ERMs. Since many natural enzyme substrates consist of specific amino acid sequences, it is not surprising that a large number of ERMs rely on peptides as ESFs. These include short peptide sequences and peptide amphiphiles for self-assembling materials,44,45 short peptides as cross-linkers in polymer hydrogels,54,55 polymer–peptide conjugates56 and the decoration of surfaces57 and particles40,58 with peptide sequences. Non peptidic substrates include polymers with functional side groups such as phenols,59 polysaccharides38 and block polymers linked via esters.60
The focus of ERMs on bond formation and breakage stands in contrast to the mechanisms employed in most other stimuli responsive materials where the stimuli typically affect equilibrium states. In pH or temperature responsive materials, the material response persists only as long as the stimulus is present but the material will often revert to its original state when the stimulus is removed. The electrochemical oxidation/reduction and deliberate degradation of the material by the stimuli are exceptions to this situation. However, even though a particular enzyme produces one product under a certain set of conditions, the enzymatic formation/cleavage of bonds is often reversible. This has been exploited in particular for the phosphatase/kinase system that allows dephosphorylation and phosphorylation (in the presence of ATP) of alcohols, respectively, providing the possibility to prepare reversible or dynamic stimuli responsive materials that are stable even in the absence of the stimulus.61,62
Even though the action of enzymes on reported ERMs relies on only two different types of reactions (bond formation/cleavage and redox reactions), the resulting changes to the ESF can be rather diverse. Upon exposure to the enzyme, the hydrophilicity of the material can be changed,48 charges may be introduced or removed,63,64 the chain length of (block-)polymers may be altered,39 cross-links may be introduced or removed from a polymer,65 steric effects may be changed,42 functional groups introduced66 or unmasked67 or bonds may be rearranged.47 If well designed, any one of these effects can be used to induce an overall change in the materials properties. They may affect the intermolecular interaction of small molecules,44,45 compromise or strengthen the structural integrity of the material,65 cause structural rearrangements such as swelling68,69 and shrinkage69 or alter the chemical functionality of a surface.70
The methods used to integrate the ESF into the material vary and depend on the material and the enzyme substrate system. In cases where a change in the functional groups, charge or hydrophilicity is desired, stable covalent attachment of the ESF to the bulk material is often sufficient. For example, short peptides have been attached to surfaces and particles via standard amide formation68,70 or thiol–metal bonds40 and peptide based cross-links were introduced in polymers using reverse Michael addition chemistry.71 Conversely, when rearrangements of the molecular or intramolecular structure are required, it is not enough to simply append the ESF to the material. In this case, a more sophisticated molecular design of the material is necessary; for example, to enzymatically trigger the self-assembly of a peptide amphiphile, it is not sufficient to change the molecular structure, the final product requires a precise balance of hydrophobic and hydrophilic components to form self-assembled architectures.13,29 The following section will discuss various ERMs in more detail. They were classed according to their predominant structural element (see Fig. 3), although the reader will be bound to find some overlap between some of the enzyme responsive systems highlighted when an ERM may fit in more than one of the five classes (e.g. polymer hydrogel particles or surface functionalised particles).
 | ||
| Fig. 3 The classification of ERMs based on their structural design elements. | ||
3. ERM systems
3.1. Polymer hydrogels
Polymer hydrogels are hydrophilic polymers that have been highly cross-linked resulting in insoluble three-dimensional networks.72 Enzyme responsiveness is introduced into these materials mainly in the form of enzyme sensitive cross-links. Hence, the most frequent result of enzyme action on enzyme responsive polymer hydrogel systems is either hydrogel formation or hydrogel degradation. An exception is the enzymatically controlled swelling of hydrogel particles. An overview of enzyme responsive polymer systems is given in Table 3. In all these examples, the ESF is simultaneously responsible for the translation of the enzymatic action into an overall change in the material properties as shown in Fig. 4. | ||
| Fig. 4 Functions performed by enzyme responsive polymer hydrogels. In all cases, the enzyme sensitive functionality (ESF) is simultaneously responsible for the translation of the enzymatic action (Transl) into a material response (MResp). | ||
| Material response | Entry | Enzyme | Enzyme sensitive functionality | Material | Ref. |
|---|---|---|---|---|---|
| Hydrogel formation | 1 | Elastase | A↓A | Poly(allylamine) | 99 |
| 2 | HRP | Hyaluronic acid/tyramine | Hyaluronic acid/tyramine | 80 | |
| Dextran/tyramine | Dextran/tyramine | 83 | |||
| Carboxymethylcellulose/tyramine | Carboxymethylcellulose/tyramine | 84 | |||
| Alginate/tyramine | Alginate/tyramine | 85 | |||
| 3 | Transglutaminase | K/Q | Gelatin | 78 | |
| Poly(KF)/[PEG] | 49 | ||||
| [PEG]/peptide conjugates | 65, 79 | ||||
| 4 | Tyrosinase | Y | Gelatine/chitosan | 86, 87 | |
| Degradation of the polymer | 5 | α-Chymotrypsin | Gelatin | Gelatin | 90 |
| 6 | Dextranase | Dextran | Dextran cross-linked with diisocyanate | 89–91 | |
| 7 | Papain | GGG | [PEG] | 91, 92 | |
| [PNIPAM]-co-[PDMA]-co-[PBMA] | |||||
| Degradation by cleavage of cross-links | 8 | α-Chymotrypsin | CY↓KC | [PDTEMA] | 98 |
| 9 | Collagenase | GGLGPAGGK | [PEG] | 55 | |
| 10 | Dextranase | Dextran | [PNIPAM] grafted on dextran and [PNIPAM]-co-[PDMA] composite | 37 | |
| 11 | Elastase | AAAAAAAAAK | [PEG] | 55 | |
| 12 | MMP-1 | APGL | [PEG] | 93 | |
| GPQG↓IAGQ | [PEG] | 65 | |||
| GPQG↓IWGQ | [PEG] | 36 | |||
| 13 | MMP-2 | GPVG↓LIGK | Pluronic© | 75 | |
| 14 | MMP-13 | PQGLA | [PNIPAM]-co-[PAAc] | 96 | |
| 15 | Papain | GFL↓G | [PEG] | 95 | |
| 16 | Plasmin | VRN | [PEG] | 93 | |
| 17 | Plasmin | D-AFK | [PEG] | 94 | |
| 18 | Subtilisin | GGL | [PHEMA]/[PEO] | 97 | |
| 19 | Trypsin | D-AFK | [PEG] | 94 | |
| Morphology control | 20 | Dextranase | Polyacrylamide/dextran | Polyacrylamide | 104 |
| 21 | Elastase | Fmoc-DA↓AR | AR-[PEGA] | 101, 102 | |
| Fmoc-A↓APV | |||||
| 22 | Glutathione-S-transferase | GR(4-Bbs)GDS | GRGDS | 103 | |
| 23 | MMP-1/12 | GPQG↓IWGQ | [PEGA] | 102 | |
| 24 | Thermolysin | Fmoc-DA↓AR | [PEGA] | 101 | |
| Fmoc-RRA↓ADD | [PEGA] | 100 | |||
| 25 | Trypsin | Fmoc-RFG | [PEGA] | 68 |
Another approach to generate cross-links between polymer chains is the enzymatic conversion of side groups into more reactive species that are subsequently able to react with moieties in neighbouring polymer chains. Kurisawa et al. showed that the oxidative coupling of phenols using horseradish peroxidase (HRP) in the presence of H2O2 is able to produce a hydrogel from hyaluronic acid/tyramine conjugates.80 The amount of H2O2 can be used to tune the mechanical properties and degradation characteristics of the hydrogel.81,82 Other polymers such as dextran83,84 and alginate85 have also been modified with tyramine and were subsequently cross-linked with HRP to form a hydrogel. A slightly different approach to hydrogelation via the enzyme induced increase in side chain reactivity was taken by Chen et al. Tyrosinase was used to convert tyrosine residues present in gelatin to quinones that were then able to react with other amino acids from neighbouring polypeptide chains.86 Subsequently, this method was used to cross-link a mixed polypeptide–polysaccharide gel consisting of both gelatine and chitosan.87
The first enzyme responsive polymer hydrogels consisted of bio-degradable materials such as dextran89–91 and gelatin90 that could be broken down by dextranase89,90 and α-chymotrypsin,90 respectively. Enzyme cleavable units were also built into the polymer.91,92 Another strategy adopted to create enzyme degradable polymer hydrogels is enzymatically cleavable cross-linkers. This was first introduced by West and Hubbell who designed PEG molecules flanked at both sides with short peptides that were terminated with polymerisable groups.93 After polymerisation, the PEG based hydrogel could be degraded with either matrix metalloprotease 1 (MMP-1) or plasmin. Another early example of enzymatically degradable PEG hydrogels is the elastase and collagenase sensitive polymer hydrogels prepared by Mann et al.55 The PEG based system was later extended to multi-armed PEG molecules cross-linked with short peptide strands54,71 responsive to MMP-1,36 plasmin,94 trypsin94 and papain.95 Other peptide cross-linked MMP sensitive systems were prepared from block copolymers from Pluronic© (an amphiphilic triblock copolymer)75 and from copolymers of N-isopropylacrylamide and acrylic acid.96 Khelfallah et al. designed a poly-2-hydroxyethylmethacrylate (pHEMA)–poly(ethylene oxide) (PEO) hydrogel that could be degraded by subtilisin via the cleavage of a peptide sequence that was incorporated into the PEO chain.97
Other strategies exist that do not involve the introduction of an enzyme degradable linker. Moore and co-workers designed polymerisable peptides that formed the basis of an α-chymotrypsin degradable hydrogel.98 Kumashiro et al. were able to demonstrate that a dually responsive system can be used to thermally control the enzymatic degradation of a polymer hydrogel.37 A composite material consisting of poly(N-isopropylacrylamide) (pNIPAM) grafted on dextran and a pNIPAM–N,N-dimethylacrylamide (DMA) copolymer was prepared that could be degraded by dextranase only between the cloud points of the two copolymer systems, effectively inhibiting enzymatic degradation below 30 °C and above 40 °C due to the steric hindrance of the enzyme.
Departing from the use of enzymatically cleavable cross-linkers, Heise and Thornton chose a different approach to generate enzyme degradable polymer hydrogels. They modified the side chains of poly(allylamine) with acetyl protected dialanine residues, thereby making the polymer less hydrophilic and causing the formation of a self-supporting polymer hydrogel.99 Exposure of the material to elastase removed the acyl group, restoring the polymers hydrophilicity and destroying the self-supporting gel.
One challenge for enzyme responsive polymer hydrogels that has not yet been fully addressed is the design of dynamic polymer hydrogel materials that are able to change morphology or chemistry reversibly upon enzymatic action. These types of materials have already been realised with other stimuli such as pH and temperature.3 While the above examples demonstrate the feasibility of triggering morphological changes in polymer hydrogels with enzymes, for a truly dynamic/reversible system further development is needed. For example, while drug release from the polymer hydrogels can be to some extent controlled by the amount of enzyme and the accessibility of the enzyme to the substrate within the hydrogel, once initiated, the release of the therapeutic will continue unhindered until the reservoir is depleted. It would be attractive to design a system where the morphological changes of the hydrogel such as swelling are only sustained while the enzyme is present but is reversed when the enzyme is removed. Similar systems can be envisioned with antagonistic pairs of enzymes whose reactions hold a balance in the material morphology; the material may be driven to one extreme or the other if the balance of enzymatic reactions is tipped, e.g. by the presence of inhibitors or the removal of cofactors for the enzymatic action.
3.2. Supramolecular materials
Supramolecular materials are based on self-assembling building blocks that are able to adapt a large variety of architectures.105 Depending on these architectures and the chemistry of the building blocks, supramolecular materials may perform various functions such as mimicking 3D environments for cells106 or serving as injectable scaffolds in biomedical applications.62 Over the last twenty years, synthetic chemists developed a vast number of molecular building blocks capable of assembling into various nanoscale architectures.105,107–110 The structural and chemical composition of these building blocks determine not only whether these molecules are able to self-assemble, they also affect the architecture of the supramolecular materials. Thus, endeavours in creating enzyme responsive supramolecular systems have not only included enzymatic formation and destruction of these materials,13,111 they are increasingly focused on designing dynamic systems where enzymes are used to trigger the conversion from one structure to another.24Even though the interactions driving the self-assembly can be somewhat complex and rely on a precise balance of intermolecular forces,105 the individual building blocks can be rather simple. In many enzyme responsive supramolecular systems, the ESF, Transl and MResp are combined and conveyed by small molecules such as short peptides or peptide amphiphiles (Fig. 5). These materials found particular consideration as precursors (gelators) to form supramolecular hydrogels13,111 and a large part of this section will be dedicated to these materials. However, non-gelling supramolecular assemblies, in particular polymer micelles, have also attracted considerable attention for drug delivery and other biological applications.112 An overview of the systems discussed here can be found in Table 4.
 | ||
| Fig. 5 The functions performed by enzyme responsive supramolecular materials. The three components of an ERM (the enzyme sensitive functionality (ESF), the translation of the enzymatic action (Transl) and the material response (MResp)) are typically combined into one functionality in the material. | ||
| Structure response | Entry | Enzyme | Precursor | Product | Ref. |
|---|---|---|---|---|---|
| a ATP: adenosine-5′-triphosphate. | |||||
| Enzyme triggered self-assembly | |||||
| Change of charge/hydrophilicity | 1 | Acid phosphatase | Phosphorylated phenols | [PEG]/poly(4-hydroxy styrene) | 59 |
| 2 | [PEO]-(pTV)5 | [PEO]-(TV)5 | 50 | ||
| 3 | [P(DEGEA-co-OPBA)]-b-[PEO]-b-[P(DEGEA-co-OPBA)] | [P(DEGEA-co-OHBA)]-b-[PEO]-b-[P(DEGEA-co-OHBA)] | 125 | ||
| 4 | Alkaline phosphatase | Fmoc-pY; Nap-FFGEpY; Nap-FFpY; Nap-GFFpY-OMe; Ac-YYYpY-OMe; Fmoc-FpY; | Fmoc-Y; Nap-FFGEY; Nap-FFY; Nap-GFFY-OMe; Ac-YYYY-OMe; Fmoc-FY; | 44, 62, 113, 116–118 | |
| 5 | MMP-9 | FFFFCG↓LDD | FFFFCG | 124 | |
| 6 | Subtilisin | Fmoc-L3-OMe | Fmoc-L3 | 52, 114 | |
| Fmoc-YL-OMe | Fmoc-YL | ||||
| Switch peptides | 7 | D-Amino acid peptidase | DA↓S(Ac); | Ac-S | 47 |
| DA↓T(Ac) | Ac-T | 47 | |||
| 8 | Dipeptidyl peptidase IV | AXXP↓S(Ac) | Ac-S | ||
| 9 | Pyroglutamate aminopeptidase | pGlu↓S(Ac) | Ac-S | 46, 47 | |
| 10 | Trypsin | R↓S(Ac) | Ac-S | 46, 47 | |
| Generation of β-sheet forming peptide amphiphiles | 11 | Lipase | Fmoc-F/FF | Fmoc-FFF | 120 |
| 12 | Thermolysin | Fmoc-X/amino acids | Fmoc-XFF; Fmoc-SF-OMe | 45, 115 | |
| Release of β-sheet forming peptide | 13 | Thrombin | VPR↓GS | L4K8L4-VPR | 122 |
| Enzyme mediated disassembly | |||||
| Change in hydrophilicity | 14 | Alkaline phosphatase | [PEG]-polylysine/[ATP] co-assemblya | PEG-polylysine | 48 |
| 15 | Lipase | Dextran-decanoate | Dextran/decanoic acid | 60 | |
| 16 | Esterase | Amphiphilic dendrimer | Hydrophilic dendrimer | 130, 131 | |
| Destruction of the building blocks | 17 | BamH I | 5′-GGATCC-3′ | Cleaved DNA strands | 129 |
| 18 | EcoR I | 5′-GAATTC-3′ | Cleaved DNA strands | 129 | |
| Cleavage of building blocks | 19 | α-Chymotrypsin | βAβAKLVFF-[PEG] | F-[PEG] | 133 |
| 20 | MMP-2 | Palmitic acid-GTAG↓LIGQERGDS; | Palmitic acid-GTAG | 126 | |
| (RADA)nPVG↓LIG(RADA)n | (RADA)nPVG and LIG(RADA)n | 127 | |||
| 21 | Urokinase plasminogen activator | KLDLKL-SGRSANA-KLDLKL | KLDLKL | 128 | |
| 22 | Type IV collagenase | QGFI↓GQPG | QGFI and GQPG | 132 | |
| Dynamic self-assemblies | |||||
| Spherical to cylindrical micelles | 23 | DNAzyme | DNA brush polymer | Truncated DNA brush polymer | 135 |
| Micelles to amorphous networks | 24 | MMP-2/9 | Peptide brush polymer (PLG↓LAG) | Truncated peptide brush polymer | 69 |
| 25 | Protein kinase A/protein phosphatase 1 | LRRASLG containing peptide brush polymer | LRRApSLG containing peptide brush polymer | 69 | |
| Micelle to fibre transition | 26 | MMP | Palmitoyl-GGGHGPLG↓LARK | Palmitoyl-GGGHGPLG | 137 |
| 27 | Phosphatase | Fmoc-FpY | Fmoc-FY | 113 | |
| Reversible self-assembly | 28 | Phosphatase/kinase | Nap-FFGEpY | Nap-FFGEY | 62 |
| 29 | KRRASVAGK-lauric acid | KRRApSVAGK-lauric acid | 61 | ||
| Loose aggregate to micelle transition | 30 | Phosphatase | Fmoc-pY/poly(2-isopropyl-2-oxazoline) | Fmoc-Y/poly(2-isopropyl-2-oxazoline) | 56 |
| Micelle to vesicle | 31 | Phosphorylase b | Amylose (n = 5)/hexadecylamine block polymer | Amylose (n = x + 5)/hexadecylamine block polymer | 39 |
Most enzyme responsive supramolecular hydrogels are based on peptides or peptide amphiphiles.44 The first record of the enzymatic formation of a supramolecular hydrogel was by dephosphorylation of the amphiphile N-(fluorenylmethyloxy carbonyl) tyrosine phosphate (Fmoc-pY) using alkaline phosphatase.44 Since this pioneering work, a number of other phosphatase responsive peptide based gelators have been designed, including Nap-FFGEpY (Nap = naphthyl),62 Nap-FFpY,116 Nap-GFFpY-OMe,117 Ac-YYYpY-OMe (Ac = acyl),118 and Fmoc-FpY.113 A non-hydrogel forming self-assembling system was introduced by Kühnle and Börner who used acid phosphatase to trigger the formation of fibres from a PEO–peptide conjugate via dephosphorylation of tyrosine residues.50
An alternative strategy to trigger self-assembly via the modification of the peptide sequence was presented by Ulijn and co-workers. Self-assembling Fmoc tripeptides were generated by coupling a non gelling dipeptide (FF) to a single Fmoc protected amino acid via reverse hydrolysis catalysed by a protease (thermolysin).45 Notably, this approach was further exploited by Hughes et al. to prepare 2D nanostructures via the enzymatic formation of Fmoc-SF-OMe from Fmoc-S and F-OMe.115,119 Palocci and co-workers expanded the reverse hydrolysis strategy by using a lipase to induce the hydrogelation of Fmoc-F3 from Fmoc-F and F2.120
The self-assembly of a molecule may also be controlled by changing the solubility or availability of the gelator. In a study by Xu and co-workers, the self-assembling Fmoc-FF molecule was coupled to a hydrophilic group via a β-lactam based linker and released upon the action of β-lactamase.121 The opposite approach was taken by Ulijn and co-workers who increased the solubility of a precursor (such as methyl esters of Fmoc protected di- or tripeptides) by enzymatic (subtilisin) ester cleavage to form a hydrogel.52 Further examples of subtilisin triggered gelation have been reported by Hirst et al.114 This work notably demonstrates that the response of the ERM (i.e. the self-assembled structure resulting from the action of the enzyme) depends on the concentration of the enzyme. It was proposed that enzyme clusters are formed from which self-assembly proceeds through enzymatic conversion of the precursor. The length and diameter of the self-assembled fibres depend on the size (and hence the mobility) of the clusters, which in turn is dependent on the enzyme concentration. This work demonstrates that different material properties may be accessed from the same building blocks by changing the concentration or activity of the enzyme.
As in the above examples on peptide amphiphiles, the self-assembly of peptide sequences may be inhibited by controlling their availability. Koga et al. accomplished this by binding the β-sheet forming sequence L4K8L4 to a PEG chain (Mw = 3000–3300) via a thrombin cleavable peptide (VPR↓GS).122 Gazit, Shabat and co-workers demonstrated the enzymatic release of a self-assembling dipeptide (FF) from a small dendritic molecule via penicillin G amidase triggered self-immolation of the dendrimer.123 Xu and co-workers designed a system where the enzymatic cleavage of a peptide results in the liberation of smaller self-assembling peptide units upon the action of MMP-9.124 In contrast to most strategies employed for the enzymatic hydrogelation of peptide amphiphiles, these studies are examples where the ESF (enzymatically cleavable units) is separate from the component that determines the materials properties (self-assembling peptide), while translation of the enzymatic action is simply accomplished by separating the self-assembling molecules from a blocking group.
In a departure from the strategies described above, Mutter et al. introduced a different type of enzyme triggered (non-hydrogel forming) self-assembly based on enzyme induced conformational changes.46 Based on ‘switch peptide’ technology, an elegant system was created where enzyme response, translation and material response are distinct and separate features of the ERM. Two oligopeptides are linked via an N-protected O/S-acyl isopeptide derived from serine, threonine or cysteine. The isopeptide interrupts the hydrogen bonding pattern between the two oligopeptides, thus preventing the formation of a secondary structure. By enzymatically cleaving the N-protection group of the isopeptide (ESF), acyl migration to the free amine occurs (Transl), restoring the default in the peptide chain and allowing the formation of secondary structures (materials response). It was shown that by using various enzyme substrates as amine protection groups, deprotection and subsequent acyl migration could be achieved with trypsin, pyroglutamate aminopeptidase, D-amino acid peptidase and dipeptidyl peptidase IV.46,47 Depending on the design of the oligomers, the acyl migration results in the formation of α-helices, β-sheets and/or self-assemblies such as fibres.
Enzyme triggered self-assembly of non-peptidic materials is relatively rare. Amir et al. reported an example of enzymatically induced micelle formation of an amphiphilic block polymer.59 To suppress micelle formation of the di-block polymer PEG–poly(4-hydroxy styrene), phosphate groups were introduced to the phenol side groups of the polymer, rendering the whole block-polymer water soluble. Exposure to acid phosphatase removed the phosphate groups and allowed the block-polymer to self-assemble into micelles. Also exploiting dephosphorylation, Woodcock et al. used acid phosphatase to trigger the formation of a micellar gel network from a triblock copolymer.125
Similar to peptide based supramolecular materials, polymer based micelles can also be enzymatically disassembled by converting the micelle forming polymer into non-self-assembling molecules. Since micelle forming polymers are typically amphiphilic block polymers, separation of the blocks is an effective way to accomplish disassembly. For example, Ge et al. used lipase to enzymatically hydrolyse the ester link between dextran and a conjugated alkyl chain, effectively cleaving the amphiphile between the two blocks.60 Similarly, Thayumanavan and co-workers used an esterase to cleave the hydrophobic moiety from an amphiphilic dendrimer, generating more hydrophilic end-groups on the dendrimer.130,131 Hennink and co-workers designed polymer amphiphiles that were linked together by a MMP sensitive peptide sequence and were thus able to enzymatically separate the two polymer blocks.132 A peptide–PEG conjugate that is able to self-assemble into micelles was prepared by Hamley and co-workers.133 These micelles were sensitive to α-chymotrypsin which degraded the peptide part of the material and thus caused the destruction of the self-assembled structure.
A new approach to enzymatically change the amphiphilic nature of the self-assembling polymer was introduced by Wang et al.48 They prepared a double-hydrophilic block polymer consisting of a PEG and a polylysine block. When mixed with ATP, electrostatic interactions allow ATP to attach to the lysine block, thus creating a ‘superamphiphile’ which is able to self-assemble into micelles. The micellar structure can be disassembled by the introduction of phosphatase which cleaves the phosphate groups from the ATP molecule, thus destroying the superamphiphile and regenerating the double hydrophilic block polymer.
Reversible supramolecular hydrogels can be realised by using enzyme pairs that catalyse the respective complementary reverse reactions. In living organisms, the phosphatase/kinase pair evolved to perform this exact task by catalysing the dephosphorylation and phosphorylation of amino acids such as serine or tyrosine. Xu and co-workers employed this enzymatic system to induce gel to sol (kinase) and sol to gel (phosphatase) transitions in a peptide amphiphile (Nap-FFGEY) where ATP levels dictate the self-assembly state.62 Webber et al. designed a self-assembling nonapeptide/lauric acid amphiphile that could be switched from a gel to a sol upon phosphorylation with protein kinase A.61 This could be reversed by dephosphorylation with alkaline phosphatase.
While the construction and destruction of self-assembled structures have applications in their own right, the conversion of one self-assembled structure to another also holds attractive possibilities as ERMs. Most examples of structural transitions of supramolecular assemblies are based on polymeric micelles. Caponi et al. showed that the weak self-assembled spherical aggregates of an Fmoc-pY terminated poly(2-isopropyl-2-oxazoline) can be driven towards the formation of micelles upon dephosphorylation with alkaline phosphatase.56,134 Gianneschi and co-workers were able to affect changes in the self-assembled structures of block polymers by altering the volume of one of the polymer blocks. The length of the DNA side chains in an amphiphilic DNA brush copolymer was shortened by exposure to DNAzyme which triggered a transition from spherical to cylindrical micelles. This approach was further developed to change the structure of self-assemblies formed from an amphiphilic block polymer that displays peptide side chains on its hydrophilic part.135 These materials could be reversibly switched from micelles to larger aggregates with the phosphatase/kinase pair and irreversibly transformed into amorphous networks using MMP 2 or 9.69
Changes in the size of micelles can be accomplished by enzymatically increasing the length of the block polymer. Morimoto et al. used phosphorylase b to extend the oligosaccharide block of an amylose based surfactant via enzymatic phosphorylation.39 When surpassing a certain length, the polymer rearranges from a micellar structure to form vesicles. Alemdaroglu et al. used a DNA polymerase (terminal deoxynucleotidyl transferase) to increase the size of the micelles formed by a DNA–polypropylene oxide (PPO) conjugate via the enzymatic elongation of the DNA sequence.136
Macroscopically observable morphology changes of supramolecular self-assemblies can be realised by converting micelles into fibres in an aqueous solution, which will subsequently form a hydrogel. Sadownik et al. demonstrated this with an aromatic peptide amphiphile (Fmoc-FpY) whose transition from a micellar to a gel forming fibrous self-assembly was triggered by dephosphorylation with alkaline phosphatase.113 Koda et al. accomplished the same transition with an aliphatic peptide amphiphile by converting the micelle forming palmitoyl-GGGHGPLGLARK into a fibre forming gelator (palmitoyl-GGGHGPLG) via proteolytic cleavage of the peptide sequence with MMP-7.137
One of the stipulated attractions to use enzymes as triggers is the fact that many of their catalysed reactions are reversible; and yet, to date only the phosphatase/kinase system has been used to accomplish such reversible morphology changes. To truly harvest the full potential of enzymatically controlled self-assembled systems, we are challenged to improve our understanding of the underpinning forces in the self-assembly processes to develop enhanced design rules that will allow us to integrate multiple ESFs within the supramolecular self-assembly system. To date, the potential of reversible peptide bond formation/cleavage to design dynamic enzyme responsive self-assemblies has been largely neglected. Maybe even more interesting is the ability of self-replication, e.g. in the case of DNA transcription where the system may not only be able to enzymatically reproduce a large number of self-assembling precursors, but is also able to self-correct defects, ultimately providing the tool to disassemble the self-assembling precursor into its starting materials.
3.3. Self-immolative materials
Self-immolative materials have been developed to mask a molecule (typically to supress the activity of a drug until delivery) by attaching it to another moiety (e.g. a peptide sequence or a polymer) via metastable covalent bonds. When stimulated by a specific triggering event, these materials fall apart into their parental components, thus releasing the masked drug. The unique trait of these materials is that exposure to the stimulus does not cleave the two components of the material directly; the triggering event occurs remote from the drug molecule and is related to the drug via the self-immolative linker. Hence, the ESF and Transl are two separate entities in the material (Fig. 6). After cleavage of the ESF, the metastable linker self-immolates (i.e. it disassembles) over a sequence of 1–4 or 1–6 eliminations through either carbonate- or carbamate-bonds, thus liberating the attached molecule as a response to the enzymatic stimulus. | ||
| Fig. 6 The function performed by an enzyme responsive self-immolative material. The enzyme sensitive functionality (ESF, red triangle) is separate from the rest of the material which consists of a polymer backbone whose disassembly simultaneously represents the translation of the enzymatic action (Transl) and the materials response (MResp). | ||
Self-immolative systems were originally developed to overcome issues related to the cleavage of direct covalent links between the drug and the masking unit that may be hindered when sterically bulky drugs are used.138 Since many self-immolative systems are intended for the release of therapeutics, the use of enzymes as stimuli gained popularity early on. A number of examples of small molecules containing self-immolative linkers exist. These include molecules sensitive to cathepsin b,139–141 trypsin,142 β-glucuronidase,143 β-lactamase,144 antibody 38C2,145 penicillin G amidase146 and azoreductase.147 In order to expand these systems to applications other than drug delivery, Waldmann and co-workers incorporated the self-immolative linker technology into the synthesis of molecules on polymeric supports where the final product was cleaved from the support with either lipase148 or penicillin G acylase.149,150
Since the above examples are based on soluble compounds that do not form any structures, they cannot be considered to be functional materials. Self-immolative systems that fit the definition of an ERM have been designed in the form of polymers that are not only able to release smaller molecules, but they are also designed to disassemble after the enzyme has triggered the self-immolation process. Only very few examples of such self-immolative enzyme responsive polymers exist (Table 5), these will be discussed in more detail below.
Similar to dendrimers, self-immolative linear polymers and polymer brushes have also been prepared. The reaction triggered by the stimulus in these materials (including self-immolative dendrimers) does not only release appended or masked molecules, it essentially disassembles the complete polymer structure into its parental units. Using similar substrates as for the dendrimers, Shabat et al. designed linear polymers and polymer brushes that disassemble when exposed to penicillin G amidase158 and catalytic antibody 38C2158 and a comb polymer sensitive to penicillin G amidase.159
Self-immolating materials are still a rather new concept whose full potential has yet to be explored. All the above examples explored these materials to either release a drug or another model compound from a support. In Section 3.2.1 we have introduced an example where the released molecules were gelators, able to form a supramolecular hydrogel.123 Further studies use enzyme responsive self-immolating materials as sensors that release fluorophores in the presence of the enzyme.160,161 The common factor in all these studies is the release of smaller molecules. To bring these materials into the same arena as other ERMs, we envision that their application may be expanded to include structural rearrangements of a material, for example as enzyme responsive cross-linkers or self-immolative polymer hydrogel particles.
3.4. Surfaces
Interfaces play an important role in biological systems, especially when artificial materials are brought in contact with living tissue. Much effort has been placed in modulating these interfaces by various means of surface modification such that the material becomes biocompatible and does not evoke an undesired foreign body response.162,163 Recently, however, the focus has shifted towards the design of responsive interfaces or surfaces that are able to interact with their biological environment9 and enzymes are emerging as attractive stimuli for the design of responsive surfaces.Compared to the systems previously described, enzyme responsive surfaces often combine enzyme sensitivity and a change in the material property in one system, making the presence of a unit responsible for the signal translation obsolete. Hence, for enzyme responsive surfaces the enzymatic action directly affects the chemical or physical properties of the material. Only when blocking groups are removed from the surface can the ESF and the translation of the enzymatic reaction be considered to occur simultaneously (Fig. 7). The main focus of this section will be on flat surfaces (Table 6). Surfaces of 3D structures—in particular particles—will be discussed shortly but treated in more detail in Section 3.5.
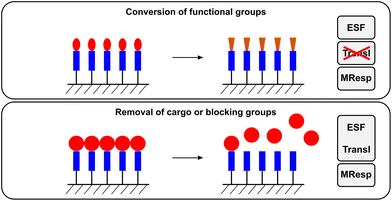 | ||
| Fig. 7 The functions performed by enzyme responsive surfaces. In the case of the conversion of functional groups on the surface, the translation of the enzymatic reaction (Transl) does not occur since the conversion of the functional group is concomitant with the material response (MResp). For the removal of compounds/materials from the surface, the enzyme sensitive functionality (ESF) is combined with the translation of the enzymatic reaction (Transl). | ||
| Structure response | Entry | Enzyme | Surfaceab | Ref. | |
|---|---|---|---|---|---|
| Before | After | ||||
| a SAM: self-assembled monolayer b PS: polystyrene | |||||
| Change in redox properties | 1 | Cutinase | 4-Hydroxyvinylvalerate-[SAM-gold] | Hydroquinone-[SAM-gold] | 35, 67, 168 |
| Change of biological properties | 2 | Abelson tyrosine kinase | Ac-AIYENPFARKC-[SAM-gold] | Ac-AIpYENPFARKC-[SAM-gold] | 165, 166 |
| 3 | Alkaline phosphatase | RGDpS-[glass] | RGDS-[glass] | 167 | |
| pYRGDpS-[glass] | YRGDS-[glass] | ||||
| 4 | Chymotrypsin | Fmoc-FRGD-[glass] | RGD-[glass] | 57 | |
| 5 | Elastase | Fmoc-AARGD-[glass] | RGD-[glass] | 70 | |
| 6 | Thermolysin | Fmoc-G↓F-[glass] | F-[glass] | 164 | |
| Release of materials | 7 | MMP | [particle]-KRGPQG↓IWGQDRCGR-[PS well plate] [particle]-KRGPQG↓IAGQDRCGR-[PS well plate] [particle]-KRGDQG↓IAGFDRCGR-[PS well plate] | [particle]-KRGPQG and IWGQDRCGR-[PS well plate]/IAGQDRCGR-[PS well plate]/IAGFDRCGR-[PS well plate] | 170 |
Non-peptide based enzyme responsive surfaces have been developed by Mrksich and co-workers. Using alkanethiol based self-assembled monolayers (SAMs), they immobilised 4-hydroxyvinylvalerate on gold substrates.35,67,168,169 Exposure to cutinase cleaves the ester of the substrate, generating hydroquinone which can electrochemically be oxidised to quinone and thus be used to determine the presence/activity of the enzyme.67 Collier and Mrksich further demonstrated that cells that are genetically engineered to express cutinase on their surface are able to initiate the same reaction.168
In addition to serving as the interfacial contact point between biomolecules or cells, surfaces have also been used as reservoirs from which materials can be enzymatically released. Segura and co-workers tethered polystyrene nanoparticles decorated with biotin to an avidin functionalised surface.170 The biotin was attached to the particle via enzyme cleavable peptide sequences such that exposure to cell expressed MMPs could cleave the peptide link and liberate the particles from the surface.
In contrast to gold particles, the surface functionalisation of mesoporous silica particles has a different function; it is used to block the pores of the material, thus preventing migration of molecules through pores until the bulky groups are removed enzymatically. Although both gold particles and mesoporous silica particles fit the description of three dimensionally structured surfaces, they will be discussed in detail in the following section on enzyme responsive particles.
3.5. Particles
Among ERMs, the interest and effort invested in the development of enzyme responsive particles is unmatched. Their main applications are focused on enzyme detection and drug delivery. Both areas draw from a wide pool of enzymes; consequently, a large number of enzyme responsive particles have been developed. The following sections class enzyme responsive particles according to the material of the particle itself (Fig. 8), which mostly also determines and limits the type of enzyme responsive mechanism that can be employed to elicit a material response. Only systems that respond directly to the enzymatic action are included in this section; there are many examples where the particle system is sensitive to the product of an enzymatic reaction. These systems are not considered to be genuinely enzyme responsive and the reader is referred to other reviews on this topic.12,27,28 | ||
| Fig. 8 The functions performed by enzyme responsive particles. The arrangement of the components (ESF, Transl and MResp) varies and depends on the type of material/function performed. | ||
| Structure response | Entry | Enzyme | Enzyme sensitive functionality | Product | Ref. |
|---|---|---|---|---|---|
| a CephS: cephalosporine | |||||
| Particle assembly in two component particle/linker systems | 1 | Alkaline phosphatase | CpYR | CYR | 63 |
| 2 | Acetylcholine-esterase | Acetylthiocholine | Thiocholine | 53 | |
| 3 | β-Lactamase | [CephS]-thiol-[linker]-thiol-[CephS] | HS-[linker]-SH | 172 | |
| [CephS]-thiol-[linker]a | HS-[linker] | 180 | |||
| 4 | Esterase | [thioester]GGRGGK-[amide] | [thiol]-GGGRGGK-[amine] | 181 | |
| Particle assembly in two component heterogeneous particle/particle systems | 5 | Abl kinase | Ac-IYAAPKKGGGGC | Ac-IpYAAPKKGGGGC | 188 |
| 6 | Src kinase | Ac-IYGEFKKKC | Ac-IpYGEFKKKC | 187, 188, 204 | |
| 7 | Kinase II | CALNNAAKKLNRTLSVA | CALNNAAKKLNRTLp(biotin)SVA | 66 | |
| 8 | Abl kinase | [particle]-SRVGEEEHVYSFPNKQKSAEC | [particle]-SRVGEEEHVpYSFPNKQKSAEC | 192 | |
| 9 | MMP-2 | [Biotin/Avidin]-GPLG↓VRGC-[PEG] | [Biotin/Avidin]-GPLG and VRGC-[PEG] | 184 | |
| 10 | MMP-2 | GKGPLG↓VRGC-[PEG]-[particle] | VRGC-[PEG]-[particle] | 186 | |
| 11 | MMP-2 | GPLG↓VRG-[PEG]-[particle] | VRG-[PEG]-[particle] | 185 | |
| 12 | MMP-7 | VPLSLTM-[PEG]-[particle] | 185 | ||
| 13 | Protein kinase A | [particle]-CALNNAALRRASLG | [particle]-CALNNAALRRApSLG | 66 | |
| Particle assembly in homogenous single particle systems | 14 | EcoRI | 5′-G↓AATTC-3′ | 5′-GAATTC-3′ | 189 |
| 3′-CTTAA↓G-5′ | 3′-CTTAAG-5′ | ||||
| 15 | Gelatinase | [particle]-gelatine | [particle] | 191 | |
| 16 | Trypsin | [particle]-gelatine | [particle] | 191 | |
| Particle dispersion by cleavage of the linker | 17 | Thermolysin | [thiol]-GGG↓FGGK-[amine] | [thiol]-GGG↓FGGK-[amine] | 181 |
| 18 | DNase I | [particle]-[DNA] | [particle] | 182 | |
| [DNA]-[particle] | |||||
| 19 | Dpn II endonuclease | [particle]-TGAG↓GATC CTCA | [particle]-TGAG | 176 | |
| ACTC CTAG↓GAGT-[particle] | |||||
| 20 | EcoRI | [particle]-G↓AATT C | [particle]-G | 175, 176 | |
| C TTAA↓G-[particle] | |||||
| 21 | EcoRV | [particle]-[DNA] | [particle] | 183 | |
| [DNA]-[particle] | |||||
| 22 | HRP | ferrocene | [ferrocene]+ | 179 | |
| 23 | Lethal factor | Ac-C(S-Ac)LRRRRVYP↓YPnorLELC(S-Ac) | Ac-C(S-Ac)LRRRRVYP and YPnorLELC(S-Ac) | 174 | |
| 24 | MMP-2 | [Biotin]-GGPLGVRGK(Biotin) | 173 | ||
| 25 | Trypsin | RRRRRR | 177 | ||
| [Biotin]-GPARLAIK(Biotin) | 173 | ||||
| 26 | Renin | RK(Biotin)IHPFHLVIHTK (Biotin)R | 173 | ||
| 27 | Thrombin | Ac-C(S-Ac)GDFPR↓GC(S-Ac) | Ac-C(S-Ac)GDFPR and GC(S-Ac) | 174 | |
| Particle dispersion by changing surface charges/hydrophilicity | 28 | Thermolysin | Fmoc-G↓FC-[particle] | Fmoc-G and FC-[particle] | 40, 190 |
| 29 | Protein kinase 2 | WGPGGPPSLPGKKGGC | WGPGGPPpSLPGKKGGC | 178 | |
| WGAVSLSRNLKKGGC | WGAVSLpSRNLKKGGC | ||||
| WGLADVSEQRRLAKKGGC | WGLADVpSEQRRLAKKGGC | ||||
| 30 | Protein kinase A | WGLSARRLAXXC | WGLpSARRLAXXC | 178 | |
| 31 | Protein kinase Cα | KKKAFSGQKKFXXC | KKKAFpSGQKKFXXC | 178 | |
| 32 | YOP protein tyrosine phosphatase | [particle]-SRVGEEEHVpYSFPNKQKSAEC | [particle]-SRVGEEEHVYSFPNKQKSAEC | 192 | |
| Release of molecules entrapped in polymer coated particles | 33 | Thrombin | DDD(PPG)2LVPRGS(PPG)3GC-[particle] | GS(PPG)3GC-[particle] | 199 |
Two component systems where a homogeneous particle population is mixed with a bivalent linker unit cause spontaneous aggregation of the particles. The two reactive units of the linker are joined via an enzymatically cleavable group. Once exposed to the enzyme this linker is broken, causing the particles to disperse. Strategies to bind the linker to the particles include biotin–avidin interactions,173 thiols174–176 and electrostatic interactions.177,178 Dispersion of the nanoparticle aggregates was accomplished with a variety of proteases (trypsin,173,177 renin,173 MMP-2,173 thrombin,174 lethal factor174 and kinases178) for peptide based linkers and endonucleases (EcoRI,175,176 Dpn II176) for DNA linkers. A host–guest based particle/linker system was recently proposed by Velders and co-workers.179 Gold nanoparticles were modified with β-cyclodextrin which selectively binds to a ferrocene dimer. Horseradish peroxidase was used to decrease the binding affinity of ferrocene for β-cyclodextrin, thus causing disassembly of the particle clusters.
The enzyme response of particle/linker systems is not restricted to particle dispersion. Enzyme induced aggregation can be accomplished when the functional end-groups of the linker are masked by enzyme sensitive groups. Thiols have been masked with β-lactamase sensitive cephalosporin172,180 or acetylcholinesterase cleavable thioesters.53 Choi et al. introduced the use of charge-induced aggregation by using a tripeptidic (CYR) linker.63 While the tyrosine in this linker is phosphorylated, the negative charge of the phosphate group engages the positively charged arginine and prevents interaction of arginine with the gold nanoparticle. Alkaline phosphatase removes the phosphate group, allowing both arginine (via electrostatic interactions) and cysteine (via formation of an S–Ag bond) to bind to the particle, thus causing aggregation. Liu et al. combined particle aggregation and dispersion.181 This was accomplished by designing a dually responsive linker. While one enzyme activates the linker to cause particle aggregation, the second enzyme cleaves the linker and thus causes re-dispersion of the particles. A peptide sequence was terminated with a masked thiol on one end and a masked amine on the other. The masking moieties were attached via thioester and amide bonds, respectively, and could be removed by exposure to esterase. The free linker then caused aggregation of the gold nanoparticles. This could be reversed by addition of thermolysin which cleaved the peptide sequence of the linker unit.
Two component systems with heterogeneous particle populations rely on the complementary interaction of the surface immobilised molecules. To cause dispersion of aggregates of a heterogeneous particle population Mirkin and co-workers prepared two sets of gold particles decorated with complementary, single stranded DNA.182 When mixing these two populations, the particles aggregated and could subsequently be dispersed by exposure to an endonuclease (DNAse I). Yu et al. used the same DNA based technology to cross-link polymer coated iron oxide particles and cause particle dispersion upon exposure to EcoRV.183 Enzyme triggered particle aggregation from heterogeneous particle systems was explored by Harris et al.184 They prepared two populations of nanoparticles, modified on their surface with either biotin or avidin. The interaction of the latter two was impeded by the attachment of PEG chains onto the biomolecules. A peptide linker between the PEG and the biotin/avidin allowed the removal of the PEG via enzymatic cleavage (MMP-2) of the peptide. This liberates the biotin and avidin molecules and causes the particles to aggregate. In further studies, these researchers used the same masking technology to create multiple enzyme responsive systems185 and particles with hidden biologically active surface functionalities that could be revealed after enzymatic action.186 Brust and co-workers designed a kinase responsive system consisting of an avidin modified gold particle population and a population of gold particles that were decorated with peptides containing substrate sequences for a kinase.66 The kinase was used to attach γ-biotin-ATP to the second particle population, causing aggregation of the particles. Another kinase responsive two component gold nanoparticle system was developed by Gupta et al. who immobilised a Src kinase substrate on one particle population and antiphosphotyrosine antibodies on a second population.187,188 Addition of the enzyme caused phosphorylation of the peptide substrate of the first population which is recognized by the antibodies on the second, thus causing particle aggregation. The feasibility of this approach to prepare Abl kinase responsive systems has also been demonstrated.188
Enzymatically triggered aggregation of a single population of gold nanoparticles was first shown by Kanaras et al.189 The particles were decorated with double stranded DNA that could be cleaved by the restriction enzyme EcoRI. The cleavage produces a self-complementary DNA fragment that is able to hybridize with a second, identical strand from another particle, thus causing aggregation of the particles. Notably the newly formed DNA strand could be covalently joined by introducing a DNA ligase to improve the stability of the construct. Stevens and co-workers demonstrated that enzyme triggered dispersion of gold particles could be accomplished when the particles are modified with short, Fmoc terminated peptide sequences.40,190 The Fmoc moiety causes the particles to aggregate due to hydrophobic interaction (π-stacking). Exposure to thermolysin detaches the Fmoc group from the particle by cleaving the peptide sequence, causing the particles to disperse. Another way of masking surface functionalities from exerting their attractive forces on neighbouring nanoparticles was introduced by Chuang et al.191 Gold particles were decorated with 6-mercaptohexan-1-ol and gelatine, the latter impeding access to the thiol functionality. Enzymatic digestion of gelatine (by either trypsin or gelatinase) removes the protective layer, unmasking the thiol and thus increasing the particle–particle interactions.
A notable advancement in enzyme responsive particle technology was the introduction of the reversible aggregation/dispersion of iron oxide nanoparticles by Maltzahn et al.192 Two populations of particles were prepared. The first was modified with a peptide sequence that could be phosphorylated by Abelson tyrosine kinase and dephosphorylated by YOP protein tyrosine phosphatase. The second population was decorated with the Src Homology 2 domain that selectively binds to the phosphorylated peptide on the first particle population. By combining both particles, aggregation and dispersion of the system could be accomplished by enzymatic phosphorylation and dephosphorylation of the peptide sequence.
A series of manuscripts describe the use of gold nanoparticles as enzyme responsive sensing agents whose material response does not rely on the aggregation or dispersion of the particles. Instead, changes in the fluorescence of attached molecules are induced by enzymatic cleavage of the fluorophore from the particle. Enzymes used for these systems include thermolysin,193,194 thrombin,193,195 cathepsin L,193 MMP,196,197 trypsin,194 chymotrypsin,194 proteinase K,194 HaeIII,198 EcoRI198 and EcoRV.198 Instead of covalent attachment of the fluorophore to the particle, Wang et al. physically entrapped the fluorophore in a peptide layer on the particle surface.199 The peptide layer was cleaved with thrombin to release the payload from the material.
| Structure response | Entry | Enzyme | Enzyme sensitive functionality | Ref. |
|---|---|---|---|---|
| Polymer capsule degradation | 1 | α-Chymotrypsin | Poly(L-lysine) | 205, 206 |
| 2 | Caspase-3 | VDEVD↓TK | 208 | |
| 3 | Chitosanase | Chitosan | 38, 205 | |
| 4 | Furin | RVRR↓SK | 210 | |
| 5 | MMP | KLGPAK | 209 | |
| 6 | Plasmin | KNRVK | 209 | |
| 7 | Trypsin | GFF | 207 | |
| Polymer particle degradation/disassociation | 8 | Cathepsin B | [PEG]-GF↓LGK-[PEG] | 202 |
| 9 | Elastase | Polypeptide | 201 | |
| 10 | Protein kinase Cα | KKKAFSGQKKF | 204 | |
| Polymer hydrogel particle swelling | 11 | Elastase, thermolysin | Fmoc-A↓APV-[PEGA] | 101, 102 |
| Fmoc-DA↓AR-[PEGA] | 68, 101, 102 | |||
| Fmoc-RRA↓ADD-[PEGA] | ||||
| 12 | MMP-1/12 | GPQG↓IWGQ | 102 |
Enzymatic degradation of polymer based particles relies on either direct degradation of the polymer itself or on the incorporation of enzyme sensitive linkers within the polymer matrix. Direct degradation of hybrid polypeptide–synthetic polymer based micellar particles with elastase was shown by Habraken et al.201 Particles with enzyme sensitive cross-links were prepared from PEG,202 and polystyrene.203 The peptidic cross-links were degraded by enzymes such as cathepsin B,202 and trypsin.203 Recently, Koga et al. presented a polymer particle formed by association of two oppositely charged components, a positively charged lipopeptide and a negatively charged polypeptide.204 Phosphorylation of the lipopeptide by a protein kinase decreases the net positive charge of the lipopeptide, thus causing disassembly of the particle.
Akashi and co-workers designed enzymatically degradable polymer capsules based on the layer by layer assembly of dextran sulfate and chitosan.38,205 These capsules could be degraded in the presence of chitosanase, thus releasing the cargo entrapped within the capsule.38 This technology was later expanded to release two different proteins sequentially upon the action of a single enzyme (chitosanase).205 A dually enzyme responsive capsule system based on dextran sulfate, chitosan and poly(L-lysine) where a first enzyme (α-chymotrypsin) was used to release one protein and a second enzyme (chitosanase) triggered the release of a second molecule was also realised.205 Wang et al. also reported a single enzyme (α-chymotrypsin) responsive poly(L-lysine) based polymer capsule,206 whereas Andrieu et al. presented a trypsin responsive capsule comprised of a polypeptide–synthetic polymer conjugate.207 Instead of producing hollow, drug loaded polymer capsules, Gu et al. enveloped proteins with a peptide cross-linked polymer shell.208 Exposure to capsase-3 cleaved the cross-link and revealed the native protein. The same strategy was used to create plasmin,209 MMP209 and furin210 degradable capsules around proteins.
| Structure response | Entry | Enzyme | Enzyme sensitive functionality | Ref. |
|---|---|---|---|---|
| Opening of pores by degradation | 1 | α-Amylase | Cyclodextrin | 213 |
| 2 | α-Chymotrypsin | Poly(L-lysine) | 215 | |
| 3 | Amylase | Various saccharides | 212 | |
| 4 | β-D-Galactosidase | Lactose | 43, 212 | |
| 5 | DNase I | Cytosine-phosphodiester-guanine oligodeoxynucleotide | 214 | |
| 6 | Elastase | Fmoc-EA↓AR | 41 | |
| 7 | Lipase | Cyclodextrin | 213 | |
| 8 | Trypsin | Avidin | 171 | |
| Opening of pores by triggering disassembly of a host–guest system | 9 | Porcine liver esterase | [adamantyl]-ester-[rotaxane] | 42 |
Degradable surfaces on mesoporous silica particles have been realised by modifying the particle surface with proteins,171 peptides41,211 and polysaccharides.212 Schlossbauer et al. attached biotin to the particle surface; subsequent exposure to avidin capped the pores and prevented leakage from within the particle.171 Trypsin was then used to degrade the protein and open the pores. Bernardos et al. used lactose43,212 and various starch derivatives212 to block the exit from the pores and enzymatically degraded the saccharide layer on the surface with β-D-galactosidase43,212 or amylases present in pancreatin.212 Coll et al. attached a peptide sequence consisting of 18 amino acids to the particle surface and demonstrated enzyme triggered release from the pores upon exposure to proteolytic enzymes from streptomyces griseus.211 By using shorter peptide sequences (4 amino acids) terminated with an Fmoc group, Thornton and Heise demonstrated that egress of molecules from the particle could also be prevented.41 In this case, elastase was used to cleave the Fmoc group from the surface and open the pores of the silica particles. A dual enzyme responsive system was presented by Park et al.213 Bulky cyclodextrins were attached to the surface via short enzyme cleavable linkers. Lipase was able to unblock the pores by cleaving the linker between the cyclodextrin and the particle, whereas α-amylase was used to digest cyclodextrin directly. Zhu and co-workers used electrostatic interactions to coat mesoporous silica particles with either a single layer of cytosine-phosphodiester-guanine oligodeoxynucleotide (CpG ODN) only214 or with CpG ODN and poly(L-lysine) via layer by layer assembly215 and thus block the pores of the particles. The former was removed by digestion of the oligonucleotide with DNase I214 whereas the latter was degraded upon exposure to α-chymotrypsin.215
In all the above examples, the enzyme sensitive group simultaneously conveyed the enzymatic action into a change in the material properties (opening of the pores). A different strategy based on a host–guest system separates the enzyme sensitive group from the translational mechanisms. A [2]rotaxane is immobilised on the particle; one end of the linear thread is capped by the particle itself, whereas the other end is capped by an enzymatically cleavable group. The presence of the bulky host of the [2]rotaxane on the particle surface prevents egress of molecules through the pores. By removing the second cap, the host of the [2]rotaxane is able to leave the surface, thus opening the pores of the mesoporous particle. This concept was first demonstrated by Patel et al. who used an α-cyclodextrin–PEG rotaxane capped with adamantyl.42 Porcine liver esterase was used to remove the cap and open the pores of the particle.
| Structure response | Entry | Enzyme | Enzyme sensitive functionality | Ref. |
|---|---|---|---|---|
| Quenching of quantum dots | 1 | Alkaline phosphatase/tyrosinase | [QD]-DADEpYLIPQQ | 228 |
| 2 | Casein kinase | [QD]-RRRADDSD | 228 | |
| 3 | MMP-2 | [luciferase]-GGPLG↓VRGG-[QD] | 227 | |
| Reduction of quantum dot quenching | 4 | BamHI | 5′-GGATCC-3′ | 222 |
| 5 | β-Lactamase | β-Lactam | 217 | |
| 6 | Caspase-1 | GL-[α-amino isobutyric acid]-AAGGWEHDSGC | 221 | |
| 7 | Caspase-3 | DEVD | 219 | |
| 8 | Chymotrypsin | GL-[α-amino isobutyric acid]-AAGGWGC | 221 | |
| 9 | Collagenase | GGLGPAGGCG | 220 | |
| PLGLCG | 224 | |||
| AL-[α-amino isobutyric acid]-AAGGPAC | 221 | |||
| 10 | MMP-7 | [gold particle]-CRPLALWRSK-[QD] | 223 | |
| [dye]-RPLALWRSK-[QD] | ||||
| 11 | Protein kinase A | LRRASLGGGGC | 64 | |
| 12 | Thrombin | GLA-[α-amino isobutyric acid]-SGFPRGRC | 221 | |
| 13 | Trypsin | RGDC | 218 | |
| 14 | uPA | SGRSANC | 225 | |
| Change of surface functionality | 15 | MMP-7 | RPLALWRS | 226 |
In a departure from the direct enzymatic cleavage of acceptors from the quantum dots, strategies to decrease the luminescence emission of quantum dots upon enzymatic action have been proposed. Yao et al. attached luciferase to a quantum dot via a MMP-2 sensitive peptide linker.227 While in proximity of the quantum dot, the radiation emitted by luciferase during its catalytic activity is transferred to the quantum dot, causing luminescence to occur from the quantum dot itself. After enzymatic detachment of the luciferase, the energy transfer is largely reduced, causing the decrease in the quantum dot luminescence intensity. Freeman et al. proposed a system where the enzyme mediated attachment of an acceptor to the quantum dot was used to lower the luminescence of the quantum dot.228 The quantum dots were modified with a serine containing peptide sequence to which a γ-adenosine triphosphate–acceptor conjugate could be attached with casein kinase.228 In a second system, Freeman et al. made the quenching of the quantum dot dependant on the subsequent action of two different enzymes. The peptide sequence with which the quantum dot was modified contained a phosphorylated tyrosine unit, which was first dephosphorylated with alkaline phosphatase. The free tyrosine unit was then oxidised with tyrosinase, producing a dopaquinone which quenches the luminescence of the quantum dots.228
Similar to the gold nanoparticle systems, the enzymatic dispersion of quantum dots has also been explored as mechanism to change their photophysical properties. Xu et al. used a positively charged peptide to mask the negative charge of quantum dots bearing carboxyl functionalities on their surface.64 Phosphorylation of the peptide reduced the overall positive charge, revealing more negative charges on the quantum dot surfaces that subsequently caused the dispersion of the particles.
4. Challenges in the development of ERMs for specific applications
4.1. Applications of ERMs
In nature, enzymes are the main control mechanisms that regulate the complex biological processes that are still unmatched by artificial systems. The vision for ERMs is to synthetically make materials that are able to replace or interact with natural biological systems in a seamless manner. The power of enzymatically controlled material properties lies in the possibility to design or pre-program a material response that is entirely controlled by the biological surrounding, ultimately achieving assimilation of the ERM into a biological process. While the level of biological complexity is still beyond our grasp, ERMs developed to date have already shown tremendous potential to perform specific tasks on cue when prompted by an enzyme.The application of enzyme responsive systems has already been demonstrated in a variety of different disciplines, including enzyme diagnostics,12,40,188,229 drug delivery51,75,76,230–232 and regenerative medicine.73,233–238 For the detection of enzymes and enzyme activity, various enzyme responsive particles have been developed (see Tables 7 and 10). Enzyme detection generally relies on a change in magnetic or spectroscopic properties due to particle aggregation or dispersion or on controlling the quenching of quantum dots.12,27 Some of these systems have been shown to be very sensitive to low enzyme concentration levels.40,190,225 The strength of enzymatically controlled drug delivery lies in the ability to allow drug release ‘on demand’ in the presence of specific enzymes. Many diseases have been associated with higher levels of specific enzymes (see Table 1), thus providing attractive markers and stimuli for the spatially and temporally targeted delivery of therapeutics. A combination of both drug delivery and enzyme detection is often desired to concomitantly treat the diseased tissue and measure the effect of the treatment. Regenerative medicine encompasses a broad spectrum of research activities with potential attraction for ERMs. Applications of ERMs in this area include the enzymatic formation and degradation of hydrogels as artificial cell supports73,234 and the enzymatic control of surface properties to control cell response57,70 in addition to the enzyme triggered release of bioactive molecules such as growth factors.238
4.2. Matching enzymes and materials to the application
One challenge in the design of ERMs is to choose the right enzyme/ESF. While model enzymes are often convenient to demonstrate the mechanism of an ERM, the system is only of value in an application if the material is able to perform under the specific conditions present in its environment. In addition, the target enzyme is required to affect only the ESF in the material, leaving the rest of the material unchanged, to avoid non specific responses. The variety of mechanisms that have been developed to realise the same material response (see for example the variety of mechanisms to create supramolecular assemblies) is reflective of the need to have a versatile repertoire of ERMs that can be adapted for a particular application.Apart from responsiveness to the marker enzyme, inertness of the ERM to other components in the system (other enzymes, pH, temperature, etc.) is essential to avoid unwanted effects of the biological environment on the material. Many ERMs have been developed to respond to an enzyme with a high specificity to a certain substrate; fewer reports have demonstrated cross-reactivity of the material with other enzymes of broader specificity (e.g. broad specificity proteases).68,101 ERMs that do not display the required specificity to a particular enzyme have limited application potential. This is of particular concern in materials where longer peptide or DNA sequences are used that may be recognised by other enzymes. In order for ERMs to be able to move forward towards high impact applications, these issues need to be addressed.
Several ERMs with simple ESF (di- or tri-peptides that are only sensitive to very specific enzymes) or intentionally broad enzyme specificity have been shown to perform well in in vitro studies. These include the formation of supramolecular hydrogels inside cells,239 and polymer hydrogel degradation.36,55,75 In these applications, the material itself (polymer, gold nanoparticles) has been chosen for its inertness in a biological surrounding (excepting the ESF), low toxicity and its ability to be metabolised after performing its function. In addition, recent trends show increasing interest in natural and renewable materials such as alginate85 and carbohydrates.39,84,86,104,240
4.3. Choosing the enzyme response mechanism
Once a target enzyme has been chosen for a particular application, the type of mechanism employed to translate enzymatic action into a material response has to be tailored to the enzymes catalytic action. While both bond formation and cleavage have been used to date, bond cleavage has been the more popular choice for existing ERMs. However, inventive methods have been employed to accomplish opposite material responses with the same enzymatic reaction. For example, enzymatic cleavage of peptide sequences on metal nanoparticles has been used to cause both aggregation and dispersion of the particles.173,185,186Recent trends in ERM development, however, point into the direction of more dynamic or reversible systems, moving away from ‘single-use’ ERMs. Incorporating functionality that can be addressed reversibly and/or on an on-demand basis is a further step towards a tighter incorporation of ERMs into a biological system. Applications where phosphatases and kinases play critical roles are ideally suited for this because of their natural design to catalyse opposite reactions. It is therefore not surprising that most reversible ERMs are based on a phosphatase/kinase system. Even though nucleases and ligases perform similar functions (cleavage and bond formation between nucleotides), only one example of an ERM is known where the DNA strands are chemically reconnected after enzymatic cleavage.189 In principle, enzymatic reactions are reversible and it has been shown that enzymes can be used to catalyse reactions opposite to their natural functions if the thermodynamic equilibrium of the reactants and products can be shifted.45,52 However, this approach has not yet found its way into the design of reversible ERMs.
4.4. Future developments
The main strength of ERMs compared to other stimuli responsive materials clearly lies in their ability to interact with a biological environment with the same communication mechanism used by nature. Ideally, ERMs will perform their function with high specificity to their target enzyme, largely undisturbed by the multitude of other processes in the biological environment. To date, research on ERMs has mainly focused on the development of enzyme responsive mechanisms and the translation into a material response. Considerable progress has been made over the last decade in the form of new translational mechanisms and the incorporation of ESFs into artificial materials. Even though the high attraction of enzyme responsive systems to act dynamically and on more than one enzymatic stimulus has been recognised, this area of ERM development still holds great potential to develop new materials with unprecedented possibilities. For example, instead of degradation based drug release mechanisms, the temporally controlled ‘on demand’ release of multiple drugs in response to multiple enzymes with integrated enzyme activity detection would be a highly attractive ERM system. But going even further, ERMs able to not simply support, but actively direct the formation of tissue in wound healing or tissue regeneration in response to marker enzymes that communicate the state of the cells/tissue to the material is thinkable. In order to accomplish these ambitious goals, the refinement of the existing mechanism will be necessary to accommodate reversible and multiple ESFs. Of equal importance will be a stronger focus on the assessment of these materials in an in vitro and in vivo setting to evaluate their performance in biological environments to be able to push ERMs to the next level and design them with the potential for real life applications in mind.Acknowledgements
MZ is supported by a Marie Curie Fellowship under the 7th Framework program. RVU acknowledges the Leverhulme Trust (Leadership Award), ERC (Starting Grant EMERgE). The material is based on research sponsored by the Air Force Laboratory, under agreement number FA9550-11-1-0263. The US Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon.References
- A. S. Hoffman, Applications of “Smart Polymers” as Biomaterials, Elsevier Academic Press, London, 2nd edn, 2004 Search PubMed.
- J. Kopecek, Eur. J. Pharm. Sci., 2003, 20, 1–16 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- R. de la Rica and H. Matsui, Chem. Soc. Rev., 2010, 39, 3499–3509 RSC.
- N. M. Alves, I. Pashkuleva, R. L. Reis and J. F. Mano, Small, 2010, 6, 2208–2220 CrossRef CAS.
- M. Delcea, H. Mohwald and A. G. Skirtach, Adv. Drug Delivery Rev., 2011, 63, 730–747 CrossRef CAS.
- A. D. Bendrea, L. Cianga and I. Cianga, J. Biomater. Appl., 2011, 26, 3–84 CrossRef CAS.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- J. E. Ghadiali and M. M. Stevens, Adv. Mater., 2008, 20, 4359–4363 CrossRef CAS.
- R. J. Williams, R. J. Mart and R. V. Ulijn, Biopolymers, 2010, 94, 107–117 CrossRef CAS.
- M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- J. E. Ghadiali, S. B. Lowe and M. M. Stevens, Angew. Chem., Int. Ed., 2011, 50, 3417–3420 CrossRef CAS.
- J. E. Ghadiali, B. E. Cohen and M. M. Stevens, ACS Nano, 2010, 4, 4915–4919 CrossRef CAS.
- M. Privman, T. K. Tam, M. Pita and E. Katz, J. Am. Chem. Soc., 2008, 131, 1314–1321 CrossRef.
- R. Bonomi, A. Cazzolaro, A. Sansone, P. Scrimin and L. J. Prins, Angew. Chem., Int. Ed., 2011, 50, 2307–2312 CAS.
- J. S. Liu, X. Z. Du and X. F. Zhang, Chem.–Eur. J., 2011, 17, 810–815 CrossRef CAS.
- W. R. Zhao, H. T. Zhang, Q. J. He, Y. S. Li, J. L. Gu, L. Li, H. Li and J. L. Shi, Chem. Commun., 2011, 47, 9459–9461 RSC.
- C. R. Gordijo, K. Koulajian, A. J. Shuhendler, L. D. Bonifacio, H. Y. Huang, S. Chiang, G. A. Ozin, A. Giacca and X. Y. Wu, Adv. Funct. Mater., 2011, 21, 73–82 CrossRef CAS.
- C. R. Gordijo, A. J. Shuhendler and X. Y. Wu, Adv. Funct. Mater., 2010, 20, 1404–1412 CrossRef CAS.
- M. E. Hahn and N. C. Gianneschi, Chem. Commun., 2011, 47, 11814–11821 RSC.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS.
- H. G. Börner, H. Kühnle and J. Hentschel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1–14 CrossRef.
- D. Aili and M. M. Stevens, Chem. Soc. Rev., 2010, 39, 3358–3370 RSC.
- K. Welser, R. Adsley, B. M. Moore, W. C. Chan and J. W. Aylott, Analyst, 2011, 136, 29–41 RSC.
- Y. Gao, Z. Yang, Y. Kuang, M.-L. Ma, J. Li, F. Zhao and B. Xu, Biopolymers, 2010, 94, 19–31 CrossRef CAS.
- J. M. Schurr, Biophys. J., 1970, 10, 717–727 CrossRef CAS.
- J. Deere, R. F. De Oliveira, B. Tomaszewski, S. Millar, A. Lalaouni, L. F. Solares, S. L. Flitsch and P. J. Halling, Langmuir, 2008, 24, 11762–11769 CrossRef CAS.
- H. J. Lee, A. W. Wark, T. T. Goodrich, S. P. Fang and R. M. Corn, Langmuir, 2005, 21, 4050–4057 CrossRef CAS.
- S. Fang, H. J. Lee, A. W. Wark, H. M. Kim and R. M. Corn, Anal. Chem., 2005, 77, 6528–6534 CrossRef CAS.
- H. J. Lee, A. W. Wark and R. M. Corn, Langmuir, 2006, 22, 5241–5250 CrossRef CAS.
- S. Nayak, W. S. Yeo and M. Mrksich, Langmuir, 2007, 23, 5578–5583 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS.
- Y. Kumashiro, T. K. Lee, T. Ooya and N. Yui, Macromol. Rapid Commun., 2002, 23, 407–410 CrossRef CAS.
- Y. Itoh, M. Matsusaki, T. Kida and M. Akashi, Biomacromolecules, 2006, 7, 2715–2718 CrossRef CAS.
- N. Morimoto, N. Ogino, T. Narita, S. Kitamura and K. Akiyoshi, J. Am. Chem. Soc., 2007, 129, 458–459 CrossRef CAS.
- A. Laromaine, L. L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS.
- P. D. Thornton and A. Heise, J. Am. Chem. Soc., 2010, 132, 2024–2028 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y. W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS.
- A. Bernardos, E. Aznar, M. Dolores Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, J. Manuel Barat and P. Amoros, Angew. Chem., Int. Ed., 2009, 48, 5884–5887 CrossRef CAS.
- Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS.
- M. Mutter, A. Chandravarkar, C. Boyat, J. Lopez, S. Dos Santos, B. Mandal, R. Mimna, K. Murat, L. Patiny, L. Saucede and G. Tuchscherer, Angew. Chem., Int. Ed., 2004, 43, 4172–4178 CrossRef CAS.
- S. Dos Santos, A. Chandravarkar, B. Mandal, R. Mimna, K. Murat, L. Saucede, P. Tella, G. Tuchscherer and M. Mutter, J. Am. Chem. Soc., 2005, 127, 11888–11889 CrossRef CAS.
- C. Wang, Q. S. Chen, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 8612–8615 CrossRef CAS.
- J. J. Sperinde and L. G. Griffith, Macromolecules, 1997, 30, 5255–5264 CrossRef CAS.
- H. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2009, 48, 6431–6434 CrossRef.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252–1256 CrossRef.
- A. K. Das, R. Collins and R. V. Ulijn, Small, 2008, 4, 279–287 CrossRef CAS.
- M. Wang, X. G. Gu, G. X. Zhang, D. Q. Zhang and D. B. Zhu, Langmuir, 2009, 25, 2504–2507 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- B. K. Mann, A. S. Gobin, A. T. Tsai, R. H. Schmedlen and J. L. West, Biomaterials, 2001, 22, 3045–3051 CrossRef CAS.
- P.-F. Caponi, X.-P. Qiu, F. Vilela, F. M. Winnik and R. V. Ulijn, Polym. Chem., 2011, 2, 306–308 RSC.
- S. J. Todd, D. Farrar, J. E. Gough and R. V. Ulijn, Soft Matter, 2007, 3, 547–550 RSC.
- L. F. Shi, V. De Paoli, N. Rosenzweig and Z. Rosenzweig, J. Am. Chem. Soc., 2006, 128, 10378–10379 CrossRef CAS.
- R. J. Amir, S. Zhong, D. J. Pochan and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 13949–13951 CrossRef CAS.
- J. Ge, D. N. Lu, C. Yang and Z. Liu, Macromol. Rapid Commun., 2011, 32, 546–550 CAS.
- M. J. Webber, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2011, 7, 9665–9672 RSC.
- Z. M. Yang, G. L. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
- Y. Choi, N. H. Ho and C. H. Tung, Angew. Chem., Int. Ed., 2007, 46, 707–709 CrossRef CAS.
- X. H. Xu, X. Liu, Z. Nie, Y. L. Pan, M. L. Guo and S. Z. Yao, Anal. Chem., 2011, 83, 52–59 CrossRef CAS.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. San Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS.
- Z. X. Wang, R. Levy, D. G. Fernig and M. Brust, J. Am. Chem. Soc., 2006, 128, 2214–2215 CrossRef CAS.
- W. S. Yeo and M. Mrksich, Angew. Chem., Int. Ed., 2003, 42, 3121–3124 CrossRef CAS.
- P. D. Thornton, G. McConnell and R. V. Ulijn, Chem. Commun., 2005, 5913–5915 RSC.
- T. H. Ku, M. P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS.
- S. J. Todd, D. J. Scurr, J. E. Gough, M. R. Alexander and R. V. Ulijn, Langmuir, 2009, 25, 7533–7539 CrossRef CAS.
- M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Bioconjugate Chem., 2001, 12, 1051–1056 CrossRef CAS.
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Biomaterials, 2010, 31, 3103–3113 CrossRef CAS.
- A. A. Aimetti, M. W. Tibbitt and K. S. Anseth, Biomacromolecules, 2009, 10, 1484–1489 CrossRef CAS.
- V. K. Garripelli, J. K. Kim, S. Son, W. J. Kim, M. A. Repka and S. Jo, Acta Biomater., 2011, 7, 1984–1992 CrossRef CAS.
- K. J. C. van Bommel, M. C. A. Stuart, B. L. Feringa and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 CAS.
- J. E. Folk and J. S. Finlayson, Adv. Protein Chem., 1977, 31, 1–133 CrossRef CAS.
- H. L. Fuchsbauer, U. Gerber, J. Engelmann, T. Seeger, C. Sinks and T. Hecht, Biomaterials, 1996, 17, 1481–1488 CrossRef CAS.
- T. J. Sanborn, P. B. Messersmith and A. E. Barron, Biomaterials, 2002, 23, 2703–2710 CrossRef CAS.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314 RSC.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- M. Kurisawa, F. Lee, L. S. Wang and J. E. Chung, J. Mater. Chem., 2010, 20, 5371–5375 RSC.
- R. Jin, C. Hiemstra, Z. Y. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS.
- Y. Ogushi, S. Sakai and K. Kawakami, J. Biosci. Bioeng., 2007, 104, 30–33 CrossRef CAS.
- S. Sakai and K. Kawakami, Acta Biomater., 2007, 3, 495–501 CrossRef CAS.
- T. H. Chen, H. D. Embree, L. Q. Wu and G. F. Payne, Biopolymers, 2002, 64, 292–302 CrossRef CAS.
- T. H. Chen, H. D. Embree, E. M. Brown, M. M. Taylor and G. F. Payne, Biomaterials, 2003, 24, 2831–2841 CrossRef CAS.
- J. F. Woessner, FASEB J., 1991, 5, 2145–2154 CAS.
- L. Hovgaard and H. Brondsted, J. Controlled Release, 1995, 36, 159–166 CrossRef CAS.
- N. Yamamoto, M. Kurisawa and N. Yui, Macromol. Rapid Commun., 1996, 17, 313–318 CrossRef CAS.
- M. Kurisawa, M. Terano and N. Yui, J. Biomater. Sci., Polym. Ed., 1997, 8, 691–708 CrossRef CAS.
- M. Kurisawa, Y. Matsuo and N. Yui, Macromol. Chem. Phys., 1998, 199, 705–709 CrossRef CAS.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- M. van Dijk, C. F. van Nostrum, W. E. Hennink, D. T. S. Rijkers and R. M. J. Liskamp, Biomacromolecules, 2010, 11, 1608–1614 CrossRef CAS.
- J. Y. Yang, M. T. Jacobsen, H. Z. Pan and J. Kopecek, Macromol. Biosci., 2010, 10, 445–454 CrossRef CAS.
- S. Kim and K. E. Healy, Biomacromolecules, 2003, 4, 1214–1223 CrossRef CAS.
- N. S. Khelfallah, G. Decher and P. J. Mesini, Macromol. Rapid Commun., 2006, 27, 1004–1008 CrossRef CAS.
- K. N. Plunkett, K. L. Berkowski and J. S. Moore, Biomacromolecules, 2005, 6, 632–637 CrossRef CAS.
- P. D. Thornton and A. Heise, Chem. Commun., 2011, 47, 3108–3110 RSC.
- T. O. McDonald, H. L. Qu, B. R. Saunders and R. V. Ulijn, Soft Matter, 2009, 5, 1728–1734 RSC.
- P. D. Thornton, R. J. Mart, S. J. Webb and R. V. Ulijn, Soft Matter, 2008, 4, 821–827 RSC.
- A. G. Patrick and R. V. Ulijn, Macromol. Biosci., 2010, 10, 1184–1193 CrossRef CAS.
- L. Perlin, S. MacNeil and S. Rimmer, Chem. Commun., 2008, 5951–5953 RSC.
- D. Klinger, E. M. Aschenbrenner, C. K. Weiss and K. Landfester, Polym. Chem., 2012, 3, 204–216 RSC.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. S. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- R. G. Weiss and P. M. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, Dordrecht, 2006 Search PubMed.
- Z. M. Yang and B. Xu, Adv. Mater., 2006, 18, 3043–3046 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728–730 RSC.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS.
- M. Hughes, H. X. Xu, P. Frederix, A. M. Smith, N. T. Hunt, T. Tuttle, I. A. Kinloch and R. V. Ulijn, Soft Matter, 2011, 7, 10032–10038 RSC.
- Q. Wang, Z. Yang, Y. Gao, W. Ge, L. Wang and B. Xu, Soft Matter, 2008, 4, 550–553 RSC.
- H. M. Wang, C. H. Ren, Z. J. Song, L. Wang, X. M. Chen and Z. M. Yang, Nanotechnology, 2010, 21 Search PubMed , Art. Nr. 225606.
- J. Gao, W. T. Zheng, D. L. Kong and Z. M. Yang, Soft Matter, 2011, 7, 10443–10448 RSC.
- M. Hughes, P. W. J. M. Frederix, J. Raeburn, L. S. Birchall, J. Sadownik, F. C. Coomer, I. H. Lin, E. J. Cussen, N. T. Hunt, T. Tuttle, S. J. Webb, D. J. Adams and R. V. Ulijn, Soft Matter, 2012, 8, 5595–5602 RSC.
- L. Chronopoulou, S. Lorenzoni, G. Masci, M. Dentini, A. R. Togna, G. Togna, F. Bordi and C. Palocci, Soft Matter, 2010, 6, 2525–2532 RSC.
- Z. Yang, P.-L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS.
- T. Koga, K. Kitamura and N. Higashi, Chem. Commun., 2006, 4897–4899 RSC.
- L. Adler-Abramovich, R. Perry, A. Sagi, E. Gazit and D. Shabat, ChemBioChem, 2007, 8, 859–862 CrossRef CAS.
- Z. Yang, M. Ma and B. Xu, Soft Matter, 2009, 5, 2546–2548 CAS.
- J. W. Woodcock, X. G. Jiang, R. A. E. Wright and B. Zhao, Macromolecules, 2011, 44, 5764–5775 CrossRef CAS.
- H. W. Jun, V. Yuwono, S. E. Paramonov and J. D. Hartgerink, Adv. Mater., 2005, 17, 2612–2617 CrossRef CAS.
- Y. Chau, Y. Luo, A. C. Y. Cheung, Y. Nagai, S. G. Zhang, J. B. Kobler, S. M. Zeitels and R. Langer, Biomaterials, 2008, 29, 1713–1719 CrossRef.
- B. Law, R. Weissleder and C. H. Tung, Biomacromolecules, 2006, 7, 1261–1265 CrossRef CAS.
- Y. Z. Xing, E. J. Cheng, Y. Yang, P. Chen, T. Zhang, Y. W. Sun, Z. Q. Yang and D. S. Liu, Adv. Mater., 2011, 23, 1117–1121 CrossRef CAS.
- M. A. Azagarsamy, P. Sokkalingam and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 14184–14185 CrossRef CAS.
- K. R. Raghupathi, M. A. Azagarsamy and S. Thayumanavan, Chem.–Eur. J., 2011, 17, 11752–11760 CrossRef CAS.
- A. J. de Graaf, E. Mastrobattista, T. Vermonden, C. F. van Nostrum, D. T. S. Rijkers, R. M. J. Liskamp and W. E. Hennink, Macromolecules, 2012, 45, 842–851 CrossRef CAS.
- V. Castelletto, J. E. McKendrick, I. W. Hamley, U. Olsson and C. Cenker, Langmuir, 2010, 26, 11624–11627 CrossRef CAS.
- P.-F. Caponi, F. M. Winnik and R. V. Ulijn, Soft Matter, 2012, 8, 5127–5130 RSC.
- M. P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS.
- F. E. Alemdaroglu, J. Wang, M. Borsch, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2008, 47, 974–976 CrossRef CAS.
- D. Koda, T. Maruyama, N. Minakuchi, K. Nakashima and M. Goto, Chem. Commun., 2010, 46, 979–981 RSC.
- C. A. Blencowe, A. T. Russell, F. Greco, W. Hayes and D. W. Thornthwaite, Polym. Chem., 2011, 2, 773–790 RSC.
- G. M. Dubowchik and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3341–3346 CrossRef CAS.
- G. M. Dubowchik, K. Mosure, J. O. Knipe and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3347–3352 CrossRef CAS.
- R. Erez, E. Segal, K. Miller, R. Satchi-Fainaro and D. Shabat, Bioorg. Med. Chem., 2009, 17, 4327–4335 CrossRef CAS.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Y. Guan, J. Med. Chem., 1999, 42, 3657–3667 CrossRef CAS.
- R. Madec-Lougerstay, J. C. Florent and C. Monneret, J. Chem. Soc., Perkin Trans. 1, 1999, 1369–1375 RSC.
- R. Satchi-Fainaro, H. Hailu, J. W. Davies, C. Summerford and R. Duncan, Bioconjugate Chem., 2003, 14, 797–804 CrossRef CAS.
- A. Gopin, N. Pessah, M. Shamis, C. Rader and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 327–332 CrossRef CAS.
- A. Gopin, C. Rader and D. Shabat, Bioorg. Med. Chem., 2004, 12, 1853–1858 CrossRef CAS.
- S. Q. Gao, Z. R. Lu, B. Petri, P. Kopeckova and J. Kopecek, J. Controlled Release, 2006, 110, 323–331 CrossRef CAS.
- B. Sauerbrei, V. Jungmann and H. Waldmann, Angew. Chem., Int. Ed., 1998, 37, 1143–1146 CrossRef CAS.
- U. Grether and H. Waldmann, Angew. Chem., Int. Ed., 2000, 39, 1629–1632 CrossRef CAS.
- U. Grether and H. Waldmann, Chem.–Eur. J., 2001, 7, 959–971 CrossRef CAS.
- M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 15688–15689 CrossRef CAS.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS.
- F. M. H. de Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490–4494 CrossRef CAS.
- K. Haba, M. Popkov, M. Shamis, R. A. Lerner, C. F. Barbas and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 716–720 CrossRef CAS.
- M. Shamis, H. N. Lode and D. Shabat, J. Am. Chem. Soc., 2004, 126, 1726–1731 CrossRef CAS.
- A. Sagi, E. Segal, R. Satchi-Fainaro and D. Shabat, Bioorg. Med. Chem., 2007, 15, 3720–3727 CrossRef CAS.
- M. Avital-Shmilovici and D. Shabat, Bioorg. Med. Chem. Lett., 2009, 19, 3959–3962 CrossRef CAS.
- R. Weinstain, P. S. Baran and D. Shabat, Bioconjugate Chem., 2009, 20, 1783–1791 CrossRef CAS.
- R. Weinstain, A. Sagi, N. Karton and D. Shabat, Chem.–Eur. J., 2008, 14, 6857–6861 CrossRef CAS.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- E. Sella, R. Weinstain, R. Erez, N. Z. Burns, P. S. Baran and D. Shabat, Chem. Commun., 2010, 46, 6575–6577 RSC.
- B. D. Ratner, Polym. Int., 2007, 56, 1183–1185 CrossRef CAS.
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS.
- R. E. Rawsterne, J. E. Gough, F. J. M. Rutten, N. T. Pham, W. C. K. Poon, S. L. Flitsch, B. Maltman, M. R. Alexander and R. V. Ulijn, Surf. Interface Anal., 2006, 38, 1505–1511 CrossRef CAS.
- X. L. Liao, J. Su and M. Mrksich, Chem.–Eur. J., 2009, 15, 12303–12309 CrossRef CAS.
- X. L. Liao, R. T. Petty and M. Mrksich, Angew. Chem., Int. Ed., 2011, 50, 706–708 CrossRef CAS.
- M. Zelzer, L. E. McNamara, D. J. Scurr, M. R. Alexander, M. J. Dalby and R. V. Ulijn, J. Mater. Chem., 2012, 22, 12229–12237 RSC.
- J. H. Collier and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2021–2025 CrossRef CAS.
- J. Li, S. Nayak and M. Mrksich, J. Phys. Chem. B, 2010, 114, 15113–15118 CrossRef CAS.
- T. Tokatlian, C. T. Shrum, W. M. Kadoya and T. Segura, Biomaterials, 2010, 31, 8072–8080 CrossRef CAS.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS.
- R. R. Liu, R. S. Liew, H. Zhou and B. G. Xing, Angew. Chem., Int. Ed., 2007, 46, 8799–8803 CrossRef CAS.
- M. Zhao, L. Josephson, Y. Tang and R. Weissleder, Angew. Chem., Int. Ed., 2003, 42, 1375–1378 CrossRef CAS.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3978–3982 CrossRef CAS.
- A. G. Kanaras, Z. X. Wang, M. Brust, R. Cosstick and A. D. Bates, Small, 2007, 3, 590–594 CrossRef CAS.
- G. T. Song, C. E. Chen, J. S. Ren and X. G. Qu, ACS Nano, 2009, 3, 1183–1189 CrossRef CAS.
- W. X. Xue, G. X. Zhang and D. Q. Zhang, Analyst, 2011, 136, 3136–3141 RSC.
- J. Oishi, Y. Asami, T. Mori, J. H. Kang, M. Tanabe, T. Niidome and Y. Katayama, ChemBioChem, 2007, 8, 875–879 CrossRef CAS.
- R. de la Rica, R. M. Fratila, A. Szarpak, J. Huskens and A. H. Velders, Angew. Chem., Int. Ed., 2011, 50, 5704–5707 CrossRef CAS.
- T. T. Jiang, R. R. Liu, X. F. Huang, H. J. Feng, W. L. Teo and B. G. Xing, Chem. Commun., 2009, 1972–1974 RSC.
- R. R. Liu, J. X. Aw, W. L. Teo, P. Padmanabhan and B. G. Xing, New J. Chem., 2010, 34, 594–598 RSC.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 3468–3470 CrossRef CAS.
- S. S. Yu, R. L. Scherer, R. A. Ortega, C. S. Bell, C. P. O'Neil, J. A. Hubbell and T. D. Giorgio, J. NanoBiotechnology., 2011, 9 DOI:10.1186/1477-3155-9-7.
- T. J. Harris, G. von Maltzahn, A. M. Derfus, E. Ruoslahti and S. N. Bhatia, Angew. Chem., Int. Ed., 2006, 45, 3161–3165 CrossRef CAS.
- G. von Maltzahn, T. J. Harris, J. H. Park, D. H. Min, A. J. Schmidt, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2007, 129, 6064–6065 CrossRef CAS.
- T. J. Harris, G. von Maltzahn, M. E. Lord, J. H. Park, A. Agrawal, D. H. Min, M. J. Sailor and S. N. Bhatia, Small, 2008, 4, 1307–1312 CrossRef CAS.
- S. Gupta, H. Andresen, J. E. Ghadiali and M. M. Stevens, Small, 2010, 6, 1509–1513 CrossRef CAS.
- S. Gupta, H. Andresen and M. M. Stevens, Chem. Commun., 2011, 47, 2249–2251 RSC.
- A. G. Kanaras, Z. X. Wang, A. D. Bates, R. Cosstick and M. Brust, Angew. Chem., Int. Ed., 2003, 42, 191–194 CrossRef CAS.
- R. C. Maher, S. A. Maier, L. F. Cohen, L. Koh, A. Laromaine, J. A. G. Dick and M. M. Stevens, J. Phys. Chem. C, 2010, 114, 7231–7235 CAS.
- Y. C. Chuang, J. C. Li, S. H. Chen, T. Y. Liu, C. H. Kuo, W. T. Huang and C. S. Lin, Biomaterials, 2010, 31, 6087–6095 CrossRef CAS.
- G. von Maltzahn, D. H. Min, Y. X. Zhang, J. H. Park, T. J. Harris, M. Sailor and S. N. Bhatia, Adv. Mater., 2007, 19, 3579–3583 CrossRef CAS.
- P. Free, C. P. Shaw and R. Levy, Chem. Commun., 2009, 5009–5011 RSC.
- X. H. Wang, J. Geng, D. Miyoshi, J. S. Ren, N. Sugimoto and X. G. Qu, Biosens. Bioelectron., 2010, 26, 743–747 CrossRef CAS.
- S. J. Zhen, Y. F. Li, C. Z. Huang and Y. F. Long, Talanta, 2008, 76, 230–232 CrossRef CAS.
- J. H. Kim and B. H. Chung, Small, 2010, 6, 126–131 CrossRef CAS.
- S. Lee, E. J. Cha, K. Park, S. Y. Lee, J. K. Hong, I. C. Sun, S. Y. Kim, K. Choi, I. C. Kwon, K. Kim and C. H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804–2807 CrossRef CAS.
- Y. Huang, S. L. Zhao, H. Liang, Z. F. Chen and Y. M. Liu, Chem.–Eur. J., 2011, 17, 7313–7319 CrossRef CAS.
- J. H. Wang, Y. A. Yue, G. J. Chen and J. Xia, Soft Matter, 2011, 7, 7217–7222 RSC.
- L. M. Randolph, M.-P. Chien and N. C. Gianneschi, Chem. Sci., 2012, 3, 1363–1380 RSC.
- G. J. M. Habraken, M. Peeters, P. D. Thornton, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 3761–3769 CrossRef CAS.
- L. C. Glangchai, M. Caldorera-Moore, L. Shi and K. Roy, J. Controlled Release, 2008, 125, 263–272 CrossRef CAS.
- M. Maier, N. Kotman, C. Friedrichs, J. Andrieu, M. Wagner, R. Graf, W. S. L. Strauss, V. Mailander, C. K. Weiss and K. Landfester, Macromolecules, 2011, 44, 6258–6267 CrossRef CAS.
- H. Koga, R. Toita, T. Mori, T. Tomiyama, J. H. Kang, T. Niidome and Y. Katayama, Bioconjugate Chem., 2011, 22, 1526–1534 CrossRef CAS.
- Y. Itoh, M. Matsusaki, T. Kida and M. Akashi, Chem. Lett., 2008, 37, 238–239 CrossRef CAS.
- Z. J. Wang, L. Qian, X. L. Wang, H. Zhu, F. Yang and X. R. Yang, Colloids Surf., A, 2009, 332, 164–171 CrossRef CAS.
- J. Andrieu, N. Kotman, M. Maier, V. Mailander, W. S. L. Strauss, C. K. Weiss and K. Landfester, Macromol. Rapid Commun., 2012, 33, 248–253 CrossRef CAS.
- Z. Gu, M. Yan, B. L. Hu, K. I. Joo, A. Biswas, Y. Huang, Y. F. Lu, P. Wang and Y. Tang, Nano Lett., 2009, 9, 4533–4538 CrossRef CAS.
- J. Wen, S. M. Anderson, J. J. Du, M. Yan, J. Wang, M. Q. Shen, Y. F. Lu and T. Segura, Adv. Mater., 2011, 23, 4549–4553 CrossRef CAS.
- A. Biswas, K. I. Joo, J. Liu, M. X. Zhao, G. P. Fan, P. Wang, Z. Gu and Y. Tang, ACS Nano, 2011, 5, 1385–1394 CrossRef CAS.
- C. Coll, L. Mondragon, R. Martinez-Manez, F. Sancenon, M. D. Marcos, J. Soto, P. Amoros and E. Perez-Paya, Angew. Chem., Int. Ed., 2011, 50, 2138–2140 CrossRef CAS.
- A. Bernardos, L. Mondragon, E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, J. M. Barat, E. Perez-Paya, C. Guillem and P. Amoros, ACS Nano, 2010, 4, 6353–6368 CrossRef CAS.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS.
- Y. F. Zhu, W. J. Meng and N. Hanagata, Dalton Trans., 2011, 40, 10203–10208 RSC.
- Y. F. Zhu, W. J. Meng, H. Gao and N. Hanagata, J. Phys. Chem. C, 2011, 115, 13630–13636 CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- C. J. Xu, B. G. Xing and H. H. Rao, Biochem. Biophys. Res. Commun., 2006, 344, 931–935 CrossRef CAS.
- L. F. Shi, N. Rosenzweig and Z. Rosenzweig, Anal. Chem., 2007, 79, 208–214 CrossRef CAS.
- K. Boeneman, B. C. Mei, A. M. Dennis, G. Bao, J. R. Deschamps, H. Mattoussi and I. L. Medintz, J. Am. Chem. Soc., 2009, 131, 3828–3829 CrossRef CAS.
- E. Chang, J. S. Miller, J. T. Sun, W. W. Yu, V. L. Colvin, R. Drezek and J. L. West, Biochem. Biophys. Res. Commun., 2005, 334, 1317–1321 CrossRef CAS.
- I. L. Medintz, A. R. Clapp, F. M. Brunel, T. Tiefenbrunn, H. T. Uyeda, E. L. Chang, J. R. Deschamps, P. E. Dawson and H. Mattoussi, Nat. Mater., 2006, 5, 581–589 CrossRef CAS.
- Y. Huang, S. L. Zhao, M. Shi, J. Chen, Z. F. Chen and H. Liang, Anal. Chem., 2011, 83, 8913–8918 CrossRef CAS.
- Y. P. Kim, Y. H. Oh, E. Oh, S. Ko, M. K. Han and H. S. Kim, Anal. Chem., 2008, 80, 4634–4641 CrossRef CAS.
- H. Y. Liu, G. X. Liang, E. S. Abdel-Halim and J. J. Zhu, Anal. Methods, 2011, 3, 1797–1801 RSC.
- S. B. Lowe, J. A. G. Dick, B. E. Cohen and M. M. Stevens, ACS Nano, 2012, 6, 851–857 CrossRef CAS.
- S. L. Sewell and T. D. Giorgio, Mater. Sci. Eng., C, 2009, 29, 1428–1432 CrossRef CAS.
- H. Q. Yao, Y. Zhang, F. Xiao, Z. Y. Xia and J. H. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346–4349 CrossRef CAS.
- R. Freeman, T. Finder, R. Gill and I. Willner, Nano Lett., 2010, 10, 2192–2196 CrossRef CAS.
- L. S. Birchall, R. V. Ulijn and S. J. Webb, Chem. Commun., 2008, 2861–2863 RSC.
- S. Sakuma, Z. R. Lu, P. Kopeckova and J. Kopecek, J. Controlled Release, 2001, 75, 365–379 CrossRef CAS.
- J. Zhou, A. L. Loftus, G. Mulley and A. T. A. Jenkins, J. Am. Chem. Soc., 2010, 132, 6566–6570 CrossRef CAS.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS.
- E. A. Phelps, N. Landazuri, P. M. Thule, W. R. Taylor and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3323–3328 CrossRef CAS.
- S. Sakai, Y. Ogushi and K. Kawakami, Acta Biomater., 2009, 5, 554–559 CrossRef CAS.
- K. Y. Lee, H. J. Kong, R. G. Larson and D. J. Mooney, Adv. Mater., 2003, 15, 1828–1832 CrossRef CAS.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- A. H. Zisch, M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla and J. A. Hubbell, FASEB J., 2003, 17, 2260–2262 CAS.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 19, 3152–3156 CrossRef CAS.
- P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932–8938 CrossRef CAS.
- E. A. MacGregor, S. Janecek and B. Svensson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1546, 1–20 CrossRef CAS.
- K. H. Park, T. J. Kim, T. K. Cheong, J. W. Kim, B. H. Oh and B. Svensson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1478, 165–185 CrossRef CAS.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Neuron, 1996, 16, 881–891 CrossRef CAS.
- Z. W. Hall and R. B. Kelly, Nature – New Biol., 1971, 232, 62–63 CAS.
- T. Ooi, T. Shibata, K. Matsumoto, S. Kinoshita and S. Taguchi, Biosci., Biotechnol., Biochem., 2009, 73, 1209–1211 CrossRef CAS.
- K. Matsumoto, Y. Mukai, D. Ogata, F. Shozui, J. M. Nduko, S. Taguchi and T. Ooi, Appl. Microbiol. Biotechnol., 2010, 86, 1431–1438 CrossRef CAS.
- M. Nakanishi, C. Yatome, N. Ishida and Y. Kitade, J. Biol. Chem., 2001, 276, 46394–46399 CrossRef CAS.
- K. Bosslet, R. Straub, M. Blumrich, J. Czech, M. Gerken, B. Sperker, H. K. Kroemer, J. P. Gesson, M. Koch and C. Monneret, Cancer Res., 1998, 58, 1195–1201 CAS.
- M. Grinda, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski and S. Papot, Med. Chem. Commun., 2012, 3, 68–70 RSC.
- M. Newman, T. Strzelecka, L. F. Dorner, I. Schildkraut and A. K. Aggarwal, Nature, 1994, 368, 660–664 CrossRef CAS.
- G. A. Wilson and F. E. Young, J. Mol. Biol., 1975, 97, 123–124 CrossRef CAS.
- F. Martinon and J. Tschopp, Cell Death Differ., 2007, 14, 10–22 CrossRef CAS.
- R. A. Black, S. R. Kronheim, J. E. Merriam, C. J. March and T. P. Hopp, J. Biol. Chem., 1989, 264, 5323–5326 CAS.
- M. J. Kostura, M. J. Tocci, G. Limjuco, J. Chin, P. Cameron, A. G. Hillman, N. A. Chartrain and J. A. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 5227–5231 CrossRef CAS.
- N. A. Thornberry and Y. Lazebnik, Science, 1998, 281, 1312–1316 CrossRef CAS.
- E. Devarajan, A. A. Sahin, J. S. Chen, R. R. Krishnamurthy, N. Aggarwal, A. M. Brun, A. Sapino, F. Zhang, D. Sharma, X. H. Yang, A. D. Tora and K. Mehta, Oncogene, 2002, 21, 8843–8851 CrossRef CAS.
- A. M. Hunter, E. C. LaCasse and R. G. Korneluk, Apoptosis, 2007, 12, 1543–1568 CrossRef CAS.
- J. Wagner, R. A. Lerner and C. F. Barbas, Science, 1995, 270, 1797–1800 CAS.
- T. Hoffmann, G. F. Zhong, B. List, D. Shabat, J. Anderson, S. Gramatikova, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 1998, 120, 2768–2779 CrossRef CAS.
- V. Turk, J. Kos and B. Turk, Cancer Cell, 2004, 5, 409–410 CrossRef CAS.
- V. Turk, B. Turk and D. Turk, EMBO J., 2001, 20, 4629–4633 CrossRef CAS.
- H. H. Heidtmann, U. Salge, M. Abrahamson, M. Bencina, L. Kastelic, N. KopitarJerala, V. Turk and T. T. Lah, Clin. Exp. Metastasis, 1997, 15, 368–381 CrossRef CAS.
- S. A. Shaikh and M. V. Deshpande, World J. Microbiol. Biotechnol., 1993, 9, 468–475 CrossRef CAS.
- W. F. Ettinger, S. K. Thukral and P. E. Kolattukudy, Biochemistry, 1987, 26, 7883–7892 CrossRef CAS.
- Y. Asano and T. L. Lubbehusen, J. Biosci. Bioeng., 2000, 89, 295–306 CrossRef CAS.
- V. R. Sinha and R. Kumria, Int. J. Pharm., 2001, 224, 19–38 CrossRef CAS.
- A. Barnett, Int. J. Clin. Pract., 2006, 60, 1454–1470 CrossRef CAS.
- B. Pro and N. H. Dang, Histol. Histopath., 2004, 19, 1345–1351 CAS.
- K. Masur, F. Schwartz, F. Entschladen, B. Niggemann and K. S. Zaenker, Regul. Pept., 2006, 137, 147–155 CrossRef CAS.
- R. Mentlein, Regul. Pept., 1999, 85, 9–24 CrossRef CAS.
- T. D. Boyer, Hepatology, 1989, 9, 486–496 CrossRef CAS.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annual Review of Biochemistry, Annual Reviews, Palo Alto, 2008, vol. 77, pp. 521–555 Search PubMed.
- N. C. Veitch, Phytochemistry, 2004, 65, 249–259 CrossRef CAS.
- R. Buettner, L. B. Mora and R. Jove, Clin. Cancer Res., 2002, 8, 945–954 CAS.
- C. A. Evans, P. J. Owenlynch, A. D. Whetton and C. Dive, Cancer Res., 1993, 53, 1735–1738 CAS.
- C. J. Auch, R. N. Saha, F. G. Sheikh, X. J. Liu, B. L. Jacobs and K. Pahan, FEBS Lett., 2004, 563, 223–228 CrossRef CAS.
- H. M. Wang, A. Davis, S. H. Yu and K. Ahmed, Mol. Cell. Biochem., 2001, 227, 167–174 CrossRef CAS.
- M. Flajolet, G. He, M. Heiman, A. Lin, A. C. Nairn and P. Greengard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4159–4164 CrossRef CAS.
- D. P. Hanger, H. L. Byers, S. Wray, K. Y. Leung, M. J. Saxton, A. Seereeram, C. H. Reynolds, M. A. Ward and B. H. Anderton, J. Biol. Chem., 2007, 282, 23645–23654 CrossRef CAS.
- J. W. Critchfield, J. E. Coligan, T. M. Folks and S. T. Butera, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6110–6115 CrossRef CAS.
- B. E. Turk, T. Y. Wong, R. Schwarzenbacher, E. T. Jarrell, S. H. Leppla, R. J. Collier, R. C. Liddington and L. C. Cantley, Nat. Struct. Mol. Biol., 2004, 11, 60–66 CAS.
- A. D. Pannifer, T. Y. Wong, R. Schwarzenbacher, M. Renatus, C. Petosa, J. Bienkowska, D. B. Lacy, R. J. Collier, S. Park, S. H. Leppla, P. Hanna and R. C. Liddington, Nature, 2001, 414, 229–233 CrossRef CAS.
- M. Therisod and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3977–3981 CrossRef CAS.
- K. E. Jaeger and T. Eggert, Curr. Opin. Biotechnol., 2002, 13, 390–397 CrossRef CAS.
- H. Birkedalhansen, W. G. I. Moore, M. K. Bodden, L. J. Windsor, B. Birkedalhansen, A. Decarlo and J. A. Engler, Crit. Rev. Oral Biol. Med., 1993, 4, 197–250 CAS.
- A. Page-McCaw, A. J. Ewald and Z. Werb, Nat. Rev. Mol. Cell Biol., 2007, 8, 221–233 CrossRef CAS.
- M. Arroyo, I. de la Mata, C. Acebal and M. P. Castillon, Appl. Microbiol. Biotechnol., 2003, 60, 507–514 CAS.
- J. Eidenmuller, T. Fath, A. Hellwig, J. Reed, E. Sontag and R. Brandt, Biochemistry, 2000, 39, 13166–13175 CrossRef CAS.
- J. C. Hutton and G. S. Eisenbarth, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8626–8628 CrossRef CAS.
- J. den Hertog, EMBO Rep., 2003, 4, 1027–1032 CrossRef CAS.
- J. N. Andersen, P. G. Jansen, S. M. Echwald, O. H. Mortensen, T. Fukada, R. Del Vecchio, N. K. Tonks and N. P. H. Moller, FASEB J., 2004, 18, 8–30 CrossRef CAS.
- C. Tamm, Pure Appl. Chem., 1992, 64, 1187–1191 CrossRef CAS.
- C. Betzel, S. Gourinath, P. Kumar, P. Kaur, M. Perbandt, S. Eschenburg and T. P. Singh, Biochemistry, 2001, 40, 3080–3088 CrossRef CAS.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS.
- T. Gonzales and J. RobertBaudouy, FEMS Microbiol. Rev., 1996, 18, 319–344 CrossRef CAS.
- G. Nguyen, Kidney Int., 2006, 69, 1503–1506 CrossRef CAS.
- R. J. Siezen and J. A. M. Leunissen, Protein Sci., 1997, 6, 501–523 CrossRef CAS.
- R. Gupta, Q. K. Beg and P. Lorenz, Appl. Microbiol. Biotechnol., 2002, 59, 15–32 CrossRef CAS.
- O. A. Adekoya and I. Sylte, Chem. Biol. Drug Des., 2009, 73, 7–16 CAS.
- P. A. Andreasen, L. Kjoller, L. Christensen and M. J. Duffy, Int. J. Cancer, 1997, 72, 1–22 CrossRef CAS.
Footnote |
| † Single letter codes will be used for amino acids in this article. |
| This journal is © The Royal Society of Chemistry 2013 |