Capillary electrophoresis combined with accelerated solvent extraction as an improved methodology for effective separation and simultaneous determination of malachite green, crystal violet and their leuco-metabolites in aquatic products
Hanwen Sun* and Haijing Qi
College of Chemistry and Environmental Science, Hebei University, Key Laboratory of Analytical Science and Technology of Hebei Province, Baoding 071002, China. E-mail: hanwen@hbu.edu.cn; Fax: +86-312-5079739
First published on 12th November 2012
Abstract
Malachite green (MG), crystal violet (CV) and their leuco-metabolites (LMG, LCV) could not be determined simultaneously by capillary zone electrophoresis because of their similar structures, low solubility in water, and easy degradation/de-methylation. A novel capillary electrophoresis (CE) method with UV detection for the effective separation and simultaneous determination of the four compounds was developed for the first time. The use of an acetonitrile–sodium dodecyl sulfate (SDS)–ammonium acetate buffer solution (pH 4.0) achieved the effective separation of the four compounds. The effects of CE conditions on separation were investigated in detail. Accelerated solvent extraction (ASE) with acetonitrile as an extractant in the presence of hydroxylamine hydrochloride and p-toluenesulfonic acid was used to extract the four compounds in aquatic products with high extraction efficiency. Under optimized separation conditions, the four analytes could be baseline separated, and there was no interference in analysis of real samples. Calibration curves have good linearity with a correlation coefficient (r) greater than 0.999. The limit of detection for MG, CV, LMG, and LCV were 0.39, 0.29, 0.08, and 0.10 μg mL−1, respectively. The intra- and inter-day variability (RSD) of peak areas were in the range of 0.3–3.4% and 1.0–4.7%, respectively. Average recoveries were 92.6–107% with RSD of 2.1% for shrimp samples, 95.4–108% with RSD of 2.3% for eel samples, and 89.1–105% with RSD of 3.1% for sardine samples. The proposed ASE–CE method has the advantages of high selectivity, good linearity and precision.
Introduction
Malachite green (MG) and crystal violet (CV) are triphenylmethane dyes. There have been many reports of the inappropriate use of MG and CV as veterinary drugs. They are readily absorbed into fish tissue from water exposure, and are reduced metabolically by fish to leucomalachite green (LMG) and leucocrystal violet (LCV). MG and CV have been banned for use as fungicides and antiseptics in aquaculture and fisheries because of their carcinogenic and mutagenic properties.1 Thus, it is necessary to develop a sensitive, rapid, inexpensive, and reliable method for the determination of MG, CV, and their leuco-metabolites in aquatic products.Spectrophotometry has been used for the determination of MG and CV in water.2,3 High-performance liquid chromatography (HPLC) has been applied for the determination of MG, CV, LMG, and LCV in water,4 and in aquatic products with MS or MS/MS detection.5–-8 The advantages of capillary electrophoresis (CE) over LC are low solvent consumption and low cost. Capillary zone electrophoresis (CZE) has been primarily carried out using an aqueous electrolyte buffer. CZE and micellar electrokinetic chromatography (MEKC) combined with Raman spectroscopy have been used for the determination of MG in water based on the stacking and sweeping modes.9 A CE method combined with conductive detection has been established for the determination of MG in fish with an limit of detection (LOD) of 1.8 μg mL−1.10 It is difficult to separate MG, LMG, CV, and LCV by CZE because of their similar structures, low solubility in water, and the easy degradation/de-methylation of MG and CV. The use of organic solvents in place of aqueous systems may provide a means of selectivity enhancement. Matysik described the potentials and application of electrochemical detection in conjunction with NACE using an acetonitrile-based buffer system for the separation and detection of MG, CV and rhodamine B.11,12 To our knowledge there has been no report on the simultaneous determination of MG, CV, LMG, and LCV by CE.
The purpose of the present work was to develop a new CE-UV detection method for the effective separation and simultaneous determination of MG, CV, LMG, and LCV. In this work, the conditions of separation were investigated and optimized. The CE method coupled with accelerated solvent extraction (ASE) has been used for the simultaneous determination of MG, CV, LMG, and LCV in aquatic products with satisfactory results.
Experimental
Instrumentation
All experiments were performed with an Agilent 3D CE system with air-cooling and a diode-array detector (Agilent, Waldbronn, Germany). Data were collected with the Agilent Chemstation version A.10.02 chromatographic data system. A 48.5 cm (40.0 cm to the detector) long, 75 μm i.d. and 365 μm o.d. uncoated fused silica capillary (Yongnian Optical Fabric Factory, Handan, China) was utilized. The extraction equipment was an APLE 2000 automatic accelerated solvent extraction apparatus (Beijing Titan Instruments Co Ltd., China). A centrifuge TGL-16M (Xiangyi centrifuge Co., Hunan, China), RE-2000A rotary evaporator (Shanghai Yarong Biochemistry Instrument Co.,) and PHS-3C pH meter (Shanghai Precision & Scientific Instrument Co., Shanghai, China) were used in sample treatment.Chemicals and solutions
MD, CV, LMG and LCV (purity: ≥ 99%) were purchased from Dr Ehrenstorfer GmbH (Hamburg, Germany). All chemicals used were of analytical grade.The stock standard solutions of MG, LMG, CV and LCV, 1000 mg L−1, were prepared by dissolving the solid chemicals in acetonitrile. These stock solutions were stored in the dark at −20 °C and were stable for at least one month. Fresh working standard solutions were prepared daily by diluting the stock solutions with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v) mixture of acetonitrile and water, and were used for different studies.
:4 (v/v) mixture of acetonitrile and water, and were used for different studies.
The buffers were prepared from ammonium acetate and the pH was adjusted to 4.0 with acetic acid. Sodium dodecyl sulfate (SDS) was used as a surfactant and p-toluenesulfonic acid was used as the organic modifier. Doubly deionized water was used throughout. Buffer solutions were prepared weekly, and filtered through 0.22 μm cellulose acetate filters (Shanghai Xinya Purification Material Factory) prior to injection.
Sample preparation
Shrimp, eel and sardine samples were purchased from a local market, and after being homogenized in a high-speed food blender, they were stored below −20 °C in a freezer until the time of analysis.Samples were extracted with APLE 2000 automatic accelerated solvent extraction apparatus. Approximately 2.000 g of the blank/spiked sample material was mixed with 2.5 g of diatomite and packed into an 11 mL stainless-steel extraction cell. Each cell was locked with stainless-steel screw caps, and circular glass microfiber filters of 1.98 cm diameter were placed above and below the packing, where diatomite was used as a filter agent to prevent the fine powder breakthrough into the collection bottle. 0.5 mL of 20% (m/v) hydroxylamine hydrochloride, 100 μL of p-toluenesulfonic acid, and 2 mL of acetonitrile were added to each cell. The solvent selected was acetonitrile. Conditions used in the extraction were: oven temperature of 60 °C with 3 min heat-up time under a pressure of 10.3 MPa and two static cycles with a static time of 5 min. The flush volume was 40% of the extraction cell volume. The extract was purged from the sample cell using pressurized nitrogen for 2 min.
Finally, each resulting extract was evaporated to dryness using the rotary evaporator. The residue was rinsed with 5 mL of acetonitrile, and evaporated to dryness under a nitrogen flow at 40 °C. The residue was re-dissolved in 1 mL of buffer solution. The solution was vortexed, and filtered through a 0.22 μm mixed cellulose ester membrane (Shanghai, China) for CE analysis.
Electrophoresis conditions
Prior to its use every day, the capillary was consecutively flushed with 0.1 M NaOH for 10 min, distilled water for 10 min and the running buffer for 10 min. After each analysis run the capillary was flushed for 3 min with the running buffer to maintain the reproducibility of the analysis. Sample introduction was carried out at the anodic capillary side using 50 mbar pressures for 5 s. The high-voltage power supply was set at 23 kV. The 50 mM ammonium acetate solution (pH 4.0) containing 50 mM SDS–acetonitrile (1:2, v/v) was used as a running buffer. The capillary temperature was kept at 25 °C. UV detection was employed at a wavelength of 580 nm for MG and CV, and 265 nm for LMG and LCV.Results and discussion
Optimization of electrophoresis conditions
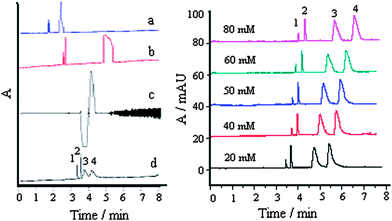 | ||
| Fig. 1 Effect of different electrolytes (left) and ammonium acetate concentration (right) on separation: a, trihydroxymethyl aminomethane; b, sodium tetraborate; c, potassium biphthalate, and d, ammonium acetate; 1, MG; 2, CV; 3, LCV, and 4, LMG; each compound, 30 μg mL−1 ; buffer, pH 4.0; buffer–acetonitrile (v/v), 1:2; SDS, 50 mM; separation voltage, 23 kV; temperature, 25 °C. | ||
The migration time and peak area of the analytes increased with an increase of electrolyte concentration. This is because the thickness of the electric double layer between the capillary walls and the buffer solution decreased with an increase of buffer concentration. Also, high buffer concentration could increase the coverage of sites on the silica surface, resulting in a decrease of the electroosmotic flow because of the decrease of the silicon hydroxyl dissociation.18 Synthetically considering migration time and response, a 50 mM ammonium acetate buffer was selected to give high sensitivity and faster analyses.
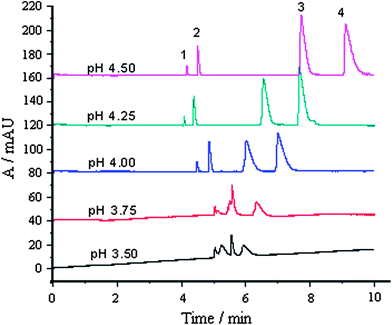 | ||
| Fig. 2 Effect of running buffer pH on separation: 1, MG; 2, CV; 3, LCV and 4, LMG; each compound, 30 μg mL−1; other conditions were as shown in Fig. 1. | ||
It is shown that the pH of the running buffer affects the resolution of the four compounds. Under higher acidity (pH 3.50 and 3.75), MG, CV, and LCV could not be separated, and had a low response. The resolution and migration time of CV, LCV and LMG increased with an increase in pH. When pH = 4.5, the migration time of LMG was up to 10 min. When pH = 4.0, the separation of the four analytes was achieved within 8 min, with highest sensitivity.
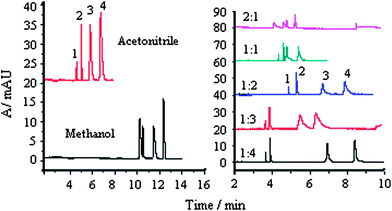 | ||
| Fig. 3 Effect of organic additive on separation buffer: 50 mM, pH 4.0; SDS: 50 mM; separation voltage, 23 kV; temperature, 25 °C; 1, MG; 2, CV; 3, LCV and 4, LMG; each compound, 30 μg mL−1; right: buffer–acetonitrile (v/v), 1.4 to 2:1. | ||
The use of an acetonitrile/methanol–ammonium acetate buffer (pH 4.0) is attractive because the four compounds show much better solubility in this medium. The organic additives affect migration of analytes in two basic ways, directly altering physicochemical properties of the separation medium (e.g. viscosity and dielectric constants) and invoking chemical equilibria involving the analytes and a buffer additive.14 Using methanol as an additive, the separation of the four compounds was achieved in 12.6 min, whereas with acetonitrile, the separation could be achieved within 8 min. This is due to the lower viscosity of acetonitrile than methanol–water mixtures and the higher dielectric constant (ε = 37.5) and autoprotolysis constant (pKauto ≥33.3) of acetonitrile than those (ε = 32.7, pKauto = 17.2) of methanol.15 Using acetonitrile with lower viscosity, higher dielectric constant, and autoprotolysis constant, better separation was achieved compared with that achieved using methanol.
The optimal proportion of acetonitrile in ammonium acetate buffer was investigated further, as shown in Fig. 3 (right). The analysis times decreased drastically with addition of acetonitrile into buffer due to the lowered viscosity. When the relative amount of acetonitrile was increased, effective separation of the four compounds could not be achieved. When the volume ratio of the buffer solution and acetonitrile was selected to be 1:2, separation was achieved, with higher response and resolution.
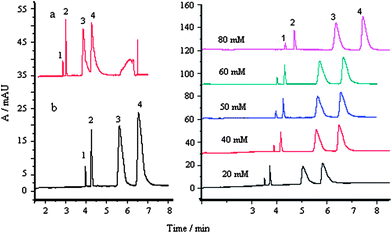 | ||
| Fig. 4 Effect of surfactants on separation: a, without SDS, b, with SDS; buffer: 50 mM, pH 4.0; buffer–acetonitrile (v/v), 1:2; separation voltage, 23 kV; temperature, 25 °C; 1, MG; 2, CV; 3, LCV and 4, LMG; each compound, 30 μg mL−1. | ||
It is evident that the use of SDS is beneficial for the separation of the analytes based on interactions between negatively charged surfactant monomers and analytes. The presence of SDS allowed the surfactant–analyte complex to migrate towards the anode,16 so that the analytes were separated in a reasonable time period. In the case of LCV and LMV some change in the peak shape after addition of SDS was observed, this is possibly due to the solubilization of poorly soluble analytes.
Fig. 4 (right) illustrates the relationship between the response of the four analytes and the concentration of the surfactants. When SDS concentrations varied from 20 to 80 mM, the resolution did not change and the migration time and peak area of the analytes increased. This indicates the occurence of stronger interactions between the analytes and the surfactants. Walbroehl and Jorgenson17 suggested a dynamic equilibrium mechanism between the associated and the unassociated forms of the analytes to account for their migration. This mechanism is also consistent with our results, as indicated by the steadily increasing peak height and peak area in Fig. 4 (right). When SDS concentration was selected to 50 mM, satisfactory separation was achieved within 7 min with higher sensitivity.
Optimization of ASE conditions
Methanol and acetonitrile have frequently been used as extractants in ASE.13 The system of McIlvaine buffer (pH 3)–acetonitrile has been used for ASE of MG, LMG, GV and LGV prior to LC–MS/MS with recovery of 82–93.5%. In this work, the extraction efficiency with methanol and acetonitrile in the presence of hydroxylamine hydrochloride and p-toluenesulfonic acid was compared. The recoveries of MG, LMG, GV and LGV from the blank shrimp sample spiked at 5 mg kg−1 for each analyte were 93–106% with acetonitrile and 74–110% with methanol (see Fig. 5). Hence, 2 mL acetonitrile–0.5 mL 20% (m/v) hydroxylamine hydrochloride–100 μL p-toluenesulfonic acid was selected for futher work.
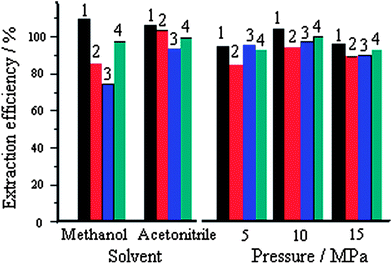 | ||
| Fig. 5 Effects of different solvents and static pressure on extraction efficiency spiked at 5 mg kg−1 for each analyte; acetonitrile/methanol volume, 2 mL; temperature, 60 °C; static time, 5 min; static cycles, 2; 1, MG; 2, CV; 3, LCV and 4, LMG; extraction efficiency data are the average value of three measurements and are less than 3% of RSD. | ||
Performance of the method
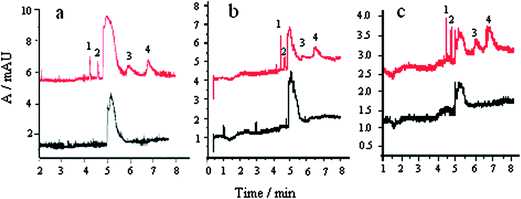 | ||
| Fig. 6 Electropherograms of blank samples and spiked samples: a, shrimp, b, eel and c, sardine; spiked level: 1, 1.5 mg kg−1 MG; 2, 0.5 mg kg−1 CV; 3, 0.5 mg kg−1 LCV, and 4, 0.5 mg kg−1 LMG. | ||
It was shown that the four analytes could be baseline separated. The unknown peak does not interfere with the separation and determination of MG, CV, LCV, and LMG. There was no interference peak in real samples.
| Analyte | Linear equation | Correlation coefficient (r) | Linear range/μg mL−1 | LOD/μg mL−1 | LOQ/μg mL−1 |
|---|---|---|---|---|---|
| MG | A = 7.5534C ¬ 26.4239 | 0.9995 | 1.32–300 | 0.39 | 1.32 |
| CV | A = 10.1398C + 1.5729 | 0.9999 | 0.98–100 | 0.29 | 0.98 |
| LMG | A = 7.3162C ¬ 10.9297 | 0.9997 | 0.27–100 | 0.08 | 0.27 |
| LCV | A = 5.9098C ¬ 8.5336 | 0.9998 | 0.34–100 | 0.10 | 0.34 |
The LOD was determined as the sample concentration that produces a peak with a height three times the level of the baseline noise, and the limit of quantification (LOQ) was calculated as the level that produced a peak with 10 times the signal-to-noise ratio. Table 1 gives the instrument LODs and LOQs. The LOD values were lower than those of the CE method.9–12 For 2 g of sample and 1 mL of final test solution, the method LOQ values for MG, CV, LCV, and LMG were 0.66, 0.49, 0.14, and 0.17 mg kg−1 in aquatic products, and could be improved by increasing the sample volume.
| Analyte | Added/mg kg−1 | Intra-day (n = 5) | Inter-day (n = 15) |
|---|---|---|---|
| RSD (%) | RSD (%) | ||
| MG | 1.5 | 1.0 | 2.0 |
| 7.5 | 1.6 | 4.0 | |
| 15.0 | 3.4 | 4.7 | |
| CV | 0.5 | 1.1 | 1.4 |
| 2.5 | 1.9 | 2.1 | |
| 5.0 | 0.9 | 3.0 | |
| LMG | 0.5 | 0.5 | 1.0 |
| 2.5 | 1.6 | 2.0 | |
| 5.0 | 1.5 | 2.9 | |
| LCV | 0.5 | 0.5 | 1.2 |
| 2.5 | 1.0 | 1.7 | |
| 5.0 | 1.7 | 2.6 |
The intra- and inter-day RSDs of the peak areas for the four analytes at near LOQ level were in the range of 0.3–3.4% and 1.0–4.7%, respectively. It was shown that the repeatability of the method is satisfactory for the residue determination of the studied compounds in aquatic products.
Sample analysis
Under the optimized conditions of ASE and CE, shrimp, eel and sardine samples were analyzed. The studied four dyes were not detected in real samples. The recovery test of the assays for the analytes was determined by adding known amounts of these dyes to the aquatic product samples. The recoveries are listed in Table 3.| Sample | Analyte | Spiked/mg kg−1 | Recovery (%) | Mean recovery (%) |
|---|---|---|---|---|
| Shrimp | MG | 1.5, 7.5, 15 | 100, 94.0, 103 | 99.0 |
| CV | 0.5, 2.5, 5.0 | 91.3, 94.0, 97.3 | 94.2 | |
| LMG | 0.5, 2.5, 5.0 | 110, 109, 102 | 107 | |
| LCV | 0.5, 2.5, 5.0 | 104, 87.3, 86.4 | 92.6 | |
| Eel | MG | 1.5, 7.5, 15 | 103, 105, 111 | 108 |
| CV | 0.5, 2.5, 5.0 | 92.0, 104, 101 | 99.0 | |
| LMG | 0.5, 2.5, 5.0 | 108, 100, 101 | 103 | |
| LCV | 0.5, 2.5, 5.0 | 99.2, 94.1, 93.0 | 95.4 | |
| Sardine | MG | 1.5, 7.5, 15 | 97.3, 89.4, 94.1 | 93.6 |
| CV | 0.5, 2.5, 5.0 | 85.7, 87.9, 93.8 | 89.1 | |
| LMG | 0.5, 2.5, 5.0 | 104, 106, 97.0 | 105 | |
| LCV | 0.5, 2.5, 5.0 | 98.0, 90.3, 92.2 | 93.5 |
The average recovery was 92.6–107% with an RSD of 2.1% for shrimp samples, 95.4–108% with an RSD of 2.3% for eel samples, and 89.1–105% with an RSD of 3.1% for sardine samples. It is indicated that the proposed method has high recovery, and can be used for the determination of the studied drugs in real samples.
Conclusion
The proposed CE method can effectively separate and simultaneously determine MG, CV, LMG, and LCV, which have similar structures. ASE with acetonitrile as an extraction solvent in the presence of hydroxylamine hydrochloride and p-toluenesulfonic acid is an effective tool for the extraction of the four compounds in aquatic products. The proposed ASE–CE method has the advantages of high selectivity, good linearity and precision.Acknowledgements
This work was supported by the Natural Science Foundation of Hebei Province (B2008000583) and the Sustainable Plan of Science and Technology of Hebei Province of China (No. 10967126D).References
- Council Directive 96/23/EC of 29 April 1996, Brussels, (23 May 1996), Official Journal Europe Communication L125, 10.
- I. Šafařík and M. Šafaříková, Water Res., 2002, 36, 196–200 CrossRef.
- L. An, J. Deng, L. Zhou, H. Li, F. Chen, H. Wang and Y. T. Liu, J. Hazard. Mater., 2010, 175, 883–888 CrossRef CAS.
- Z. Zhang, K. Zhou, Y. Q. Bu, Z. J. Shan, J. F. Liu, X. Y. Wu, L. Q. Yang and Z. L. Chen, Anal. Methods, 2012, 4, 429–433 RSC.
- Y. F. Tao, D. M. Chen, X. Q. Chao, H. Yu, Y. H. Pan, Z. L. Liu, L. L. Huang, Y. L. Wang and Z. H. Yuan, Food Control, 2011, 22, 1246–1252 CrossRef CAS.
- M. Dowling, R. Duffy, D. G. Smyth, P. P. Mulder, C. Duffy, L. Regan and M. R. Smyth, Anal. Chim. Acta, 2007, 58, 411–419 CrossRef.
- X. Wu, G. Zhang, Y. Wu, X. Hou and Z. Yuan, J. Chromatogr., A, 2007, 1172, 121–126 CrossRef CAS.
- G. Chen and S. Miao, J. Agric. Food Chem., 2010, 58, 7109–7114 CrossRef CAS.
- C. H. Tsai, J. D. Lin and C. H. Lin, Talanta, 2007, 72, 368–372 CrossRef CAS.
- B. M. Huang, C. W. Yao and H. Cheng, Chin. J. Appl. Chem., 2007, 24, 327–330 CAS.
- F. M. Matysik, J. Chromatogr., A, 1998, 802, 349–354 CrossRef CAS.
- F. M. Matysik, Electrochim. Acta, 1998, 43, 3475–3482 CrossRef CAS.
- H. W. Sun, X. S. Ge, Y. K. Lv and A. B. Wang, J. Chromatogr., A, 2012, 1237, 1–23 CrossRef CAS.
- Q. Yang, L. M. Benson, K. L. Johnson and S. Naylor, J. Biochem. Biophys. Methods, 1999, 38, 103–121 CrossRef CAS.
- M. L. Riekkola, M. Jussila, S. P. Porras and I. E. Valko, J. Chromatogr., A, 2000, 892, 155–170 CrossRef CAS.
- J. Li and J. S. Fritz, Electrophoresis, 1999, 20, 84–91 CrossRef CAS.
- Y. Walbroehl and J. W. Jorgenson, Anal. Chem., 1986, 58, 479–481 CrossRef CAS.
- N. Chen, L. Wang and Y. K. Zhang, J. Microcolumn Sep., 1995, 7, 193–198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |