Sensitive analysis of trace water analytes using colourimetric cavity ringdown spectroscopy
Cathy M.
Rushworth
a,
Yathukulan
Yogarajah
a,
Yan
Zhao
b,
Hywel
Morgan
*b and
Claire
Vallance
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: claire.vallance@chem.ox.ac.uk
bSchool of Electronics and Computer Science, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: hm@ecs.soton.ac.uk
First published on 4th October 2012
Abstract
The application of colourimetric cavity ringdown spectroscopy to the detection of trace compounds in water has been investigated using nitrite and iron(II) as test analytes. Samples were contained within one of three commercially available flow cells ranging in optical path length from 0.1 mm to 2.0 mm, and positioned within a two-mirror ringdown cavity. A measurement of the decay rate of the intensity of an optical pulse introduced into the cavity allows an ultrasensitive determination of optical absorption by the sample. A calibration using the known absorption coefficient of potassium permanganate at 532 nm was first carried out in order to determine the detection sensitivity in terms of minimum detectable absorption per unit path length when using each flow cell. The detection of nitrite and iron was then carried out by using well-known colour reactions, namely the Griess reaction for nitrite and the bathophenanthroline method for iron(II), to convert the analytes into strongly absorbing derivatives, which were quantified by a cavity ringdown measurement. In this first application of colourimetric cavity ringdown spectroscopy to the liquid phase, detection limits of 1.9 nM for nitrite and 3.8 nM for Fe(II) were demonstrated in a flow cell of path length 1.0 mm. The volume of sample analysed is only 196 nL, so that detection limits of this order correspond to the detection of less than 1 billion molecules. The detection method is therefore suitable for integration into a microfluidic sensing platform.
Introduction
Sensitive and selective chemical analysis of trace analytes in water remains a challenge. A plethora of colour reactions have been developed in order to convert colourless or weakly absorbing species into strongly absorbing, visible products.1,2 Yet, even with such strong signal amplification, the detection of extremely low concentrations of even these strongly absorbing products remains a challenge. This is especially true when employing microfluidic sensing platforms. Sensors based on microfluidic platforms are attractive, consuming less power and fewer reagents than their larger-scale counterparts, and opening the way to remote sensors able to operate for long periods of time without human intervention. However, the tiny sample volumes and short optical path lengths inherent in microfluidic sensors necessitates the development of highly sensitive detection techniques. In this paper, we apply cavity ringdown spectroscopy (CRDS) to trace chemical analysis in water. Cavity ringdown spectroscopy is a highly sensitive spectroscopic technique, used most commonly for the analysis of weak optical absorptions of trace gas species.3 The experimental setup is straightforward: a pulse of light (usually from a laser) is injected into an optical cavity formed from two highly reflective mirrors, and the exponential decay of the light intensity within the cavity is recorded, usually by monitoring the small amount of light that ‘leaks out’ through one of the mirrors. The exponential decay constant, or ‘ringdown time’ of the cavity provides a sensitive measure of the cavity losses, in particular any optical absorption by a sample placed within the cavity. The high sensitivity of cavity ringdown techniques has two sources. Firstly, light trapped within the cavity undergoes multiple passes through the sample, vastly increasing the effective absorption path length relative to single-pass methods; for gas-phase species, path lengths of several kilometres are typically achieved in an experimental footprint of a few tens of centimetres. Secondly, since the rate of decay of the light intensity within the cavity is measured, rather than the light intensity itself, the signal is decoupled from the initial light intensity injected into the cavity, and is virtually immune to shot-to-shot variations in the laser intensity.Over the past decade, there has been considerable interest in exploring the potential scope of CRDS for liquid-phase measurements, and we refer the interested reader to recent review articles4,5 for a detailed overview of the field. We have recently demonstrated the use of CRDS for analysing liquids within microfluidic channels by integrating a microfluidic chip into a two-mirror cavity.6 In this paper, we demonstrate that by combining CRDS with colourimetry we can achieve high-sensitivity detection of trace analytes in water samples, using the quantitative detection of dissolved nitrite and Fe(II) as examples. While the present work employs a high-optical-quality flow cell to contain the liquid samples, in the longer term it is envisaged that the methodology could be implemented on a microfluidic platform for ship-based or remote sensing applications.
Nitrite (NO2−) is a reactive chemical species which plays an important part in the global carbon and nitrogen cycles, as well as being a key marine nutrient required for phytoplankton growth.7,8 In sea water, nitrite levels range from 0.1–50 μM, but are most commonly below 0.2 μM.9 In river water, European Union guidelines set a maximum of 0.01 mg L−1 (0.22 μM) for rivers supporting salmon fisheries,10 but significantly higher river values have been reported across Europe (see, for example, ref. 11). At excess levels, nitrite is harmful to both humans and aquatic life, being implicated in methaemoglobinaemia (‘blue baby syndrome’), and formation of several known carcinogens.12 The current guidelines for short-term exposure of nitrates and nitrites provided by WHO are 3 mg L−1 (62.5 μM) for nitrite, and 50 mg L−1 (807 μM) for nitrate.13
Nitrite (and nitrate) detection has recently been the subject of two excellent reviews.14,15 Spectroscopic methods, including direct UV absorption,16 indirect visible absorption,17 and fluorescence18,19 are the most common, owing to their excellent limits of detection and relative experimental simplicity.14 Of the spectroscopic methods, determination of nitrite with the Griess Assay,20 first published in 1879, is by far the most common detection technique.14 In the Griess Assay, nitrite is acidified and undergoes diazotisation with a primary amine (such as sulfanilamide), followed by coupling to an aromatic molecule to produce an intensely coloured azo dye, which has an absorption maximum2,17 in water of around 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 520 nm. It should be noted that throughout this paper we use the natural log form of the Beer–Lambert law, and thus the quoted absorption coefficients are approximately 2.303 times larger than those commonly cited using the decadic log form of the law. The dye is formed in a 1:1 ratio from nitrite in the sample. Thus, by monitoring the reaction using spectrophotometry at a wavelength at or close to the absorption maximum for the azo dye, measurement of nitrite concentrations in water samples can be made. The Griess Assay owes its popularity to the high sensitivity that can be achieved owing to the dye's high peak absorbance, and to the high selectivity of the reaction, owing to the nitrite-specificity of diazotisation. Taken together, these factors mean that sample pre-treatment (e.g. pre-concentration, distillation, or scrubbing of other analytes) is not usually required. It should be noted that nitrate can also be detected using the Griess reaction, after reduction to nitrite, using, for example, copperised cadmium.21
000 M−1 cm−1 at 520 nm. It should be noted that throughout this paper we use the natural log form of the Beer–Lambert law, and thus the quoted absorption coefficients are approximately 2.303 times larger than those commonly cited using the decadic log form of the law. The dye is formed in a 1:1 ratio from nitrite in the sample. Thus, by monitoring the reaction using spectrophotometry at a wavelength at or close to the absorption maximum for the azo dye, measurement of nitrite concentrations in water samples can be made. The Griess Assay owes its popularity to the high sensitivity that can be achieved owing to the dye's high peak absorbance, and to the high selectivity of the reaction, owing to the nitrite-specificity of diazotisation. Taken together, these factors mean that sample pre-treatment (e.g. pre-concentration, distillation, or scrubbing of other analytes) is not usually required. It should be noted that nitrate can also be detected using the Griess reaction, after reduction to nitrite, using, for example, copperised cadmium.21
Iron is an essential element for all known living organisms.22 The concentration of dissolved iron (Fe2+ and Fe3+) in marine and aquatic environments varies widely, depending to a large extent on the dissolved oxygen concentration. The median iron concentration in rivers has been reported13 to be 12.5 μM, with a range22 of 3–25 μM. In contrast, in the open ocean, iron is present at such low concentrations (0.02–2 nM) that it is thought to be one of the limiting factors for phytoplankton production.22 The development of methods for quantifying iron in surface sea water is therefore of growing importance. In drinking water, iron levels are considered primarily in terms of their consequences for taste and appearance, rather than in the context of any health issues.13 In the UK, the maximum level of iron permitted in tap water is 3.6 μM.13
Iron concentrations can be determined colourimetrically using several reactions,1,23,24 including complexation with thiocyanate25 for Fe3+, complexation with 4,7-diphenyl-1,10-phenanthroline26 (known as bathophenanthroline) for Fe2+, and complexation with 3-(2-pyridyl)-5,6-bis(4-phenylsulfonic acid)-1,2,4-triazine27 (known as ferrozine) for Fe2+. The maximum optical absorption coefficient achieveable using the thiocyanate method25 is reported to occur at 480 nm. However, in order to minimise absorption from hydrated Fe3+, which occurs at short wavelengths, detection is often carried out at 500 nm where the absorption coefficient is reported to be 18150 M−1 cm−1.28 The maximum absorption coefficient when using the ferrozine method is reported to be 64300 M−1 cm−1 at 562 nm,27 falling to an estimated 40200 M−1 cm−1 around 532 nm. In contrast, the maximum absorption coefficient of the bathophenanthroline method26 is reported to be 51600 M−1 cm−1 at 533 nm. We chose to use the bathophenanthroline method as this method had the highest absorption coefficient at the wavelength of our laser (532 nm). It should be noted that Fe3+ can also be detected using Fe2+-specific reagents after reduction to Fe2+ (see, for example, ref. 29).
Methods
Instrumentation
A schematic of the experimental set-up used is shown in Fig. 1. The system is similar to that reported in reference,6 except that a commercially available flow cell replaces the microfluidic chip used in the previous work. The characteristics of the three different flow cells employed in this work are listed below.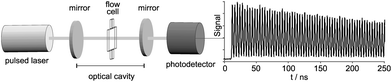 | ||
| Fig. 1 Schematic of experimental set-up. A laser pulse enters the 0.585 m long cavity through the first cavity mirror, and undergoes multiple passes through the sample. Light leaking out through the second mirror is recorded using a photodetector, and the resulting exponential decay is analysed to determine the cavity losses, and therefore the sample absorption. The cavity output signal shown on the right consists of a series of decaying pulses: here only a small section of the trace is shown for clarity. | ||
(1) Starna Scientific Ltd 45/Q/2 rectangular flow cell, optical path length 2 mm, internal width 10 mm, nominal volume 0.90 mL.
(2) Starna Scientific Ltd 45/Q/1 rectangular flow cell, optical path length 1 mm, internal width 10 mm, nominal volume 0.45 mL.
(3) Starna Scientific Ltd 45/Q/0.1 rectangular flow cell, optical path length 0.1 mm, internal width 10 mm, nominal volume 0.045 mL.
The light source employed in all of the cavity ringdown measurements is a microchip Nd:YAG laser (NP-10620-100, Teem Photonics) which produces 0.9 ns, 6 μJ pulses at 1064 nm with a repetition rate of 7.4 kHz. The laser beam is frequency doubled to produce 532 nm light, which is separated from the fundamental by a dichroic mirror before being injected into the cavity. The optical cavity is formed from two concave 25 mm dielectric mirrors (CVI Melles Griot, radius of curvature 1 m, reflectivity 99.8% at 532 nm) positioned at a separation of 0.585 m. The flow cell is positioned vertically in the centre of the cavity on a double-rotation (Thorlabs, RP01) and three-axis translation stage (Newport Corporation, 443), with micrometer resolution actuators (Newport Corporation, SM-25). This allows the angle of the flow cell with respect to the laser beam polarization to be optimised in order to maximise the ringdown time, and hence the sensitivity of the absorption measurement. Liquid was delivered to the flow cell using syringe pumps (Fusion 400, Chemyx).
On each experimental cycle, a light pulse is coupled into the cavity through the back of the first mirror, and undergoes repeated reflections within the cavity. A small amount of light is coupled out of the cavity through the mirrors on each reflection. Behind the second mirror, the cavity output is collected by a 3 mm diameter liquid light guide (NT53-428, Edmund Optics), which directs the light to a photomultiplier tube (PMT, Hamamatsu, H6780-20). The signal from the PMT is displayed on a digital oscilloscope (Tektronix, TDS 3044B), which is interfaced to a personal computer via a GPIB-USB interface (National Instruments, 778927-01). Ringdown traces (averaged up to 512 times on the oscilloscope) are acquired and analyzed continuously via a LabVIEW interface (National Instruments, LabVIEW 10). As the laser pulse duration is shorter than the cavity round-trip time, the signal consists of a train of pulses with exponentially decaying intensities, I(t).
Data analysis and cavity characterisation
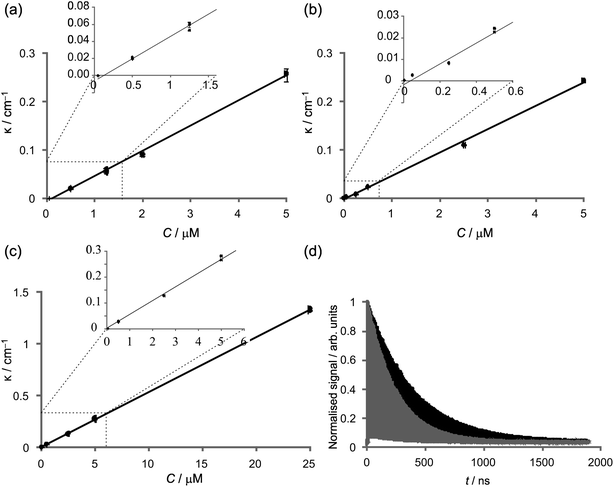
Determination of the ringdown time τ is carried out automatically by the same LabVIEW interface that acquires the data. The electronic response of the photomultiplier contributes a non-zero baseline to the signal, which is subtracted from the data before further processing. Next, a threshold is applied to the ringdown pulse train, which specifies the minimum pulse height to include in the determination of the ringdown time. A peak-finding algorithm then determines the amplitude of each peak in the exponentially decaying pulse train, and the ringdown time, τ, is determined from the slope of a log plot of peak intensity vs. time i.e. ln I(t) = ln I(0) − t/τ. The ringdown time depends on the intrinsic properties of the cavity and on the absorption by the sample as follows.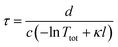 | (1) |
Here, d is the cavity length in m, c is the speed of light in the cavity medium in m s−1, and Ttot is the total round-trip transmission of the cavity (in the absence of sample). The sample absorption is given by κl, where l is the optical path length through the sample in cm−1 and κ = αC is the absorption per unit path length, with dimensions of cm−1, which is dependent on the molecular (natural) absorption coefficient α (dm3 mol−1 cm−1) and the concentration C (mol dm−3). Recording ringdown traces in the absence (τ0) and presence (τ) of an absorbing sample allows κ to be determined:
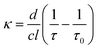 | (2) |
In our cavity set-up, the cavity losses L = 1 − T arise from losses at the mirrors, determined by the mirror reflectivity R, and also from the losses occasioned by the insertion of a flow cell into the cavity. Before insertion of the flow cell into the cavity, the ringdown time of a 532 nm light pulse is measured to be 952 ns (corresponding to a mirror reflectivity of 99.795% at this wavelength, close to the manufacturer's specification). After insertion of the flow cell, the average ringdown time for flow cells filled with deionised water (MilliQ, 18 MΩ cm, filtered and irradiated to reduce organics) were 415 ns, 458 ns, and 584 ns for the 2 mm, 1 mm, and 0.1 mm cells, respectively. The flow cell introduces reflection, scattering, and absorption losses into the ringdown cavity. Reflection losses for the (linearly polarised) laser light at the flow cell surfaces can be minimised by inserting the flow cell at Brewster's angle (for more detail see ref. 6), and the optimum ringdown times quoted above were observed when the flow cells were positioned at an angle of ∼56° to the cavity axis. By measuring the cavity transmission T in the presence and absence of the flow cell, the round-trip losses associated with the cell can be quantified, and are found to be around 0.25%. Table 1 lists the total cavity loss and the loss associated with the flow cell per round trip, the number of passes through the sample during one ringdown time τ, and the associated effective path length per unit ringdown time, lτ, for each flow cell arrangement. We see that the 1 mm and 2 mm cells yield effective path lengths of several tens of centimetres, while even the 0.1 mm cell has an effective path length of 3.5 cm. We typically record the ringdown time over three to five ringdown times, yielding total path lengths of around 2 m, 1 m, and 15 cm for the 2 mm, 1 mm, and 0.1 mm cells.
| Flow cell | L tot / % | L cell / % | N τ | l τ / mm |
|---|---|---|---|---|
| 2.0 mm | 0.469 | 0.265 | 213 | 440 |
| 1.0 mm | 0.425 | 0.221 | 235 | 289 |
| 0.1 mm | 0.333 | 0.129 | 299 | 35 |
The focus of this work is on demonstrating the application of colourimetric CRDS to the detection of trace analytes in water, determining the achievable detection limits, and investigating the factors limiting the detection sensitivity. For each of the three chemical species considered, we have determined the detection limit in two different ways, firstly from the signal to noise ratio of the baseline ringdown measurement, and secondly from the uncertainty in the y intercept of a plot of measured optical absorption κ vs. analyte concentration. To estimate the detection limit based on the signal-to-noise ratio of the baseline ringdown signal, we rewrite eqn (2) as
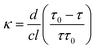 | (3) |
At the detection limit, corresponding to the minimum detectable change in ringdown, we have τ − τ0 = Δτmin and ττ0 ∼ τ02, giving
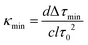 | (4) |
In the present work, we define Δτmin to be three times the standard deviation in the baseline ringdown time τ0i.e. Δτmin = 3σ(τ0). Determination of the detection limit using this method accounts for limitations on the detection sensitivity imposed by the intrinsic noise associated with the measurement, and represents the potential detection limit of the CRDS measurements using the experimental setup described in Section 2.1. However, it does not account for any other sources of experimental uncertainty e.g. impurities in the reagents used. To determine the detection limit when all sources of error are taken into account, we have measured the absorption per unit path length, κ, as a function of concentration, C, for each of the analytes studied. When κ is plotted as a function of C, the uncertainty in the κ intercept gives a good measure of the limit of uncertainty in the measurement of κ, which is equivalent to the detection limit.
Potassium permanganate calibration
As noted previously, the flow cells are positioned between the cavity mirrors at an angle to the cavity axis so as to maximise the measured ringdown time. This increases the optical path length slightly relative to the manufacturer's specifications for light incident normal to the flow cell surface. To obtain an accurate determination of the optical path length through each flow cell when positioned at the optimum angle within the ringdown cavity, a calibration was carried out based on the known absorption of a series of dilute aqueous solutions of potassium permanganate, KMnO4. The absorption coefficient, α, for KMnO4 at 532 nm was determined (with three-standard deviation error) to be (4805 ± 10) M−1 cm−1 in a conventional single-pass absorption measurement carried out using a commercial UV-vis spectrometer (Varian Cary-100 Bio UV-vis spectrometer) employing a 1 cm cuvette. For each flow cell, a series of measurements were made for solutions spanning the dynamic range of the cell, with the minimum concentration corresponding to the minimum detectable change in ringdown, and the maximum to the onset of signal saturation. These ranges spanned 10 nM to 25 μM for the 2 mm cell, 13 nM to 130 μM for the 1 mm cell, and 200 nM to 500 μM for the 0.1 mm cell. Ringdown times of the KMnO4 solutions were measured in alternation with distilled water ‘blanks’, in order to ensure that complete flushing of the flow cell with the solution of interest had been achieved. Each measurement was repeated three times to ensure reproducibility. Flushing of the cells for between one and two minutes at flow rates of 0.5–2.0 mL min−1 was required to ensure complete flushing. Measurement of the ringdown time was performed under stopped flow conditions, although no noticeable difference was observed when recording under flow conditions.The calibration data for each flow cell, together with sample ringdown traces, are shown in Fig. 2. The path lengths for the 2 mm, 1 mm, and 0.1 mm flow cells were determined to be 2.07 mm, 1.23 mm, and 0.117 mm, respectively. Nitrite measurements were made in the 1 mm flow cell only, as the limit of detection was found to be well within the required range after the initial set of measurements using this cell. Iron measurements were made in all three flow cells in order to investigate the effect of the path length on the limit of detection.
 | ||
| Fig. 2 Plot of κ versus permanganate concentration C / μM, determined for (a) 2 mm flow cell, with path length 0.207 cm, (b) 1 mm flow cell, with path length 0.123 cm and (c) 0.1 mm flow cell, with path length 0.0117 cm. The error bars shown represent three-standard-deviation errors in κ, determined from 30 measurements of the ringdown time (y axis), and 1% errors in the KMnO4 concentrations (x axis). (d) Example ringdown traces recorded in the 0.1 mm flow cell. The black trace corresponds to the ringdown time when the flow cell is filled with water, τ0 = 588 ns, and the overlain grey trace corresponds to the ringdown time when the flow cell is filled with 100 μM permanganate solution, τ = 232 ns. | ||
Nitrite detection
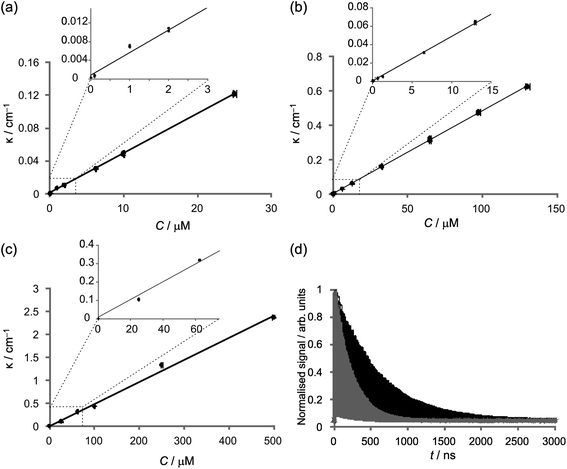
Nitrite was quantified using the well-known Griess Assay.20 Several versions of this reaction exist, and the method we chose to use was that described by Sieben et al.17 and Tovar et al.9 Reagents were purchased from Sigma Aldrich and used without further purification. The “Griess reagent” was created by dissolving 0.5 g of sulfanilamide (Sigma Aldrich, S9251) in 50 mL of deionised water and 5 mL of concentrated hydrochloric acid (analytical reagent grade) in a 500 mL volumetric flask. To this was added 0.05 g of N-(1-naphthyl)ethylenediamine dihydrochloride (Sigma Aldrich, 33461), and the resulting solution was made up to 500 mL with deionised water. Standard nitrite solutions were created by serial dilution in deionised water from a standard 1 M sodium nitrite solution (Sigma Aldrich, 35271).To perform a nitrite determination measurement, the Griess reagent and nitrite solution were mixed in a beaker and left to stand for at least 10 minutes to ensure that the Griess reaction had reached completion before the azo dye absorption was measured. Rather than using deionised water to determine the ‘blank’ τ0 value for our ringdown measurements, a 1:1 mixture of deionised water and the Griess reagent was employed. This was found to be essential in order to eliminate contributions to the signal from trace quantities of nitrite present in the deionised water and/or the reagents used to formulate the Griess reagent. Absorption measurements were made using the 1 mm flow cell. For each measurement, a series of ringdown traces were acquired at time intervals of 2 s while the flow cell was flushed firstly with deionised water, then a 1:1 mixture of Griess reagent and water (constituting the τ0 measurement), and finally a 1:1 mixture of Griess reagent and the nitrite solution of interest. A flow rate of 0.5 mL min−1 was employed, with each measurement requiring a total sample volume of 1 mL. The measurements were repeated three times for each concentration, with seven different nitrite concentrations investigated, in the range 25 nM to 10 μM. The x and y error bars associated with each data point in Fig. 3 represent 1% errors in the concentrations of the standard solutions, and three-standard-deviation errors in the value of κ determined from 30 separate measurements of the ringdown time.
 | ||
| Fig. 3 (a) Example graph of ringdown time versus time, as water, then 1:1 Griess–water, then 1:1 Griess–100 nM nitrite is flowed through the 1 mm cell. (b) Plot of κ / cm−1versus nitrite concentration C / μM, determined in 1 mm flow cell. The gradient of the graph is (96400 ± 1600) M−1 cm−1. | ||
Iron detection
Colourimetric determination of iron(II) was achieved using sulfonated bathophenanthroline (Sigma Aldrich, 11890), which reacts with Fe(II) in a 3:1 ratio to produce a strongly absorbing product with peak absorption at 533 nm. The reagent is readily soluble in aqueous solutions,30 and thus avoids the time-consuming organic steps traditionally required for the use of non-sulphonated bathophenanthroline. Iron(II) standard solutions were prepared by dissolving iron(II) chloride (Sigma Aldrich, 372870) in deionised water, followed by serial dilution to produce a range of iron(II) concentrations in the millimolar to nanomolar range. The reaction is usually carried out at low pH. However, acidifying the samples using 1 mM hydrochloric acid (analytical reagent grade) to produce a final solution of pH ∼3 led to a very poor blank ringdown measurement (<100 ns), presumably owing to significant levels of iron in the hydrochloric acid. Instead, iron(II) and bathophenanthroline were prepared in deionised water, without attempting to control the pH. Blair and Diehl31 investigated the pH dependence of the formation of iron(II) tribathophenanthroline disulphonic acid, and found that the complex was stable within the pH range 3–11. Although acidic reaction conditions are thus not required for complex formation, Bruland and Rue note in Chapter 6 of ref. 22 that the oxidation kinetics of Fe(II) to Fe(III) can be markedly slowed by acidifying the sample to a pH near 3. To prevent decrease in iron(II) concentrations through oxidation to iron(III), we prepared fresh iron(II) solutions twice daily: during this time, under our experimental conditions, oxidation was not noticeable.
A 3 mM stock solution of bathophenanthroline was used in all cases, in a large excess compared to the iron concentrations. The characteristic red colour of the product formed instantly, and did not fade, as has been previously reported,1 thus the solutions were flowed into the flow cell immediately after thorough mixing. In analogy to the nitrite measurements, a 1:1 mixture of water and bathophenanthroline solution was used to record the blank τ0 value, with the data acquisition sequence following that outlined above for the nitrite measurements. Measurements were made using all three flow cells over a range of Fe(II) concentrations. A flow rate of 2 mL min−1 was employed when using the 2 mm flow cell, with a measurement requiring a total sample volume of 5 mL, while a flow rate of 0.5 mL min−1 was employed when using the 1 mm and 0.1 mm flow cells, requiring total sample volumes of 1 mL and 0.5 mL, respectively. Errors were determined as above for the nitrite measurements.
Results and discussion
As noted in Section 2, the detection limit for each of the three analytes studied was determined using two different methods, the first based on the 3σ uncertainty in the baseline τ0 measurement (around 0.4% in our current experimental setup), and the second based on the uncertainty in the intercept of a plot of κ, the absorption per unit path length, vs. analyte concentration C. Table 2 lists the results of this analysis for the potassium permanganate measurements, which were used both to calibrate the absorption path length per pass (see Section 2) and to quantify the minimum detectable absorption per unit pass using each of the three flow cells. The table summarises the geometrical properties of each flow cell (optical path length when inserted into the cavity at the appropriate angle to optimise the ringdown time, and total probed volume of liquid), the baseline ringdown time for a flow cell filled with deionised water, and the detection limit expressed both in terms of minimum detectable absorbance per unit pass and minimum detectable permanganate concentration, as determined from the uncertainty in the τ0 measurement using eqn (4).| Flow cell | l / mm | Illuminated volume / nL | τ 0 / ns | κ min / cm−1 | C min / nM |
|---|---|---|---|---|---|
| 2.0 mm | 2.07 | 393 | 415 ± 1.6 | 8.8 × 10−5 | 18 |
| 1.0 mm | 1.23 | 196 | 458 ± 1.8 | 1.4 × 10−4 | 29 |
| 0.1 mm | 0.117 | 20 | 584 ± 2.3 | 1.1 × 10−3 | 240 |
The minimum detectable absorbances determined from the baseline τ0 measurement are 8.8 × 10−5 cm−1, 1.4 × 10−4 cm−1, and 1.1 × 10−3 cm−1 for the 2.0 mm, 1.0 mm, and 0.1 mm cells, respectively. If we instead estimate the detection limits based on the 3σ uncertainty in the y intercept of a κ vs. concentration plot (similar to Fig. 2), we obtain values of 8.1 × 10−4 cm−1, 2.5 × 10−3 cm−1, and 5.8 × 10−2 cm−1 for the three cells. As noted in Section 3, the fact that these values are higher than those estimated simply from the signal-to-noise ratio of the ringdown measurements is unsurprising, since they include all sources of experimental uncertainty, including variations in analyte and/or colourimetric reagent concentrations when preparing the standard solutions.
Fig. 3(a) shows a typical dataset for a nitrite determination measurement, illustrating the different ringdown times recorded for deionised water, a 1:1 mixture of deionised water and Griess reagent, and finally a 1:1 mixture of a nitrite solution and Griess reagent. Fig. 3(b) shows the κ vs. concentration plot obtained after carrying out a series of such measurements for nitrite solutions spanning a range of concentrations in the nanomolar to millimolar range. Given the known absorption path length for the 1 mm flow cell, the slope of the plot yields an absorption coefficient for the azo dye product of the Griess assay of (96400 ± 1600) M−1 cm−1. This, together with our previously determined minimum detectable absorption per unit path length for this cell of κmin = 1.4 × 10−4 cm−1, would predict a nitrite detection limit of 1.4 nM, based solely on the signal-to-noise ratio in our measurement of τ. The alternative determination of the detection limit, based on the 3σ uncertainty in the y-intercept of the plot in Fig. 3(b), yields a value of κmin = 3.9 × 10−3 cm−1, or 39 nM nitrite. Unfortunately, these concentrations are significantly exceeded by the concentration of nitrite present in the deionised water, complicating the verification of the detection limit through measurements on standard solutions. Based on the difference in ringdown times measured for pure deionised water and a 1:1 mixture of deionised water and Griess reagent (see Fig. 3(a)), we estimate the total nitrite concentration in the deionised water to be 170 ± 70 nM. We believe that the uncertainty in this measurement is largely due to a real variation in the nitrite concentration within the deionised water source over the several days during which measurements were made. To reduce the effects of this variation on our nitrite measurements, within each three-stage measurement of the type shown in Fig. 3(a), the same batch of deionised water was used. This seems to be a valid approach, since a redetermination of κmin based on the 3σ uncertainty in the Griess–water mixture yields a value of 1.8 × 10−4 cm−1, or 1.9 nM nitrite, only slightly higher than the value obtained using deionised water as the blank. We have verified detection of nitrite down to a concentration of 12.5 nM. At these levels, a clear difference could be seen in the ringdown time of the Griess–nitrite solution (357 ns) relative to the Griess–water solution (367 ns). It is likely that the actual detection limit is close to the predicted 1.4 nM (with the higher value determined from the plot being an artefact of the purity of the deionised water available to us), but we are hampered in accessing lower concentrations by the high blank concentration of nitrite. Even so, this detection limit is significantly below the ambient nitrite concentration in marine and aquatic environments, such that CRDS represents a promising approach for detecting low levels of nitrite in such environments.
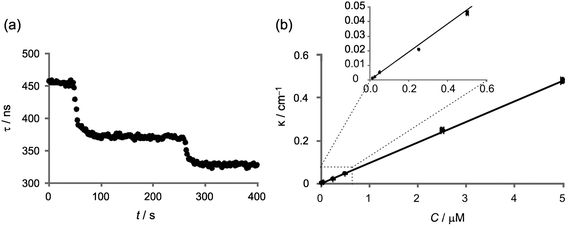
Fe(II) is present at considerably lower levels than nitrite in marine environments, and is consequently much more difficult to detect. For this reason, detection limits were determined using all three flow cells in order to determine the optimum path length for such measurements. Plots of κ vs. Fe(II) concentration for each flow cell are shown in Fig. 4. Based on the signal-to-noise in the baseline τ0 measurement recorded for a 1:1 mixture of bathophenanthroline and deionised water, the minimum detectable absorptions per pass, κmin, for the 2.0 mm, 1.0 mm, and 0.1 mm flow cells were determined to be 5.2 × 10−4 cm−1, 1.9 × 10−4 cm−1 and 8.1 × 10−4 cm−1, respectively. Given that the absorption coefficient of the absorbing species is 52400 ± 600 M−1 cm−1 (the weighted mean and error of the values for α determined in each flow cell and given in the caption to Fig. 4), this corresponds to the detection of 10 nM, 4 nM and 16 nM iron in each case. Based on the uncertainty in the y-intercept of the κ vs. C plot, the detection limits are 5.2 × 10−3 cm−1 for the 2 mm flow cell, 1.5 × 10−3 cm−1 for the 1 mm flow cell and 8.0 × 10−3 cm−1 for the 0.1 mm flow cell. Qualitatively, we detected a reproducible ca. 2 ns change in ringdown time using the 1 mm flow cell when measuring 5 nM iron (the lowest concentration measured in this dataset), and a reproducible ca. 3 ns change in ringdown time using the 0.1 mm flow cell when measuring 50 nM iron. In contrast, using the 2 mm flow cell, 50 nM (the lowest concentration measured) was not detectable. The higher absolute variation in the blank signal arising from the higher absorbance within the longer path length cell caused the minimum detectable change in ringdown time to be higher than measured with the 1 mm and 0.1 mm flow cells. Thus, we can see that the 1 mm flow cell has the most favourable limit of detection under our current experimental conditions.
 | ||
| Fig. 4 Plot of κ/cm−1versus Fe(II) concentration C/μM, where the value of the absorption coefficient α is determined from the gradient of the graph for (a) 2 mm flow cell, α = (52400 ± 2100) M−1 cm−1, (b) 1 mm flow cell, α = (48200 ± 1800) M−1 cm−1, and (c) 0.1 mm flow cell, α = (53100 ± 700) M−1 cm−1. (d) Example ringdown traces recorded using the 1 mm flow cell. The black trace is the ringdown time for the water-filled cell, τ0 = 383 ns, and the overlain grey trace is the ringdown time for 0.5 μM iron, τ = 241 ns. | ||
The results described above demonstrate that CRDS has sufficient sensitivity to detect nitrite and iron at concentrations in the nanomolar range, for optical path lengths of 1 mm or less. Our best detection limits based on the signal-to-noise ratio of the measurements are 1.9 nM for nitrite and 3.8 nM for Fe(II), with slightly higher limits recorded in practice due to problems with preparing a ’blank’ sample containing a sufficiently low concentration of the analyte of interest. When the illuminated volume is taken into account, this corresponds to the detection of around 1 billion molecules. At this stage, we should compare the detection sensitivities achieved using CRDS with those obtained using existing techniques.
King et al. state that using a standard spectrophotometer with 10 cm cells, the practical detection limit for Fe(II) using the ferrozine assay is 20 nM.32 By placing a flow cell in a cavity, we have improved on this detection limit by around a factor of five, while also reducing the optical path length by two orders of magnitude. Similarly, for nitrite, Raimbault et al. achieved a detection limit of 1.2 nM nitrite using a 5 cm cell.33 By optimising a 2.5 cm long microfluidic absorption cell, Sieben et al. achieved a detection limit of 14 nM nitrite.17 Using a similar absorption cell, also 2.5 cm in length, Floquet et al. measured a detection limit of 21 nM iron.34
An alternative method of achieving greatly increased absorption path lengths is to employ a liquid core waveguide (LCW).35 LCWs have been applied to both nitrite36 and iron detection,37,38 and detection limits in the 0.1 nM concentration range have been demonstrated when using LCWs several metres in length.38,39 LCWs provide an attractive approach, using relatively small volumes of reagents, and achieving high sensitivity in a relatively small experimental footprint which is easily interfaced with flow systems. However, they suffer from high blank signals, significant dead volume at the liquid–light interface, and the measurements are also prone to contamination from air bubbles. Though we have not yet demonstrated detection limits on the sub-nanomolar level, for measurements in which a slightly poorer detection limit is tolerable, cavity ringdown measurements provide a long effective absorption path length while maintaining a short physical path length that avoids most of the problems associated with LCWs.
Because we do not currently have the required sensitivity to be able to detect iron in the sub-nanomolar concentrations present in the open ocean, we conclude with a brief discussion of possible improvements to the set-up which may lower the detection limit further. In a cavity ringdown measurement, the detection limit can be improved either by increasing the ringdown time (i.e. reducing the cavity loses), by increasing the path length per pass, or by minimising the shot-to-shot variation in τ. We will consider each of these factors in turn.
Increasing the ringdown time can be achieved either by increasing the reflectivity of the mirrors or by minimising the flow cell losses. Since our cavity losses are currently dominated by losses associated with the flow cell, increasing the mirror reflectivity will have a limited effect. For example, based on the current losses associated with our 1.0 mm flow cell, increasing the mirror reflectivity from the current relatively modest value of 99.8% to the ‘best available’ value of 99.999%, assuming we could still measure the ringdown time to an accuracy of ∼0.4% as at present, would only improve our detection sensitivity by around a factor of two. This improvement would come at the cost of greatly reduced signal levels, since a much smaller amount of light can be coupled into and out of the cavity on each pass. Losses associated with the flow cell could be reduced by the use of custom-designed Brewster's angle flow cells. Such an approach was demonstrated in 2003 by Snyder and Zare, who used a specially designed Brewster's angle flow cell which allowed for light to strike each surface at the correct Brewster's angle for each interface.40 The loss through this 300 μm path length flow cell (0.06%) is roughly half the loss we determined through our 100 μm flow cell, despite the three times longer path length, demonstrating the improvement possible through the use of a better flow cell design. In our case, if we inserted Snyder and Zare's flow cell into our present cavity, we would potentially achieve a detection limit of κmin = 3.6 × 10−4 cm−1, corresponding to a detection limit of 7.0 nM iron in a 300 μm path length. If we combined Snyder and Zare's flow cell with 99.99% mirrors, we could potentially achieve κmin = 1.9 × 10−5 cm−1, corresponding to a detection limit of 1.9 nM iron in 300 μm path length.
Though eqn (1) implies that an increased path length should improve the detection sensitivity in a cavity ringdown measurement, this is unfortunately not always the case. We have already shown that increasing the path length will not necessarily increase the sensitivity if the blank concentration is high. Additionally, increasing the path length increases the scattering and absorption losses through the solvent significantly, and thus lowers the ringdown value. The relatively high blank signal could certainly be improved by the use of higher purity reagents/solvents; in the present work we used all reagents as purchased without further purification. It is worth noting that in the first application of CRDS to liquid-phase analysis, Xu et al. inserted single 1 cm or double 1 cm quartz cells into a ringdown cavity formed from two 99.97% reflectivity mirrors, and κmin = 5 × 10−5 cm−1 was achieved through measurement of benzene in the pure liquid and in hexane.41 This is comparable with the values obtained in the present work in the 2 mm flow cell.
The detection limit can also be improved by reducing the variation in the measured ringdown time, τ. This would most readily be achieved by moving from a pulsed laser source to a CW source. For example, Zare and coworkers42 improved on their minimum detectable absorbance measured in 2005, by replacing the pulsed light source with a single-mode, continuous-wave light source which allowed excitation of only a single cavity mode. This reduced the shot-to-shot variation in τ from 1% (ref. 40) to 0.04%, allowing determination of κmin = 7.8 × 10−6 cm−1.42 In our case, the variation in τ is already only 0.4%, and we believe that our measurements are more significantly affected by the degree of flushing of the flow cell, and volume errors in making up the stock solutions.
Finally, as for any analytical technique, the detection limit can be improved by incorporating a preconcentration step into the analysis. For example, King et al. found a 40-fold improvement in the detection limit for iron when using a ferrozine-coated C18 column.32 Blain and Tréguer achieved a detection limit of 0.1 nM Fe(II) and 0.3 nM Fe(III), by carrying out analysis in a 3 cm optical path length cell, after preconcentration using a similar column.43 Manzoori and Soflaee found a detection limit of 3.8 nM nitrite in a 1 cm cuvette, gaining a 70-fold improvement through preconcentration using a sodium-dodecyl-sulphate-coated alumina column.44 These preconcentration factors would allow us to determine iron at close to the lowest ambient seawater levels in our 1 mm flow cell.
The Griess assay for nitrite is very well established, highly specific to nitrite, and the absorption coefficient of the azo dye product is so large that over the range of ambient concentrations of nitrite found in waterways and oceans, our nitrite quantification protocol is unlikely to suffer significantly from interference due to other absorbing species present in a sample. We have used our method to quantify nitrite concentrations in a range of bottled waters and river water samples and obtained nitrite concentrations in line with those expected, but have not included the data in the present manuscript as we currently have no independent means of verifying the measured concentrations. In contrast with the Griess assay, the iron complexation reaction used in this work is not immune to interference from other analytes. In particular, copper ions can also bind to the ligand, though with lower efficiency. We are currently carrying out studies to determine the extent of this interference and developing a method for quantifying both iron and copper based on their relative binding efficiencies to two different ligands.
In summary, we have demonstrated the application of CRDS to the detection of both nitrite and iron in path lengths similar to those encountered in microfluidic systems, and have shown that the method could potentially be useful for developing ship-based or remote sensors for monitoring trace species in marine and aquatic environments. The detection sensitivity for nitrite is already sufficient to monitor ambient levels in all such environments. The detection sensitivity for Fe(II) is sufficient for monitoring levels in river or drinking water, but not yet sufficiently low to monitor Fe(II) in marine environments. Potential approaches for improving the detection limit into the range required for such measurements have been discussed.
Acknowledgements
This work was funded by the United Kingdom Engineering and Physical Sciences Research Council through grant EP/G027838/1. We would like to thank the Physical and Theoretical Chemistry Department's workshops for fabricating the flow cell mounts used in this experiment, and also Jason. W. L. Lee for writing the LabVIEW data acquisition and analysis software.References
- E. B. Sandell, Colorimetric Determination of Traces of Metals, John Wiley & Sons Inc., 1959 Search PubMed.
- D. F. Boltz and J. A. Howell, Colorimetric Determination of Nonmetals, John Wiley & Sons Inc., 1978 Search PubMed.
- A. O'Keefe and D. A. G. Deacon, Rev. Sci. Instrum., 1988, 59, 2544–2555 CrossRef CAS.
- C. Vallance, New J. Chem., 2005, 29, 867–874 RSC.
- L. van der Sneppen, F. Ariese, C. Gooijer and W. Ubachs, Annu. Rev. Anal. Chem., 2009, 2, 13–35 CrossRef CAS.
- D. James, B. Oag, C. M. Rushworth, J. W. L. Lee, J. Davies, J. T. Cabral and C. Vallance, RSC Adv., 2012, 5376–5384 RSC.
- K. Grasshoff, K. Kremling and M. Ehrhardt, Methods of Seawater Analysis, Wiley-VCH, 3rd edn, 2007 Search PubMed.
- F. J. Millero, Chemical Oceanography, CRC Press, 3rd edn, 2005 Search PubMed.
- A. Tovar, C. Moreno, M. P. Mánuel-Vez and M. García-Vargas, Anal. Chim. Acta, 2002, 469, 235–242 CrossRef CAS.
- EEC, A Directive of the Quality of Freshwater Needing Protection or Improvement in Order to Support Fish Life. 78/659/EEC, Official J. European Union, 1978, vol. L222 Search PubMed.
- B. H. L. Kelso, R. Smith, R. J. Laughlin and S. D. Lennox, Appl. Environ. Microbiol., 1997, 63, 4679–4685 CAS.
- C. S. Bruning-Fann and J. B. Kaneene, Vet. Hum. Toxicol., 1993, 35, 521–538 CAS.
- WHO, Guidelines for Drinking-Water Quality, World Health Organization, Geneva, Switzerland, 4th edn, 2011 Search PubMed.
- M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785–803 CrossRef CAS.
- J. Dutt and J. Davis, J. Environ. Monit., 2002, 4, 465–471 RSC.
- T. F. Rozan and G. W. Luther, Mar. Chem., 2002, 77, 1–6 CrossRef CAS.
- J. V. Sieben, C. F. A. Floquet, I. R. G. Ogilvie, M. C. Mowlem and H. Morgan, Anal. Methods, 2010, 2, 484–491 RSC.
- S. H. Lee and L. R. Field, Anal. Chem., 1984, 56, 2647–2653 CrossRef CAS.
- A. Büldt and U. Karst, Anal. Chem., 1999, 71, 3003–3007 CrossRef.
- P. Griess, Ber. Dtsch. Chem. Ges., 1879, 12, 426–428 CrossRef.
- J. F. Van Staden, Anal. Chim. Acta, 1982, 138, 403–408 CrossRef CAS.
- D. R. Turner and K. A. Hunter, The Biogeochemistry of Iron in Seawater, John Wiley & Sons, 2001 Search PubMed.
- E. P. Achterberg, T. W. Holland, A. R. Bowie, R. F. C. Mantoura and P. J. Worsfold, Anal. Chim. Acta, 2001, 442, 1–14 CrossRef CAS.
- S. Pehkonen, Analyst, 1995, 120, 2655–2663 RSC.
- J. T. Woods and M. G. Mellon, Ind. Eng. Chem., 1941, 13, 551–554 CAS.
- G. F. Smith, W. H. McCurdy and H. Diehl, Analyst, 1952, 77, 418–422 RSC.
- L. L. Stookey, Anal. Chem., 1970, 42, 779–781 CrossRef CAS.
- H. E. Bent and C. L. French, J. Am. Chem. Soc., 1941, 63, 568–572 CrossRef CAS.
- W. B. Fortune and M. G. Mellon, Ind. Eng. Chem., 1938, 10, 60–64 CAS.
- P. Trinder, J. Clin. Pathol., 1956, 9, 170 CrossRef CAS.
- D. Blair and H. Diehl, Talanta, 1961, 7, 163–174 CrossRef CAS.
- D. W. King, J. Lin and D. R. Kester, Anal. Chim. Acta, 1991, 247, 125–132 CrossRef CAS.
- P. Raimbault, G. Slawyk, B. Coste and J. Fry, Mar. Biol., 1990, 104, 347–351 CrossRef CAS.
- C. F. A. Floquet, V. J. Sieben, A. Milani, E. P. Joly, I. R. G. Ogilvie, H. Morgan and M. Mowlem, Talanta, 2011, 84, 235–239 CrossRef CAS.
- L. J. Gimbert and P. J. Worsfold, TrAC, Trends Anal. Chem., 2007, 26, 914–930 CrossRef CAS.
- E. T. Steimle, E. A. Kaltenbacher and R. H. Byrne, Mar. Chem., 2002, 77, 255–262 CrossRef CAS.
- R. D. Waterbury, W. Yao and R. H. Byrne, Anal. Chim. Acta, 1997, 357, 99–102 CrossRef CAS.
- J.-Z. Zhang, C. Kelbe and F. J. Millero, Anal. Chim. Acta, 2001, 438, 49–57 CrossRef CAS.
- J.-Z. Zhang, Deep-Sea Res., Part I, 2000, 47, 1157–1171 CrossRef CAS.
- K. L. Snyder and R. N. Zare, Anal. Chem., 2003, 75, 3086–3091 CrossRef CAS.
- S. C. Xu, G. H. Sha and J. C. Xie, Rev. Sci. Instrum., 2002, 73, 255–258 CrossRef CAS.
- K. L. Bechtel, R. N. Zare, A. A. Kachanov, S. S. Sanders and B. A. Paldus, Anal. Chem., 2005, 77, 1177–1182 CrossRef CAS.
- S. Blain and P. Tréguer, Anal. Chim. Acta, 1995, 308, 425–432 CrossRef CAS.
- J. L. Manzoori and S. Soflaee, Anal. Lett., 2001, 34, 231–237 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |