A liquid chromatography-electrospray tandem mass spectrometry method for the determination of multiple pesticide residues involved in suspected poisoning of non-target vertebrate wildlife, livestock and pets
Michael Jeffrey Taylor*, Laura Marie Melton, Elizabeth Ann Sharp and Jennifer Elizabeth Watson
SASA, Roddinglaw Road, Edinburgh, UK. E-mail: Michael.Taylor@sasa.gsi.gov.uk; Fax: +44 (0)131 2448926; Tel: +44 (0)1312448864
First published on 26th October 2012
Abstract
A liquid chromatography tandem mass spectrometry (LC/MS/MS – triple quadrupole) method that facilitates the qualitative and quantitative determination of multiple pesticide residues that could be present in wild and domestic vertebrate animals has been developed. The method involves the direct analysis of crude extracts from a variety of test specimens taken from (non-target) birds and mammals suspected as being victims of accidental or deliberate exposure to pesticides. Performance of the method was validated following replicate analysis of pseudo-matrices (chicken muscle and chicken liver tissue) that had been fortified with 102 different pesticides and their degradation products at two different concentration levels (0.1 mg kg−1 and 1.0 mg kg−1). Residues detected were quantified following interpolation against external calibration curves obtained using matrix-matched standards that covered a residue concentration range between 0.025 μg ml−1 and 0.5 μg ml−1. Application of the method is demonstrated through inclusion of a few examples that show screening and confirmation results of pesticide residues detected in animals involved in suspected poisoning incidents.
Introduction
The United Kingdom's Wildlife Incident Investigation Scheme (WIIS) is operated in Scotland by SASA, a scientific division of the Scottish Government's Agriculture, Food and Rural Communities Directorate. The WIIS1,2 investigates poisoning of non-target vertebrate wildlife, beneficial insects such as honeybees, livestock and pets whenever accidental or deliberate exposure to pesticides is suspected. The scheme provides a means of post-registration surveillance of pesticide use and a measure of the success of the pesticide registration process. Incidents resulting from approved use or misuse can highlight problems with the conditions of approval or label instructions and provide valuable feedback into the regulatory process. In contrast, operation of the scheme can also reveal the sinister practice of deliberate and illegal attempts to poison animals. Evidence from such incidents can be used by the Scottish Government and police to enforce a variety of legislation relating to the safe use of pesticides and the protection of the environment and animals.3–5The chemical diagnosis of suspected pesticide exposure or poisoning incidents involving wildlife, livestock, domestic animals, beneficial insects (e.g. honeybees) or baits i.e. animal carcases deliberately laced with pesticides, is extremely challenging primarily because:
• Hundreds of professional and amateur plant protection and biocidal products are commercially available and currently approved for use in the UK.
• Products withdrawn from use in the UK are still accessible.
• New products are regularly introduced to the market.
• Analytical methods, techniques and instrumentation used should be capable of the detection, identification and quantitation of multiple pesticide residues in a variety of sample types and complex mixtures even if they are present at ultra-low levels.
• Analytical methods, data and records must comply with rigorous quality assurance procedures and data can be presented, scrutinised and challenged in associated enforcement actions or legal proceedings.
• Targeted chemicals must be extracted from a variety of test specimens such as liver tissue, stomach contents or viscera.
There are several examples of how Liquid Chromatography-Electrospray Tandem Mass Spectrometry (LC/MS/MS) has been successfully applied to determine the presence of multiple pesticide residues that remain in or on food of animal origin including various animal tissues e.g. red/white meat or offal. In 2009, Pang et al.6 combined gas chromatography mass spectrometry (GC/MS) and LC/MS/MS techniques following gel permeation chromatography (GPC) clean up, for the quantitative determination of several hundred pesticides in animal muscles (cow, sheep, chicken and rabbit). They concluded that LC/MS/MS was more sensitive than GC/MS for the determination of over 200 pesticides. Mol et al. utilised Ultra Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC/MS/MS) in the development of a generic extraction method for the simultaneous determination of pesticides, mycotoxins, plant toxins and veterinary drugs in feed and food matrices which included meat.7 Advanced LC/MS based residue methods for the determination of multiple pesticides and their degradation products in foodstuffs which included animal tissues were reviewed by Fernandez-Alba et al. in 2008. They described the advantages and pitfalls of these methods, particularly relating to large-scale screening, identification and quantitation of multiple pesticide residues.8
However, examples of the use of LC/MS/MS for routine monitoring of multiple pesticide residues that may remain in or on specimens taken from vertebrate wildlife or pets suspected of being accidentally or deliberately exposed to pesticides are rare. In 1996, Brown et al. published details of analytical methods used for the determination of multiple pesticide residues and single pesticide residues in wildlife specimens submitted to the WIIS operating in England and Wales. The different methods involved a variety of sample clean-up and derivatisation techniques, combined with GC, GC/MS and/or High Performance Liquid Chromatography (HPLC) and covered a wide range of pesticides and their metabolites.9 The same author updated this review in 2005 and LC/MS and LC/MS/MS were included as front-line techniques for the determination of single pesticide residues (imidacloprid, paraquat and diquat), multiple (carbamate) pesticide residues and anticoagulant rodenticides.10 Hunter et al. published details of a single residue method in 2004 that used LC/MS/MS (negative ion mode) for the quantitative determination of chloralose in specimens collected from suspected poisoned birds of prey.11 Wang et al. (2006) carried out a 6-year retrospective review of pesticide poisoning in domestic animals and livestock in Austria whereby pesticides were characterised by non-MS based chromatographic methods only.12
Most recently (2011), Novotný et al. published details of case studies involving carbamate intoxication of animals in the Czech Republic.13 The authors only noted the use of ‘chromatographic techniques’ for the analysis of tissue (liver), solids (bait, unidentified material) or liquid samples (gastric content).
At SASA, before the method reported here was developed in 2010, we depended almost exclusively on GC/MS/MS multiple pesticide residue methods to screen biological specimens for organochlorine, organophosphate and pyrethroid pesticides or to confirm the presence of a few carbamate pesticides such as carbofuran and aldicarb (initially determined by HPLC-UV). This situation combined with the dearth of relevant multiple pesticide residue methods based on LC/MS/MS highlighted the need for us to significantly and quickly extend our WIIS-Scotland ‘target’ pesticide inventory to include pesticides that were amenable to LC/MS/MS. In an effort to optimise the efficiency of sample preparation procedures, it was decided to employ the same extraction solvent (ethyl acetate) procedures for LC/MS/MS as that used to extract multiple pesticides from specimens prepared for existing GC/MS/MS methods. The ambition was to be able to simply split a common sample extract prior to LC/MS/MS (no clean-up) and GC/MS/MS (GPC clean-up) experimentation. The advantages of using a common sample extract for subsequent LC/MS/MS and GC/MS/MS analyses have been demonstrated by Pihlström14 and Lehotay.15 Both authors utilised this approach for the determination of multiple pesticide residues in various foodstuffs following extraction by ethyl acetate and QuEChERS, respectively.
Experimental
High performance liquid chromatography
HPLC gradient separation was achieved using an 1100 Series Liquid Chromatograph system comprising a column heater module, vacuum degasser, quaternary pump and auto injector/sampler (Agilent Technologies, Santa Clara, USA). A 3 μm Hypersil Gold C18 BDS column 100 × 4.6 mm (Thermo Fisher Scientific, Loughborough, UK) fitted with a Security Guard™ guard cartridge (Phenomenex, Macclesfield, UK) was operated at 35 °C and at a flow rate of 0.5 ml min−1. This flowrate proved optimum for the LCMS system used, eliminated the need for post-column flow splitting and did not compromise peak resolution. The overall run-time and solvent consumption would be reduced through the use of e.g. 2.1 mm internal diameter columns. However, narrow-bore columns were not readily compatible with this particular LCMS system. The gradient elution timetable using A: methanol (5 mM ammonium acetate) and B: water, methanol (5 mM ammonium acetate) 95/5, v/v was:| Time (min) | %A | %B |
|---|---|---|
| 0.00 | 30.0 | 70.0 |
| 1.00 | 30.0 | 70.0 |
| 2.00 | 60.0 | 40.0 |
| 5.00 | 60.0 | 40.0 |
| 22.00 | 85.0 | 15.0 |
| 34.00 | 85.0 | 15.0 |
| 35.00 | 30.0 | 70.0 |
| 40.00 | 30.0 | 70.0 |
An injection volume of 10 μl was used throughout.
An isocratic HPLC separation utilising the above instrument configuration, general operating parameters and mobile phase solutions was used for confirmation purposes i.e. 80% A: 20% B; flow rate: 0.5 ml min−1; stop time: 12 minutes; injection volume: 10 μl.
Mass spectrometry
Electrospray ionisation was achieved using a Quattro Ultima Tandem (triple quadrupole) Mass Spectrometer (Waters Corporation (Micromass), Manchester, UK). The instrument was operated in positive ionisation mode and 102 different pesticides, metabolites and degradation products were monitored during the chromatographic separation using multiple reaction monitoring (MRM) combined with time-scheduled data acquisition sequences. An interchannel delay of 0.02 s, an interscan time of 0.1 s, dwell times between 0.05 and 0.25 seconds and span corresponding to 0.2 Da were used. Argon of 99.9% purity (BOC, Manchester, UK) was used as collision gas (1.4 × 10−3 mbar gas cell pressure).Optimum cone voltage and collision energy values were determined for each analyte following direct infusion (Harvard syringe pump) of individual pesticide solutions. The molecular ion species was identified i.e. [M + H]+, [M + Na]+, [M + NH4]+ or in the case of 3-hydroxy carbofuran [M-H2O + H]+and selected as the precursor ion. The precursor ion → product ion transitions listed in Table 1 were used for screening, confirmation and construction of associated calibration curves. However, the data acquisition capability of the LCMS system employed limited the number of MRM transitions that could be adequately monitored per data acquisition function/channel, especially when time-scheduled data acquisition windows overlapped. Consequently, it was not possible to effectively combine screen and confirmation MRMs in a single experiment for all 102 pesticides sought. Therefore, whenever a positive result was indicated following the initial screen a separate confirmation experiment was performed whereby the original screen and an alternative precursor ion → product ion transitions were monitored (where possible) in combination with the much faster isocratic HPLC separation. In addition, our strategy was to prepare a fresh set of matrix-matched standards and re-analyse the sample extract as soon as possible in order to minimise any instability of analytes in sample matrix solution (ideally within 24 h). The quantitative results obtained from the two separate MRMs monitored in the confirmation experiment had to agree within ±20% in order to satisfy in-house quality control procedures. Analytical method performance criteria and guidelines for confirmation of organic residues or contaminants present in animal products are specified in Commission Decision 2002/657/EC16 whereby the number of (chromatographic/mass spectrometric) identification points (IPs) is specified i.e. at least 3 IPs depending upon the pesticide. We believe that this confirmatory approach accumulates a comparable (minimum) number of IPs.
| Pesticide [precursor ion assignment] | Rt min | MRM screen | CV V | CE eV | MRM confirmation |
|---|---|---|---|---|---|
| a Rt = retention time (minutes); CV = cone voltage; CE = collision energy. | |||||
| Acetamiprid [M + H] | 6.31 | 223 > 126 | 25 | 15 | 223 > 90 |
| Aldicarb [M + Na] | 7.20 | 213 > 89 | 8 | 8 | 213 > 116 |
| Aldicarb sulphone [M + NH4] | 3.28 | 240 > 86 | 13 | 22 | 240 > 148 |
| Aldicarb sulphoxide [M + H] | 3.49 | 207 > 89 | 16 | 15 | 207 > 143 |
| Atrazine [M + H] | 9.44 | 216.1 > 74 | 30 | 20 | 216.1 > 104 |
| Azoxystrobin [M + H] | 10.98 | 404 > 372 | 21 | 16 | 404 > 344 |
| Bendiocarb [M + H] | 7.90 | 224.2 > 109 | 20 | 20 | 224 > 167 |
| Benfuracarb [M + H] | 20.19 | 411 > 194.9 | 20 | 25 | 411 > 251.9 |
| Bitertanol [M + H] | 17.86 | 338 > 269 | 25 | 8 | 338 > 70 |
| Boscalid [M + H] | 11.74 | 343 > 307 | 25 | 25 | 343 > 140 |
| Bromuconazole [M + H] | 15.68 | 378.1 > 158.8 | 30 | 20 | 378.1 > 69.8 |
| Bupirimate [M + H] | 15.23 | 317 > 272 | 25 | 25 | 317 > 166 |
| Carbaryl [M + H] | 8.32 | 202 > 145 | 20 | 15 | 202 > 127 |
| Carbendazim [M + H] | 6.77 | 192 > 160 | 35 | 15 | 192 > 132 |
| Carbofuran [M + H] | 7.95 | 222 > 165 | 20 | 10 | 222 > 123 |
| Carbofuran-3 hydroxy [M-H2O + H] | 6.24 | 220 > 162.9 | 26 | 12 | 220 > 181 |
| Carbosulfan [M + H] | 27.43 | 381 > 118 | 25 | 25 | 381 > 160 |
| Chlorotoluron [M + H] | 9.12 | 213.1 > 72.1 | 30 | 20 | 213.1 > 140 |
| Clofentizene [M + H] | 17.33 | 303 > 138 | 25 | 14 | 303 > 102 |
| Clothianadin [M + H] | 5.93 | 250 > 168.8 | 20 | 15 | 250 > 131.9 |
| Cyazofamid [M + H] | 14.41 | 325 > 108 | 20 | 10 | 325 > 261 |
| Cymoxanil [M + H] | 6.62 | 199.1 > 128 | 15 | 10 | 199.1 > 111 |
| Cyproconazole [M + H] | 13.56 | 292.3 > 125 | 25 | 30 | 294 > 127 |
| Cyprodinil [M + H] | 16.04 | 226 > 108 | 25 | 25 | 226 > 93 |
| Demeton-S-methyl [M + H] | 6.31 | 231 > 89 | 15 | 10 | 231 > 199.8 |
| Demeton-S-methyl sulphone [M + H] | 4.02 | 263 > 169 | 26 | 18 | 263 > 109 |
| Difenoconazole [M + H] | 18.91 | 406.2 > 250.9 | 30 | 25 | 408 > 252.9 |
| Diflubenzuron [M + H] | 15.05 | 311 > 158 | 25 | 10 | 311 > 141 |
| Dimethoate [M + H] | 6.31 | 230 > 125 | 16 | 22 | 230 > 199 |
| Dimethomorph [M + H] | 12.44 | 388 > 301 | 25 | 15 | 388 > 165 |
| Dimoxystrobin [M + H] | 15.50 | 349 > 260 | 25 | 18 | 349 > 228 |
| Disulfoton [M + H] | 17.81 | 275 > 89 | 25 | 18 | 349 > 61.1 |
| Disulfoton sulphoxide [M + H] | 9.08 | 291.1 > 184.9 | 25 | 10 | 291.1 > 213 |
| Diuron [M + H] | 10.07 | 233 > 159.9 | 30 | 25 | 235 > 161.9 |
| Epoxiconazole [M + H] | 14.28 | 330.2 > 121 | 20 | 25 | 330.2 > 101 |
| Famoxadone [M + NH4] | 17.15 | 392.2 > 331 | 15 | 10 | 392.2 > 238 |
| Fenamidone [M + H] | 11.46 | 312.1 > 236 | 25 | 15 | 312.1 > 264 |
| Fenarimol [M + H] | 14.01 | 331 > 268 | 25 | 25 | 331 > 81 |
| Fenbuconazole [M + H] | 14.87 | 337 > 125 | 25 | 25 | 337 > 70 |
| Fenhexamid [M + H] | 13.56 | 302 > 97 | 36 | 26 | 302 > 55 |
| Fenpropimorph [M + H] | 26.94 | 304 > 147 | 25 | 30 | 304 > 116 |
| Fenpyroximate [M + H] | 24.66 | 422 > 138 | 25 | 25 | 422 > 366 |
| Fenthion [M + H] | 16.18 | 279.1 > 168.9 | 25 | 15 | 279.1 > 246.9 |
| Fenthion sulphone [M + H] | 8.44 | 311.1 > 278.9 | 40 | 15 | 310.8 > 124.9 |
| Fenthion sulphoxide [M + H] | 8.23 | 294.9 > 279.9 | 35 | 20 | 294.9 > 264 |
| Fludioxonil [M + H] | 12.51 | 266 > 158 | 25 | 25 | 266 > 180 |
| Flufenacet [M + H] | 13.83 | 364.1 > 194 | 20 | 10 | 364.1 > 152 |
| Fluopicolide [M + H] | 12.37 | 385 > 174.8 | 20 | 25 | 385 > 194 |
| Fluoxastrobin [M + H] | 13.47 | 459.1 > 426.8 | 30 | 20 | 461.1 > 428.9 |
| Fluquinconazole [M + H] | 13.23 | 376.2 > 349 | 25 | 20 | 376.2 > 306.9 |
| Flusilazole [M + H] | 15.32 | 316 > 165 | 20 | 25 | 316 > 247 |
| Flutriafol [M + H] | 9.23 | 302.2 > 69.8 | 20 | 25 | 302.2 > 122.8 |
| Imazalil [M + H] | 16.98 | 297 > 159 | 32 | 25 | 297 > 201 |
| Imidacloprid [M + H] | 5.60 | 256 > 175 | 25 | 25 | 256 > 209 |
| Indoxacarb [M + H] | 19.26 | 528 > 203 | 25 | 35 | 528 > 150 |
| Isofenphos [M + H] | 17.55 | 346 > 245 | 20 | 10 | 346 > 217 |
| Isoproturon [M + H] | 9.75 | 207 > 165 | 20 | 12 | 207 > 72 |
| Kresoxim-methyl [M + H] | 15.50 | 314 > 222 | 25 | 10 | 314 > 116 |
| Linuron [M + H] | 11.46 | 249.7 > 160 | 26 | 20 | 249.7 > 182 |
| Malathion [M + H] | 12.30 | 331 > 127 | 30 | 20 | 331 > 285 |
| Mepanipyrim [M + H] | 13.37 | 224 > 106 | 25 | 30 | 224 > 77 |
| Metconazole [M + H] | 17.37 | 320.3 > 69.8 | 25 | 25 | 320.3 > 124.7 |
| Methiocarb [M + H] | 11.81 | 226 > 121 | 25 | 15 | 226 > 169 |
| Methomyl [M + H] | 4.13 | 185 > 128 | 22 | 10 | 163 > 88 |
| Metolcarb [M + H] | 7.66 | 166 > 109 | 25 | 9 | 166 > 194 |
| Metrafenone [M + H] | 17.64 | 409.1 > 209 | 25 | 15 | 409.1 > 229 |
| Monocrotophos [M + H] | 4.67 | 241 > 127 | 18 | 18 | 224 > 127 |
| Myclobutanil [M + H] | 13.09 | 289 > 70 | 25 | 20 | 289 > 125 |
| Ofurace [M + H] | 7.90 | 282 > 254 | 25 | 10 | 284 > 160 |
| Omethoate [M + H] | 3.23 | 214 > 155 | 20 | 12 | 214 > 125 |
| Oxamyl [M + NH4] | 3.56 | 237 > 72 | 25 | 12 | 237 > 90 |
| Oxydemeton methyl [M + H] | 3.82 | 247 > 109 | 18 | 15 | 247 > 169 |
| Penconazole [M + H] | 16.31 | 284 > 159 | 30 | 30 | 284 > 70 |
| Pencycuron [M + H] | 18.08 | 329 > 218 | 30 | 15 | 329 > 125 |
| Phorate [M + H] | 17.24 | 261 > 75 | 25 | 25 | 261 > 171 |
| Phorate sulphone [M + H] | 9.18 | 293 > 115 | 25 | 25 | 293 > 171 |
| Picoxystrobin [M + H] | 15.41 | 368 > 145 | 25 | 20 | 368 > 205 |
| Pirimicarb [M + H] | 9.02 | 239 > 72 | 28 | 21 | 239 > 181.9 |
| Prochloraz [M + H] | 17.94 | 376 > 308 | 25 | 10 | 376 > 266 |
| Propamocarb [M + H] | 6.77 | 189 > 102 | 30 | 15 | 189 > 144 |
| Propiconazole [M + H] | 16.98 | 342 > 159 | 25 | 30 | 342 > 69 |
| Pymetrozine [M + H] | 4.01 | 218 > 105 | 25 | 30 | 218 > 79 |
| Pyraclostrobin [M + H] | 17.15 | 388.1 > 194 | 25 | 10 | 388.1 > 163.9 |
| Pyrethrins [M + H] summed | 24.26 | 361 > 149 | 25 | 8 | 361 > 107 |
| 329 > 161 | 329 > 133 | ||||
| 317 > 149 | 317 > 121 | ||||
| 373 > 161 | 373 > 133 | ||||
| 375 > 163 | 375 > 121 | ||||
| 331 > 163 | 331 > 121 | ||||
| Pyrifenox [M + H] | 14.91 | 295.1 > 93 | 30 | 25 | 297 > 93 |
| Pyrimethanil [M + H] | 11.46 | 200 > 107 | 40 | 28 | 200 > 82 |
| Quinoxyfen [M + H] | 22.21 | 308 > 197 | 20 | 30 | 308 > 162 |
| Simazine [M + H] | 8.00 | 202.1 > 131.9 | 30 | 20 | 202.1 > 124 |
| Spinosad [M + H] summed | 27.69 | 732.5 > 142 | 25 | 25 | 732.5 > 98.5 |
| 746 > 142 | 746 > 95 | ||||
| Spiromesifen [M + H] | 23.34 | 371 > 273 | 25 | 22 | 371 > 255 |
| Spiroxamine [M + H] | 22.48 | 298.2 > 143.9 | 30 | 22 | 298.2 > 100 |
| Tebuconazole [M + H] | 16.31 | 308 > 70 | 30 | 24 | 308 > 125 |
| Tebufenpyrad [M + H] | 20.98 | 334 > 145 | 25 | 25 | 334 > 117 |
| Tetraconazole [M + H] | 14.32 | 372 > 70 | 25 | 15 | 374 > 161 |
| Thiabendazole [M + H] | 7.60 | 202 > 175 | 30 | 25 | 202 > 131 |
| Thiacloprid [M + H] | 6.70 | 253 > 126 | 25 | 20 | 253 > 186 |
| Thiamethoxam [M + H] | 4.38 | 292.1 > 210.9 | 20 | 10 | 292.1 > 181 |
| Thiodicarb [M + H] | 8.63 | 355 > 88 | 25 | 20 | 355 > 108 |
| Triazamate [M + H] summed | 6.62 | 287 > 198, 315 > 224 | 25 | 15 | 315 > 72 |
| Trichlorfon [M + H] | 7.90 | 256.9 > 108.7 | 25 | 25 | 256.9 > 220.9 |
| Trifloxystrobin [M + H] | 19.26 | 409 > 186 | 20 | 25 | 409 > 206 |
| Triticonazole [M + H] | 13.83 | 318 > 69.8 | 30 | 35 | 318 > 124.8 |
A nitrogen generator (Peak Scientific, Renfrew, UK) and compressor system (Atlas Copco, Cumbernauld, UK) were used to supply nitrogen as the desolvation, cone and nebuliser gas. These were set at universally applied values of approximately 500 l h−1 (desolvation gas flow rate) and 80 l h−1 (cone gas flow rate). The ion source was operated at 150 °C, the desolvation temperature held at 350 °C and the capillary voltage was maintained at 3 kV. The LC/MS/MS instrument was controlled and data processed using MassLynx 4.0 and QuanLynx Application Manager software (Waters Corporation (Micromass), Manchester, UK).
Materials and apparatus
Certified pesticide reference standards (purity ≥98%) were purchased from Qmx (Thaxted UK) and pure solvent standard solutions were prepared individually then as mixtures using methanol (HPLC grade, Rathburn, Walkerburn, UK). High purity laboratory water used for HPLC mobile phase was generated in-house and AnalaR grade ammonium acetate (VWR, Lutterworth, UK) was used to prepare aqueous mobile phase ‘buffer’ solution. Ethyl acetate (glass distilled grade) was the extraction solvent (Rathburn, Walkerburn, UK) and anhydrous sodium sulphate used to absorb any moisture released during sample processing was purchased from Sigma-Aldrich (Gillingham, UK). HPLC (0.45 μm PTFE) syringe filters (Chromacol Ltd, Welwyn Garden City, UK) were used to filter the final extract.Sample extraction
Liver tissue was chopped and a portion (≤5 g) weighed into a beaker (100 ml). Sufficient anhydrous sodium sulphate was added to the sample in order to absorb any moisture. The mixture was allowed to dry for 20–30 minutes until friable then transferred into a conical flask (250 ml) and 100 ml of ethyl acetate added. Digestive tract material (≤10 g of a representative subsample) was weighed directly into a 150 ml or 250 ml conical flask, sodium sulphate added and left for 20–30 minutes then 100 ml of ethyl acetate added. The flask was securely stoppered and tumbled for approximately 1 hour. The resulting extract was filtered through a Whatman no. 1 filter paper (18.5 cm) into a round-bottomed flask (150 ml). The extract was evaporated to low volume (<1 ml) by rotary evaporation (bath temperature was kept below 30 °C). The extract was quantitatively transferred to a 5 ml volumetric flask containing 2.5 ml of cyclohexane and made to volume with ethyl acetate. 1 ml of this solution was then transferred into a round-bottomed (25 ml) flask along with 10 ml methanol (the remaining ethyl acetate–cyclohexane solution was retained for additional GCMS sample preparation). The methanolic solution was evaporated to low volume (approximately 1 ml) before it was quantitatively transferred into a 2 ml volumetric flask and made up to volume with methanol containing 5 mM ammonium acetate. An appropriate aliquot of the final solution was simply filtered into an auto sampler vial using a 1 ml disposable syringe fitted with a 0.45 μm PTFE syringe filter. The sample was then ready for LC/MS/MS analysis.Suspected bait materials were either dried with sodium sulphate and the whole sample was soaked in ethyl acetate or a representative subsample was taken and extracted in the same way as described for digestive tract material. When sample amounts were limited the extraction method was the same but reagent amounts were adjusted proportionately.
Preparation of pure solvent standards, matrix-matched standards and fortified samples
Stock solutions of individual pesticides were prepared from certified reference materials into methanol (≈400 μg ml−1) and aliquots taken to compose standard mixtures (5 μg ml−1). Serial dilution with methanol generated a range of mixture solutions which were subsequently used to prepare matrix-matched calibration standards and for the fortification of ‘spiked’ samples. Chicken muscle and liver tissue ideally described as organic, were purchased from local retail outlets in order to generate ‘blank’ pseudo-matrices using the same extraction/solvent exchange procedures described above. The matrix solutions were prepared so that 1 ml ≡ 10 g of tissue in methanol. A range of matrix-matched calibration standards were subsequently prepared by admixing aliquots of pure solvent standard mixture and matrix solution to ensure a matrix concentration of 0.5 g ml−1. The matrix-matched pesticide concentrations used were 0.025 μg ml−1; 0.1 μg ml−1; 0.2 μg ml−1; 0.5 μg ml−1. Matrix solution ‘blanks’ were screened during the validation process in order to ensure that they were free from pesticide residues. Even though none of the pesticides included in this method were found, it is recommended that matrix is similarly screened whenever matrix supplies are replenished.Recovery and repeatability were determined following analysis of ‘spiked’ samples that were fortified with known amounts of each pesticide. Mixed standards were used to fortify chicken muscle tissue or chicken liver with amounts consistent with the majority of lower residue levels found in liver tissue from animals known to have been accidentally or deliberately exposed to pesticides i.e. 0.4–1 mg kg−1. Two fortification levels of 0.1 mg kg−1 and 1.0 mg kg−1 were selected for chicken muscle tissue and chicken liver.
Results and discussion
The pesticides chosen to extend the WIIS-Scotland target inventory were readily available from an existing and comprehensive target inventory (several hundred pesticides) assembled to facilitate SASA's participation in UK and EU ‘Pesticide Residues in Food’ annual surveillance programs.17 It was extremely important that the selection criteria presented the most efficient and cost-effective provision of an enhanced monitoring service. This was achieved by filtering the ‘foodstuffs’ inventory so that, in the first instance, the new and extended WIIS-Scotland target inventory reflected past and present pesticide usage patterns in Scotland.18 It was also deemed vital to include some pesticides that were no longer or had never been approved for use in the UK because ‘illegal’ compounds such as carbofuran, aldicarb and isofenphos were known to be accessible and were being used to deliberately poison animals. Some other pesticides that showed no appreciable use in Scotland but had been detected in non-target animals elsewhere in the UK were also included e.g. benfuracarb, fenthion, indoxacarb, methomyl or myclobutanil.19 Finally all pesticides selected were amenable to LC/MS/MS. This process allowed us to quickly identify candidate pesticides and improve our capability to ‘capture’ pesticides known to have been used throughout Scotland in particular.Multi-point (n = 4) calibration curves were generated from (MRM) ion-chromatogram peak area measurements for each analyte present in matrix-matched standards (chicken muscle or liver). Matrix-matched standards were chosen in order to compensate for any signal suppression/enhancement compared to their relative response in pure solvent.11 The calibration acceptance criteria were that determination coefficients (r2) obtained were ≥0.96 for each analyte and that the signal:noise (S:N) value of the lowest calibration level (LCL) was >5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. All pesticides that yielded r2 values ≥0.96 and S:N values ≥5:1 for the LCL were subsequently considered for generic extraction and method optimisation. Pesticides that failed repeatedly to satisfy these criteria (i.e. despite additional optimisation of general experimental parameters such as preparation of fresh standards and/or minor changes to universally applied instrument parameters) were deferred in order to minimise the method development effort and avoid any compromise in the response of those pesticides that passed.
:1. All pesticides that yielded r2 values ≥0.96 and S:N values ≥5:1 for the LCL were subsequently considered for generic extraction and method optimisation. Pesticides that failed repeatedly to satisfy these criteria (i.e. despite additional optimisation of general experimental parameters such as preparation of fresh standards and/or minor changes to universally applied instrument parameters) were deferred in order to minimise the method development effort and avoid any compromise in the response of those pesticides that passed.
Further assessment of the utility of the generic ethyl acetate extraction process was achieved following analysis of muscle and liver tissue that had been fortified at two different fortification levels i.e. 0.1 mg kg−1 and 1.0 mg kg−1 of each pesticide prior to sample extraction. The efficiency of the ethyl acetate extraction was considered acceptable if mean recoveries following replicate analysis (n = 5 or n = 6) of the target analyte in each matrix fell within the range 60–140% and yielded a relative standard deviation (RSD) ≤20%. These analytical performance measures were comparable with European Union guidelines for method validation and quality control procedures for the determination of pesticide residues in food and feed20 where mean recoveries should fall within the range 70–120% with a RSD of ≤20%. The guidelines also specify that a generalised mean recovery range of 60–140% for routine multi-residue analysis may be used (the guidelines used to establish in-house performance criteria during method development were superseded in 2011). The majority of pesticides listed in Table 1, complied with the above performance criteria with some minor exceptions. These are highlighted in italics in Tables 2 and 3 which contain the analytical performance data for chicken muscle tissue and chicken liver tissue fortified at each level, respectively.
| Pesticide | Fortification level 0.1 mg kg−1 | Fortification level 1 mg kg−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | n | Mean | CV (%) | Min | Max | n | Mean | CV (%) | |
| a Mean recoveries <60% or >140% and RSDs >20% are shown in italics. | ||||||||||
| Acetamiprid | 73 | 84 | 6 | 80 | 4.7 | 75 | 89 | 6 | 81 | 6.3 |
| Aldicarb | 77 | 96 | 6 | 90 | 7.7 | 66 | 92 | 6 | 81 | 12.3 |
| Aldicarb sulphone | 82 | 92 | 6 | 88 | 5.0 | 77 | 90 | 6 | 84 | 6.1 |
| Aldicarb sulphoxide | 66 | 86 | 6 | 77 | 10.3 | 83 | 92 | 6 | 87 | 3.7 |
| Atrazine | 79 | 87 | 6 | 82 | 4.0 | 71 | 81 | 6 | 75 | 4.7 |
| Azoxystrobin | 73 | 79 | 6 | 76 | 2.9 | 70 | 84 | 6 | 77 | 5.9 |
| Bendiocarb | 78 | 92 | 6 | 85 | 6.9 | 80 | 86 | 6 | 84 | 3.0 |
| Benfuracarb | 72 | 79 | 6 | 76 | 3.3 | 68 | 77 | 6 | 73 | 5.7 |
| Bitertanol | 75 | 83 | 6 | 79 | 3.9 | 70 | 81 | 6 | 76 | 6.1 |
| Boscalid | 64 | 81 | 6 | 72 | 8.8 | 75 | 85 | 6 | 80 | 5.1 |
| Bromuconazole | 80 | 92 | 6 | 85 | 5.2 | 68 | 84 | 6 | 73 | 7.8 |
| Bupirimate | 60 | 86 | 6 | 74 | 14.6 | 77 | 98 | 6 | 86 | 9.2 |
| Carbaryl | 71 | 86 | 6 | 80 | 7.3 | 79 | 93 | 6 | 85 | 5.9 |
| Carbendazim | 75 | 87 | 6 | 82 | 5.6 | 81 | 88 | 6 | 85 | 3.6 |
| Carbofuran | 91 | 101 | 6 | 94 | 3.9 | 88 | 118 | 6 | 101 | 10.2 |
| Carbofuran (3 hydroxy) | 71 | 91 | 6 | 84 | 8.7 | 77 | 92 | 6 | 84 | 6.4 |
| Carbosulfan | 66 | 75 | 6 | 69 | 5.8 | 61 | 72 | 6 | 67 | 6.0 |
| Chlorotoluron | 79 | 95 | 6 | 85 | 6.4 | 74 | 81 | 6 | 77 | 3.4 |
| Clofentizene | 72 | 84 | 6 | 80 | 5.6 | 71 | 81 | 6 | 76 | 5.4 |
| Clothianadin | 68 | 90 | 6 | 79 | 11.4 | 64 | 91 | 6 | 74 | 12.4 |
| Cyazofamid | 78 | 88 | 6 | 82 | 5.1 | 72 | 83 | 6 | 78 | 5.0 |
| Cymoxanil | 75 | 86 | 6 | 80 | 5.9 | 80 | 92 | 6 | 86 | 5.1 |
| Cyproconazole | 62 | 93 | 6 | 83 | 13.3 | 71 | 83 | 6 | 77 | 5.0 |
| Cyprodinil | 75 | 93 | 6 | 84 | 8.3 | 66 | 77 | 6 | 70 | 6.1 |
| Demeton-S-Methyl | 54 | 153 | 6 | 84 | 44.6 | 45 | 90 | 6 | 79 | 21.7 |
| Demeton-S-methyl sulphone | 76 | 81 | 6 | 78 | 2.7 | 78 | 91 | 6 | 86 | 5.2 |
| Difenoconazole | 72 | 86 | 6 | 80 | 7.1 | 72 | 80 | 6 | 75 | 4.9 |
| Diflubenzuron | 71 | 85 | 6 | 77 | 7.3 | 71 | 82 | 6 | 76 | 5.2 |
| Dimethoate | 71 | 88 | 6 | 80 | 8.1 | 74 | 89 | 6 | 81 | 6.6 |
| Dimethomorph | 78 | 87 | 6 | 81 | 4.2 | 65 | 76 | 6 | 72 | 5.6 |
| Dimoxystrobin | 75 | 92 | 6 | 82 | 8.4 | 71 | 93 | 6 | 79 | 10.4 |
| Disulfoton | 59 | 79 | 6 | 68 | 11.3 | 58 | 85 | 6 | 72 | 13.2 |
| Disulfoton sulphoxide | 75 | 94 | 6 | 82 | 7.9 | 81 | 89 | 6 | 84 | 3.8 |
| Diuron | 62 | 95 | 6 | 76 | 15.6 | 66 | 99 | 6 | 77 | 16.0 |
| Epoxiconazole | 73 | 89 | 6 | 83 | 7.5 | 72 | 84 | 6 | 78 | 5.8 |
| Famoxadone | 71 | 78 | 6 | 76 | 3.6 | 68 | 80 | 6 | 73 | 6.2 |
| Fenamidone | 74 | 83 | 6 | 78 | 5.0 | 69 | 78 | 6 | 74 | 4.1 |
| Fenarimol | 62 | 100 | 6 | 79 | 16.3 | 70 | 90 | 6 | 80 | 9.0 |
| Fenbuconazole | 71 | 90 | 6 | 79 | 9.5 | 73 | 84 | 6 | 78 | 5.3 |
| Fenhexamid | 81 | 91 | 6 | 85 | 4.2 | 74 | 82 | 6 | 78 | 4.0 |
| Fenpropimorph | 94 | 107 | 6 | 100 | 5.1 | 68 | 79 | 6 | 73 | 5.5 |
| Fenpyroximate | 75 | 84 | 6 | 80 | 4.6 | 66 | 84 | 6 | 74 | 8.4 |
| Fenthion | 74 | 89 | 6 | 79 | 6.7 | 59 | 75 | 6 | 71 | 8.7 |
| Fenthion sulphone | 53 | 85 | 6 | 69 | 18.3 | 69 | 104 | 6 | 89 | 16.4 |
| Fenthion sulphoxide | 74 | 84 | 6 | 79 | 6.1 | 77 | 89 | 6 | 82 | 6.2 |
| Fludioxonil | 86 | 108 | 6 | 97 | 7.2 | 75 | 92 | 6 | 83 | 8.3 |
| Flufenacet | 76 | 86 | 6 | 81 | 4.6 | 72 | 81 | 6 | 76 | 4.6 |
| Fluopicolide | 76 | 87 | 6 | 83 | 5.6 | 74 | 79 | 6 | 77 | 2.7 |
| Fluoxastrobin | 65 | 76 | 6 | 71 | 5.3 | 71 | 80 | 6 | 75 | 5.0 |
| Fluquinconazole | 63 | 109 | 6 | 83 | 22.1 | 97 | 115 | 6 | 104 | 6.6 |
| Flusilazole | 82 | 96 | 6 | 88 | 5.6 | 69 | 84 | 6 | 76 | 6.5 |
| Flutriafol | 74 | 85 | 6 | 80 | 4.8 | 49 | 83 | 6 | 71 | 17.6 |
| Imazalil | 64 | 82 | 6 | 77 | 8.7 | 74 | 86 | 6 | 78 | 6.1 |
| Imidacloprid | 71 | 87 | 6 | 77 | 7.6 | 67 | 85 | 6 | 76 | 9.6 |
| Indoxacarb | 65 | 88 | 6 | 72 | 12.0 | 64 | 81 | 6 | 72 | 10.5 |
| Isofenphos | 42 | 75 | 6 | 59 | 18.2 | 56 | 121 | 6 | 81 | 29.4 |
| Isoproturon | 74 | 90 | 6 | 81 | 7.3 | 68 | 79 | 6 | 76 | 5.4 |
| Kresoxim methyl | 77 | 94 | 6 | 86 | 8.0 | 72 | 100 | 6 | 81 | 12.8 |
| Linuron | 70 | 124 | 6 | 93 | 23.6 | 77 | 89 | 6 | 83 | 5.6 |
| Malathion | 73 | 82 | 6 | 78 | 3.9 | 74 | 78 | 6 | 76 | 2.3 |
| Mepanipyrim | 77 | 86 | 6 | 82 | 3.9 | 70 | 82 | 6 | 77 | 6.3 |
| Metconazole | 91 | 98 | 6 | 93 | 2.8 | 57 | 86 | 6 | 74 | 14.7 |
| Methiocarb | 75 | 90 | 6 | 81 | 6.8 | 75 | 84 | 6 | 80 | 4.2 |
| Methomyl | 97 | 117 | 6 | 102 | 7.6 | 94 | 119 | 6 | 107 | 8.3 |
| Metolcarb | 67 | 82 | 6 | 73 | 8.2 | 75 | 90 | 6 | 80 | 6.9 |
| Metrafenone | 74 | 89 | 6 | 81 | 7.1 | 74 | 80 | 6 | 76 | 3.5 |
| Monocrotophos | 79 | 87 | 6 | 83 | 3.3 | 76 | 91 | 6 | 83 | 7.0 |
| Myclobutanil | 77 | 88 | 6 | 84 | 4.8 | 74 | 86 | 6 | 80 | 5.4 |
| Ofurace | 66 | 83 | 6 | 77 | 8.5 | 77 | 90 | 6 | 85 | 5.0 |
| Omethoate | 67 | 75 | 6 | 72 | 5.5 | 85 | 96 | 6 | 91 | 5.3 |
| Oxamyl | 80 | 87 | 6 | 85 | 3.0 | 78 | 89 | 6 | 84 | 5.4 |
| Oxydemeton methyl | 73 | 85 | 6 | 79 | 6.8 | 79 | 89 | 6 | 84 | 4.9 |
| Penconazole | 68 | 86 | 6 | 79 | 9.4 | 74 | 84 | 6 | 78 | 4.9 |
| Penycycuron | 77 | 90 | 6 | 86 | 5.6 | 68 | 80 | 6 | 75 | 5.5 |
| Phorate | 74 | 83 | 6 | 78 | 4.0 | 68 | 78 | 6 | 74 | 5.5 |
| Phorate sulphone | 75 | 87 | 6 | 82 | 5.9 | 77 | 91 | 6 | 83 | 6.4 |
| Picoxystrobin | 77 | 92 | 6 | 87 | 6.6 | 74 | 86 | 6 | 79 | 6.1 |
| Pirimicarb | 83 | 91 | 6 | 87 | 3.5 | 72 | 81 | 6 | 78 | 4.2 |
| Prochloraz | 72 | 82 | 6 | 76 | 5.3 | 73 | 80 | 6 | 77 | 3.7 |
| Propamocarb | 33 | 43 | 6 | 38 | 9.5 | 51 | 60 | 6 | 55 | 10.1 |
| Propiconazole | 77 | 81 | 6 | 79 | 1.9 | 77 | 87 | 6 | 83 | 5.3 |
| Pymetrozine | 63 | 74 | 6 | 70 | 5.4 | 72 | 80 | 6 | 76 | 4.3 |
| Pyraclostrobin | 75 | 87 | 6 | 80 | 6.0 | 72 | 83 | 6 | 77 | 5.4 |
| Pyrethrins | 77 | 87 | 6 | 81 | 5.1 | 72 | 81 | 6 | 76 | 4.5 |
| Pyrifenox | 80 | 88 | 6 | 84 | 3.7 | 72 | 81 | 6 | 77 | 5.0 |
| Pyrimethanil | 73 | 96 | 6 | 87 | 9.2 | 74 | 85 | 6 | 78 | 5.0 |
| Quinoxyfen | 68 | 81 | 6 | 76 | 6.1 | 65 | 80 | 6 | 72 | 8.7 |
| Simazine | 67 | 87 | 6 | 80 | 9.0 | 75 | 84 | 6 | 79 | 5.5 |
| Spinosad | 56 | 64 | 6 | 59 | 5.1 | 71 | 98 | 6 | 86 | 10.8 |
| Spiromesifen | 75 | 97 | 6 | 83 | 9.6 | 74 | 94 | 6 | 82 | 9.3 |
| Spiroxamine | 78 | 86 | 6 | 81 | 3.7 | 70 | 80 | 6 | 74 | 4.9 |
| Tebuconazole | 78 | 89 | 6 | 85 | 5.2 | 71 | 83 | 6 | 78 | 5.7 |
| Tebufenpyrad | 76 | 90 | 6 | 82 | 7.3 | 67 | 78 | 6 | 74 | 6.7 |
| Tetraconazole | 78 | 101 | 6 | 85 | 10.1 | 68 | 96 | 6 | 80 | 11.7 |
| Thiabendazole | 72 | 85 | 6 | 78 | 7.3 | 75 | 85 | 6 | 80 | 4.6 |
| Thiacloprid | 74 | 82 | 6 | 78 | 3.8 | 77 | 89 | 6 | 83 | 6.0 |
| Thiamethoxam | 78 | 88 | 6 | 83 | 4.8 | 74 | 84 | 6 | 81 | 5.0 |
| Thiodicarb | 42 | 49 | 6 | 46 | 6.6 | 35 | 49 | 6 | 40 | 11.8 |
| Triazamate | 63 | 73 | 6 | 67 | 6.7 | 60 | 70 | 6 | 65 | 5.4 |
| Trichlorfon | 56 | 101 | 6 | 77 | 22.3 | 72 | 128 | 6 | 93 | 21.9 |
| Trifloxystrobin | 75 | 86 | 6 | 80 | 5.0 | 73 | 83 | 6 | 76 | 4.5 |
| Triticonazole | 84 | 95 | 6 | 91 | 4.3 | 51 | 88 | 6 | 70 | 17.8 |
| Pesticide | Fortification level 0.1 mg kg−1 | Fortification level 1 mg kg−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | n | Mean | CV (%) | Min | Max | n | Mean | CV (%) | |
| a Mean recoveries <60% or >140% and RSDs >20% are shown in italics. | ||||||||||
| Acetamiprid | 68 | 84 | 5 | 76 | 7.7 | 81 | 108 | 6 | 91 | 11.0 |
| Aldicarb | 76 | 86 | 5 | 81 | 5.9 | 71 | 107 | 6 | 92 | 14.1 |
| Aldicarb sulphone | 37 | 50 | 5 | 47 | 11.9 | 62 | 94 | 6 | 73 | 16.2 |
| Aldicarb sulphoxide | 34 | 47 | 5 | 40 | 13.1 | 58 | 79 | 6 | 68 | 11.5 |
| Atrazine | 69 | 81 | 5 | 74 | 6.6 | 77 | 100 | 6 | 84 | 10.3 |
| Azoxystrobin | 71 | 79 | 5 | 76 | 4.2 | 82 | 100 | 6 | 86 | 8.0 |
| Bendiocarb | 79 | 94 | 5 | 84 | 7.7 | 87 | 110 | 6 | 92 | 9.8 |
| Benfuracarb | 56 | 68 | 5 | 63 | 7.3 | 75 | 97 | 6 | 81 | 10.4 |
| Bitertanol | 73 | 77 | 5 | 75 | 2.0 | 80 | 99 | 6 | 85 | 8.3 |
| Boscalid | 65 | 77 | 5 | 70 | 6.6 | 81 | 98 | 6 | 87 | 6.9 |
| Bromuconazole | 74 | 99 | 5 | 86 | 10.6 | 77 | 116 | 6 | 89 | 16.6 |
| Bupirimate | 70 | 80 | 5 | 75 | 6.6 | 76 | 94 | 6 | 85 | 7.3 |
| Carbaryl | 70 | 79 | 5 | 75 | 4.1 | 83 | 109 | 6 | 92 | 10.8 |
| Carbendazim | 70 | 79 | 5 | 74 | 5.6 | 82 | 115 | 6 | 93 | 13.4 |
| Carbofuran | 87 | 95 | 5 | 91 | 3.4 | 93 | 115 | 6 | 100 | 8.1 |
| Carbofuran (3 hydroxy) | 78 | 85 | 5 | 81 | 3.7 | 79 | 111 | 6 | 91 | 12.3 |
| Carbosulfan | 29 | 34 | 5 | 31 | 6.2 | 58 | 65 | 6 | 62 | 3.8 |
| Chlorotoluron | 74 | 88 | 5 | 81 | 6.4 | 75 | 107 | 6 | 87 | 14.3 |
| Clofentizene | 69 | 77 | 5 | 73 | 4.3 | 81 | 101 | 6 | 86 | 9.1 |
| Clothianadin | 61 | 83 | 5 | 74 | 10.9 | 69 | 106 | 6 | 88 | 13.5 |
| Cyazofamid | 76 | 84 | 5 | 80 | 4.6 | 75 | 100 | 6 | 83 | 10.7 |
| Cymoxanil | 67 | 79 | 5 | 74 | 6.5 | 83 | 118 | 6 | 92 | 13.9 |
| Cyproconazole | 66 | 86 | 5 | 75 | 9.9 | 76 | 104 | 6 | 91 | 14.2 |
| Cyprodinil | 67 | 86 | 5 | 78 | 9.3 | 68 | 100 | 6 | 82 | 17.3 |
| Demeton-S-methyl | 33 | 89 | 5 | 63 | 38.2 | 93 | 106 | 6 | 99 | 5.6 |
| Demeton-S-methyl sulphone | 71 | 87 | 5 | 80 | 7.4 | 83 | 112 | 6 | 92 | 11.3 |
| Difenoconazole | 70 | 77 | 5 | 75 | 3.9 | 77 | 95 | 6 | 82 | 8.6 |
| Diflubenzuron | 66 | 83 | 5 | 74 | 11.6 | 75 | 100 | 6 | 84 | 10.2 |
| Dimethoate | 79 | 83 | 5 | 81 | 2.5 | 79 | 105 | 6 | 87 | 10.7 |
| Dimethomorph | 75 | 81 | 5 | 78 | 3.3 | 74 | 110 | 6 | 86 | 15.7 |
| Dimoxystrobin | 55 | 87 | 5 | 74 | 18.2 | 80 | 107 | 6 | 92 | 11.7 |
| Disulfoton | 64 | 77 | 5 | 70 | 7.1 | 66 | 107 | 6 | 83 | 17.7 |
| Disulfoton sulfoxide | 73 | 80 | 5 | 77 | 3.6 | 82 | 106 | 6 | 91 | 9.1 |
| Diuron | 64 | 96 | 5 | 76 | 17.0 | 80 | 109 | 6 | 92 | 11.2 |
| Epoxiconazole | 93 | 106 | 5 | 99 | 4.7 | 81 | 109 | 6 | 89 | 11.9 |
| Famoxadone | 69 | 79 | 5 | 75 | 5.4 | 77 | 93 | 6 | 84 | 6.6 |
| Fenamidone | 69 | 83 | 5 | 74 | 7.6 | 76 | 98 | 6 | 83 | 9.8 |
| Fenarimol | 67 | 88 | 5 | 78 | 12.0 | 81 | 99 | 6 | 87 | 7.7 |
| Fenbuconazole | 64 | 73 | 5 | 68 | 4.9 | 80 | 107 | 6 | 88 | 11.6 |
| Fenhexamid | 72 | 85 | 5 | 77 | 7.9 | 74 | 112 | 6 | 87 | 16.6 |
| Fenpropimorph | 81 | 90 | 5 | 84 | 4.5 | 82 | 116 | 6 | 94 | 13.4 |
| Fenpyroximate | 54 | 69 | 5 | 61 | 9.8 | 72 | 102 | 6 | 82 | 14.1 |
| Fenthion | 67 | 93 | 5 | 75 | 14.2 | 71 | 103 | 6 | 84 | 12.8 |
| Fenthion sulphone | 63 | 79 | 5 | 68 | 10.1 | 81 | 115 | 6 | 94 | 14.6 |
| Fenthion sulphoxide | 73 | 80 | 5 | 77 | 3.5 | 87 | 110 | 6 | 92 | 9.8 |
| Fludioxinil | 64 | 125 | 5 | 89 | 26.4 | 67 | 116 | 6 | 87 | 21.3 |
| Flufenacet | 75 | 83 | 5 | 78 | 5.1 | 79 | 102 | 6 | 86 | 9.8 |
| Fluopicolide | 72 | 85 | 5 | 79 | 6.1 | 79 | 106 | 6 | 86 | 12.0 |
| Fluoxastrobin | 69 | 78 | 5 | 74 | 4.8 | 77 | 90 | 6 | 81 | 5.8 |
| Fluquinconazole | 67 | 91 | 5 | 79 | 11.9 | 84 | 121 | 6 | 109 | 12.3 |
| Flusilazole | 71 | 91 | 5 | 80 | 9.4 | 81 | 106 | 6 | 90 | 10.9 |
| Flutriafol | 66 | 92 | 5 | 77 | 13.8 | 54 | 150 | 6 | 89 | 41.7 |
| Imazalil | 69 | 82 | 5 | 75 | 6.7 | 80 | 98 | 6 | 86 | 7.3 |
| Imidacloprid | 55 | 125 | 5 | 80 | 34.3 | 77 | 111 | 6 | 90 | 12.9 |
| Indoxacarb | 67 | 90 | 5 | 77 | 11.1 | 73 | 92 | 6 | 83 | 9.9 |
| Isofenphos | 30 | 46 | 5 | 38 | 16.9 | 46 | 71 | 5 | 58 | 20.0 |
| Isoproturon | 78 | 88 | 5 | 81 | 5.2 | 71 | 104 | 6 | 84 | 14.5 |
| Kresoxim methyl | 76 | 91 | 5 | 82 | 7.1 | 83 | 109 | 6 | 89 | 11.3 |
| Linuron | 76 | 95 | 5 | 87 | 9.1 | 75 | 87 | 6 | 82 | 7.1 |
| Malathion | 68 | 84 | 5 | 77 | 8.4 | 79 | 96 | 6 | 86 | 6.4 |
| Mepanipyrim | 73 | 87 | 5 | 78 | 6.7 | 73 | 104 | 6 | 85 | 13.2 |
| Metconazole | 73 | 85 | 5 | 79 | 5.4 | 56 | 131 | 6 | 89 | 35.6 |
| Methiocarb | 73 | 81 | 5 | 76 | 4.3 | 81 | 105 | 6 | 88 | 10.2 |
| Methomyl | 111 | 131 | 5 | 123 | 7.1 | 110 | 123 | 6 | 113 | 9.0 |
| Metolcarb | 83 | 115 | 6 | 93 | 13.4 | 78 | 86 | 6 | 81 | 4.1 |
| Metrafenone | 73 | 82 | 5 | 78 | 4.9 | 80 | 100 | 6 | 84 | 9.1 |
| Monocrotophos | 77 | 108 | 6 | 89 | 12.6 | 66 | 87 | 6 | 75 | 11.0 |
| Myclobutanil | 74 | 83 | 5 | 77 | 4.8 | 75 | 112 | 6 | 89 | 15.1 |
| Ofurace | 74 | 79 | 5 | 77 | 2.5 | 84 | 119 | 6 | 96 | 12.8 |
| Omethoate | 38 | 48 | 5 | 44 | 8.4 | 62 | 78 | 6 | 68 | 8.4 |
| Oxamyl | 43 | 57 | 5 | 52 | 10.1 | 65 | 95 | 6 | 76 | 14.7 |
| Oxydemeton methyl | 57 | 73 | 5 | 67 | 9.5 | 74 | 111 | 6 | 88 | 14.6 |
| Penconazole | 60 | 77 | 5 | 71 | 9.6 | 80 | 104 | 6 | 88 | 10.2 |
| Penycycuron | 74 | 87 | 5 | 78 | 7.0 | 72 | 113 | 6 | 86 | 17.1 |
| Phorate | 67 | 80 | 5 | 72 | 7.3 | 75 | 95 | 6 | 83 | 9.0 |
| Phorate sulfone | 72 | 80 | 5 | 76 | 4.7 | 84 | 101 | 6 | 92 | 6.2 |
| Picoxystrobin | 76 | 80 | 5 | 78 | 1.9 | 77 | 104 | 6 | 88 | 11.8 |
| Pirimicarb | 70 | 88 | 5 | 78 | 8.3 | 73 | 110 | 6 | 87 | 15.5 |
| Prochloraz | 72 | 80 | 5 | 75 | 4.3 | 83 | 98 | 6 | 87 | 6.4 |
| Propamocarb | 35 | 40 | 5 | 38 | 5.4 | 76 | 110 | 6 | 89 | 14.3 |
| Propiconazole | 69 | 82 | 5 | 77 | 6.4 | 85 | 104 | 6 | 91 | 7.3 |
| Pymetrozine | 62 | 79 | 5 | 72 | 8.7 | 74 | 108 | 6 | 85 | 15.1 |
| Pyraclostrobin | 74 | 81 | 5 | 77 | 3.6 | 78 | 97 | 6 | 84 | 7.9 |
| Pyrethrins | 82 | 89 | 5 | 86 | 3.5 | 75 | 96 | 6 | 82 | 9.3 |
| Pyrifenox | 76 | 81 | 5 | 78 | 2.7 | 74 | 113 | 6 | 88 | 16.7 |
| Pyrimethanil | 66 | 79 | 5 | 71 | 8.0 | 72 | 109 | 6 | 87 | 16.7 |
| Quinoxyfen | 57 | 84 | 5 | 68 | 16.5 | 74 | 91 | 6 | 82 | 7.2 |
| Simazine | 71 | 86 | 5 | 77 | 7.8 | 74 | 115 | 6 | 90 | 17.1 |
| Spinosad | 43 | 46 | 5 | 45 | 3.0 | 47 | 57 | 6 | 51 | 9.0 |
| Spiromesifen | 79 | 90 | 5 | 84 | 4.8 | 81 | 116 | 6 | 91 | 14.0 |
| Spiroxamine | 73 | 85 | 5 | 77 | 6.1 | 76 | 108 | 6 | 87 | 13.9 |
| Tebuconazole | 72 | 83 | 5 | 77 | 5.7 | 73 | 115 | 6 | 89 | 17.6 |
| Tebufenpyrad | 79 | 98 | 5 | 87 | 8.8 | 77 | 99 | 6 | 85 | 10.4 |
| Tetraconazole | 72 | 75 | 5 | 74 | 1.5 | 66 | 121 | 6 | 85 | 25.3 |
| Thiabendazole | 74 | 82 | 5 | 78 | 5.0 | 85 | 113 | 6 | 94 | 11.5 |
| Thiacloprid | 65 | 76 | 5 | 72 | 6.1 | 80 | 112 | 6 | 90 | 12.7 |
| Thiamethoxam | 70 | 74 | 5 | 72 | 2.1 | 77 | 102 | 6 | 86 | 11.5 |
| Thiodicarb | 54 | 59 | 5 | 56 | 3.7 | 54 | 66 | 6 | 61 | 7.3 |
| Triazamate | 69 | 78 | 5 | 73 | 4.4 | 67 | 94 | 6 | 76 | 12.5 |
| Trichlorfon | 42 | 87 | 5 | 70 | 28.2 | 67 | 141 | 6 | 106 | 34.6 |
| Trifloxystrobin | 72 | 79 | 5 | 76 | 4.3 | 79 | 97 | 6 | 84 | 8.0 |
| Triticonazole | 72 | 85 | 5 | 78 | 6.8 | 60 | 116 | 6 | 86 | 24.5 |
In chicken muscle tissue, target performance criteria were achieved at both fortification levels for 95 compounds. Seven compounds yielded mean recovery or RSD values outwith the target ranges. In chicken liver tissue, target performance criteria were achieved for 85 compounds at both fortification levels. Seventeen compounds yielded mean recovery or RSD values, respectively outwith the target ranges. The magnitude of these failures was such that they were either just below or just above the recovery and/or RSD values set by us. There were a couple of exceptions but even the lowest mean recovery values (30–50%) showed acceptable RSDs. Therefore, these anomalies were not considered to be prohibitive for screening purposes or to present a significant risk of generating false positives or negatives. Consequently, all 102 pesticides listed in Table 1 were incorporated into the method.
LC/MS/MS ion chromatograms of analytes selected from throughout the experimental mass and retention time ranges for the lowest calibration level (LCL) matrix-matched standard (chicken muscle) are shown in Fig. 1. The MRM peaks yielded by the matrix-matched LCL are annotated with retention time (Rt) and peak area (PA) values and serve to illustrate the selectivity and sensitivity of the method. There is an occasional requirement to re-extract and re-analyse samples e.g. in cases where results are disputed. However, this approach is not possible with ‘single-shot’ samples due to finite sample availability. Such samples may have to be retained for corroboration by third parties involved in e.g. enforcement investigations or actions. Consequently, it was important to assess the stability of the selected pesticides in crude extract. This was achieved by simply comparing the analyte peak retention time and peak area measurement following analysis of a LCL matrix-matched (chicken muscle) standard at 0 days and following storage at 5 °C for 7 days. Table 4 shows the results obtained for the 8 analytes presented in Fig. 1. A 46% reduction in the peak area response of carbosulfan between 0 and 7 days was most dramatic. The data were extremely limited from this basic exercise and must be interpreted with caution. However, the study serves to illustrate that quantitation of certain pesticides may be significantly compromised and should be considered if and when a sample extract needs to be re-analysed.
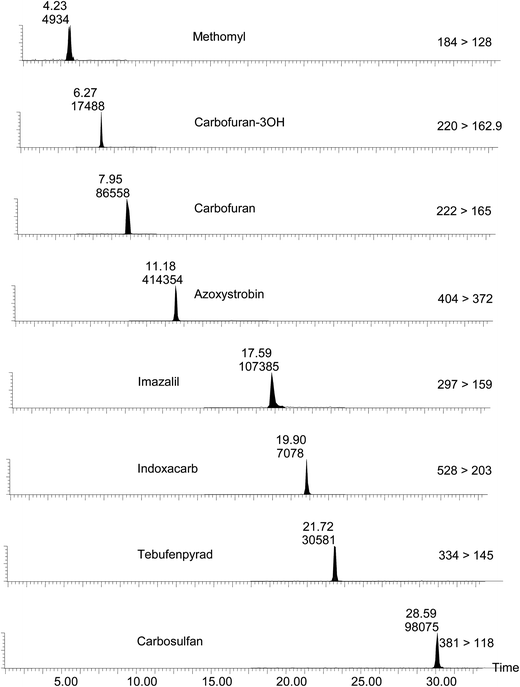 | ||
| Fig. 1 MRM ion chromatograms (gradient separation) selected throughout the experimental mass range and retention time range. Peaks of the LCL (chicken muscle matrix – 0.025 μg ml−1) are annotated with retention time (Rt – minutes) and peak area values. | ||
| Analyte | Rt (min) | Rt (min) | Peak area | Peak area | Ratio |
|---|---|---|---|---|---|
| (0-days) | (7-days) | (0-days) | (7-days) | ||
| Methomyl | 4.23 | 4.19 | 4934 | 6191 | 0.80 |
| Carbofuran – 3OH | 6.27 | 6.27 | 17488 | 25391 | 0.69 |
| Carbofuran | 7.95 | 7.95 | 86558 | 87781 | 0.99 |
| Azoxystrobin | 11.18 | 11.25 | 414354 | 450930 | 0.92 |
| Imazalil | 17.59 | 17.68 | 107385 | 106967 | 1.00 |
| Indoxacarb | 19.90 | 19.99 | 7078 | 6173 | 1.15 |
| Tebufenpyrad | 21.72 | 21.81 | 30581 | 29473 | 1.04 |
| Carbosulfan | 28.59 | 28.68 | 98075 | 45479 | 2.16 |
Application of the method has established the utility of LC/MS/MS for the determination of multiple pesticide residues in investigations into suspected poisoning. Residues ranging from 0.01 to several thousand mg kg−1 (not corrected for recovery) have been detected in specimens from a variety of wild and domestic animals. It is important to ensure that any dilution factors are applied whenever extracts have been diluted to fall within the narrow but linear experimental concentration range. Results obtained indicated that exposure to pesticides was responsible for the death or illness of the animal since other potential causes of death such as disease, trauma or starvation were ruled out following post mortem examination.
A detailed presentation of typical results obtained is shown in Fig. 2. This figure contains the total ion chromatogram (TIC), screen, confirmation and proximate standard MRM chromatograms generated following detection of a residue of carbofuran in the crude extract of liver taken from a golden eagle (Aquila chrysaetos) found dead in the Grampian region of Scotland (March 2011). Carbofuran is the active ingredient of insecticidal products used to control ‘soil dwelling’ and ‘foliar feeding’ insects. Carbofuran (pure chemical), classified by the World Health Organisation as ‘highly hazardous’, is extremely toxic to birds and mammals21 particularly when used incorrectly. Furthermore, products containing carbofuran have not been approved for use in the United Kingdom since December 2001 (although they are approved for use in other countries). Carbofuran (2,3-dihydro-2,2-dimethylbenzofuran-7-yl methylcarbamate) yielded an intense [M + H]+ molecular ion at m/z 222 and a product-ion mass spectrum rich in structural detail, when subject to positive ion mode MS/MS. The MRM transition used for screening (m/z 222 → m/z 165) and transitions used for confirmation (m/z 222 → m/z 165 and m/z 222 → m/z 123) were used in conjunction with gradient and isocratic HPLC separation, respectively. In this particular example, it was necessary to dilute the original extract for the confirmation experiment, as the response from the screening experiment indicated that it was outwith the linear range.
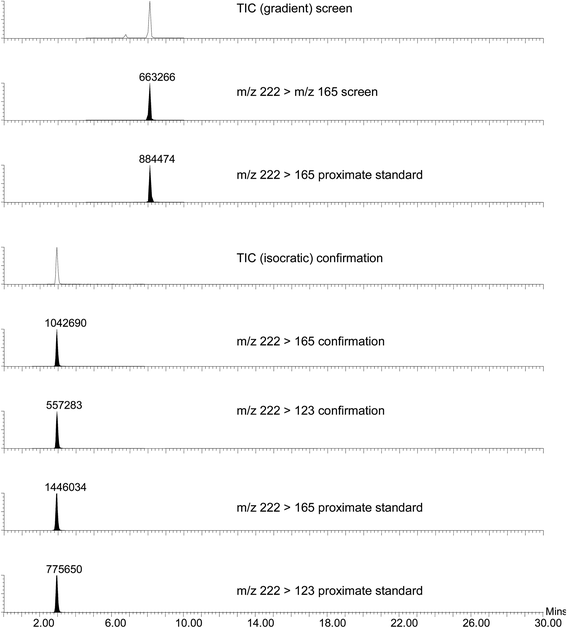 | ||
| Fig. 2 Total ion chromatograms (TIC), screen, confirmation and proximate standard ion chromatograms obtained following analysis of crude extract from the liver of a golden eagle (Aquila chrysaetos) found dead in Grampian, Scotland (March 2011) and confirmed to be a victim of carbofuran poisoning. | ||
It is important to recognise that application of the method can refute or confirm any suspicion that a dead or sick animal has been exposed to any of the pesticides included in the LC/MS/MS target inventory. Consequently, results presented in this paper represent a select and small proportion of the numerous animal species and specimens that have been and continue to be analysed using this method. It would be straightforward to assess the incorporation of additional pesticides, e.g. identified from annual pesticide usage information, into the method.
Conclusions
The chemical diagnosis of suspected animal poisonings or environmental contamination following accidental or deliberate exposure to pesticides has been significantly enhanced following the development and routine application of a relatively simple and quick LC/MS/MS method. It is possible to examine crude extract directly i.e. without laborious sample clean-up, for the presence of multiple pesticide residues due to the high selectivity and sensitivity associated with LC/MS/MS. We have also found the method to be suitable for the analysis of digestive tract content and also blood, vomit and faecal specimens e.g. from sick animals believed to be victims of sub-lethal exposure. In addition, the method is routinely and successfully applied to the characterisation of suspicious chemicals/substances and analysis of suspected baits i.e. carcases, meat or eggs that may have been deliberately laced with toxic pesticides.Ultimately, the results obtained provide robust scientific evidence that confirms or refutes any suspicion that non-target vertebrate animals have been exposed to any of the pesticides included following their approved use, misuse, abuse or due to the persistence of some of these chemicals in the environment. The evidence collected can be used to initiate investigations into how and why the exposure occurred and to identify any violation of legislation designed to protect animals and the environment.
References
- The Wildlife Incident Investigation Scheme (Scotland). http://www.sasa.gov.uk/pesticide_wildlife/wiis/index.cfm.
- M. J. Taylor, E. A. Sharp, L. M. Melton and J. E. Watson, Pesticide Poisoning of Animals in 2010: A Report of Investigations in Scotland, Scottish Government, Edinburgh, Scotland, 2011 Search PubMed.
- Food and Environment Protection Act 1985, http://www.opsi.gov.uk/RevisedStatutes/Acts/ukpga/1985/cukpga_19850048_en_1.
- The Wildlife and Countryside Act 1981 (Amendment) (Scotland) Regulations 2001, Scottish Statutory Instrument 2001 No. 337, http://www.opsi.gov.uk/legislation/scotland/ssi2001/20010337.htm.
- Possession of Pesticides (Scotland) Order 2005, Scottish Statutory Instrument 2005 No. 66, http://www.opsi.gov.uk/legislation/scotland/ssi2005/20050066.htm.
- G. F. Pang, Y. Z. Cao, C. L. Fan, G. Q. Jia, J. J. Zhang, X. M. Li, Y. M. Liu, Y. Q. Shi and Z. Y. Li, J. AOAC Int., 2009, 92(3), 933–940 CAS.
- H. G. Mol, P. Blaza-Bolanas, P. Zomer, T. C. de Rijk, A. A. Stolker and P. P. Mulder, Anal. Chem., 2008, 80(24), 9450–9459 CrossRef CAS.
- A. R. Fernandez-Alba and J. F. Garcia-Reyes, TrAC, Trends Anal. Chem., 2008, 27(11), 973–990 CrossRef CAS.
- P. M. Brown, A. Charlton, M. Cuthbert, L. Barnett, L. Ross, M. Green, L. Gillies, K. Shaw and M. Fletcher, J. Chromatogr., A, 1996, 754, 463–478 CrossRef CAS.
- P. M. Brown, G. Turnbull, S. Charman, A. J. A. Charlton and A. Jones, J. AOAC Int., 2005, 88(1), 204–220 CAS.
- K. Hunter, M. J. Taylor, E. A. Sharp, L. M. Melton and S. LeBouhellec, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 805, 303–309 CrossRef CAS.
- Y. Wang, P. Kruzik, A. Helsberg, I. Helsberg and R. Wolf-Dieter, Forensic Sci. Intl., 2006, 169(2–3), 157–160 Search PubMed.
- L. Novotný, J. Misík, A. Honzlová, P. Ondráček, K. Kuča, O. Vávra, V. Rachač and P. Chloupek, J. Appl. Biomed., 2011, 9, 157–161 CrossRef.
- T. Pihlström, Anal. Bioanal. Chem., 2007, 389, 1773–1789 CrossRef.
- S. J. Lehotay, J. AOAC Int., 2007, 90(2), 485–520 CAS.
- European Union CD 2002/657/EC.
- Making Sure Our Food is Safe: Measuring, Monitoring and Assessing Residues, http://www.pesticides.gov.uk/guidance/industries/pesticides/topics/food-safety/pesticide-residues/keeping-our-food-safe-measuring-monitoring-assessing-residues.
- Pesticide Usage Surveys in Scotland, http://www.sasa.gov.uk/pesticides/pesticide-usage.
- WIIS–UK Quarterly Reports, http://www.pesticides.gov.uk/guidance/industries/pesticides/topics/reducing-environmental-impact/wildlife/WIIS-Quarterly-Reports-2010-2011.
- Method Validation and Quality Control Procedures for Pesticide Residues in Food and Feed, Document No. SANCO/10684/2009, http://www.google.co.uk/url?q=http://ec.europa.eu/food/plant/protection/resources/qualcontrol_en.pdf&sa=U&ei=3sGWT5e4EMHd8AOoh4GdCg&ved=0CBQQFjAA&usg=AFQjCNE8O54RRhFteszoG37cXNu8QQGVAA.
- Carbofuran and Wildlife Poisoning: Global Perspectives and Forensic Approaches, ed. N. Richards, John Wiley and Sons, New York, 2012 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |