 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measuring and modelling cell-to-cell variation in uptake of gold nanoparticles†
J. Charles. G. Jeynes*, Christopher Jeynes, Michael J. Merchant and Karen J. Kirkby
Ion Beam Centre, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: j.c.jeynes@surrey.ac.uk
First published on 26th September 2013
Abstract
The cell-to-cell variation of gold nanoparticle (GNP) uptake is important for therapeutic applications. We directly counted the GNPs in hundreds of individual cells, and showed that the large variation from cell-to-cell could be directly modelled by assuming log-normal distributions of both cell mass and GNP rate of uptake. This was true for GNPs non-specifically bound to fetal bovine serum or conjugated to a cell penetrating peptide. Within a population of cells, GNP content varied naturally by a factor greater than 10 between individual cells.
Gold nanoparticles (GNP) have been studied intensively for their possible application in medicine; either as drug delivery vehicles or by using their physical properties as photothermal or radiosensitisation agents in cancer therapy.1 Research has focused on many factors relating to cell uptake of GNPs including using various peptides or ligands to increase uptake, the effects of size and shape, and how these different factors affect kinetic parameters.2 It is accepted that the surface layer of GNPs is of utmost importance to cell uptake, with various proteins or peptides increasing uptake many times. Measuring the gold in cells with commonly used analytical techniques such as inductively coupled plasma mass spectrometry (ICP-MS) or atomic absorption spectrometry (AAS)2 average the mass of gold in thousands of cells. Other techniques like flow cytometry,3 X-ray microscopy,4,5 and electron microscopy6 have also all been used to investigate GNP uptake, but none of these methods can directly do a full mass analysis of individual cells. Particle Induced X-ray Emission (PIXE) on its own has been used recently to weigh and analyse cells incubated with GNPs, but at reduced accuracy since cell thickness is ignored,7 and the same comment applies to X-ray fluorescence8 (XRF). However it is important to quantify the cell-to-cell variation and distribution in GNP uptake, if GNPs are to be used as therapeutics. This is because the dose of a drug attached to a nanoparticle will be directly related to how many nanoparticles (NP) enter the cell. The same applies if the GNPs themselves are used in photothermal or X-ray enhancement therapy. It is also interesting to relate the mass of the cell to how many GNPs are in it, as this can help explain the nature of uptake on a cell-by-cell basis. Statistical aspects of quantum dot uptake have been explored by Summers et al. using flow cytometry; they showed that the entry of NPs into cells is dominated by the Poisson distribution.9 Related work shows that cell-to-cell variability in virus particle uptake is due to factors including receptor molecule distributions, cell size and local cell density.10
So far, the GNP content along with the corresponding mass of individual cells has not been reported quantitatively. Here, we use spectrometric methods (ion beam analysis, IBA11) to count the various atomic species per cell: thus, each measured cell is effectively weighed. The great advantage of IBA (PIXE + Elastic Backscattering Spectrometry; EBS) is that depth information is available directly from the EBS signal, unlike XRF or PIXE alone, making accurate quantification directly available. With a 2.5 MeV scanned proton microprobe and a spot size of 2 μm, the cells are imaged using the phosphorus signals from the PIXE signal (see Fig. 1), and measured quantitatively using the EBS signal (see Fig. S1b† for comparable data at lower accuracy from PIXE). Nuclear microprobes have a long history of elemental mapping of biological tissues and cells on a micron scale12–14 with high sensitivity and accurate quantification available when PIXE and EBS are used self-consistently.15
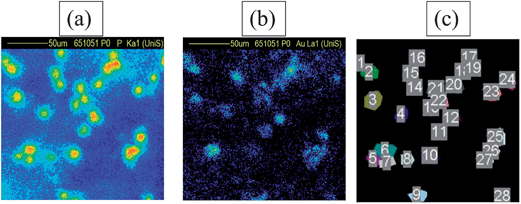 | ||
| Fig. 1 Elemental PIXE maps of cells incubated with GNPs (50 nm + TAT) in a 0.04 mm2 scanned region. (a) Phosphorus signal from cells (b) corresponding gold signal from the same cells (c) “Masks” were created over the cells so that individual spectra could be reconstructed off-line for each cell (in most cases only the mask labels can be seen in (c)). In this particular region, there were 28 cells, giving 28 individual EBS spectra for analysis. | ||
This work investigates the uptake of GNPs by RT112 cancer cells, where the GNPs were coated with Fetal Bovine Serum (FBS) or with a transactivator of transcription (TAT) peptide before incubation with the cells. It is important to first conjugate the TAT peptide to the GNPs because the media which the cells require for healthy growth also contains FBS. The conjugated TAT effectively screens the FBS in the media from non-specifically binding to the GNPs.2 The cells were incubated with the coated GNPs for more than two doubling times (48 hours) to ensure that measurements were well averaged over the cell-cycle and that the effect of the rapid initial GNP uptake was marginalised.2 It must be noted that this is not a realistic scenario in vivo as nanoparticles have a typical half life of a few hours in the bloodstream. TAT is a peptide sequence isolated from the HIV virus which is thought to translocate the virus into the cell nucleus.16 TAT was chosen as the conjugation peptide as it has been shown to increase NP uptake into cells.17 FBS is comprised of essential proteins needed for growth and is known to bind non-specifically to GNPs in solution, aiding uptake.2 The conjugation of GNPs to TAT is described in the ESI,† following methods developed by Patel et al.18 Dynamic light scattering measurements shows that the size of the GNP increases by about 20 nm with addition of TAT peptide, independent on the initial core size of the GNP, and the Zeta potential becomes less negative by about 10 mV.18 For non-specific binding of fetal bovine serum to GNPs see Chithrani et al.,2 while Tsai et al. shows that serum increases the initial core size of a GNP by about 10 nm.19
RT112 cells (a cancerous human urinary bladder cell line: doubling time 20 h) was used for these experiments, as previous experience has shown good uptake of GNPs. Briefly, 1 × 105 cells were seeded in 35 mm diameter Petri dishes and incubated with GNPs of either 30 nm + FBS, 50 nm + FBS, 30 nm + TAT and 50 nm + TAT for 48 hours in RPMI media (which contained 1 mM L-glutamine and 10% FBS essential for cell growth; see ESI† methods). For analysis preparation, the cells were washed with PBS, trypsinised so that they were in suspension, fixed with paraformaldehyde, washed with PBS, resuspended in deionised water and finally pipetted onto a polypropylene film 4 μm thick at a concentration of 5 × 103 cells per mm2. After air drying, the samples were placed in a vacuum chamber, where a 2.5 MeV scanning proton microbeam of about 300 pA with a spot size of about 2 μm was delivered from a 2 MV tandem accelerator.20 At this energy, the protons can traverse about 150 μm in water, so easily penetrate the cell with small energy loss. 200 × 200 μm regions of the samples were probed, each region containing 30–100 cells, with data collection (scanning) times of up to 10 hours per region. A list of the energies of all detected photons or scattered particles is kept with the coordinates of their origin so that spectra from arbitrary areas can be reconstructed off-line. Further details are in the ESI.†
To investigate the GNP uptake at an individual cell level, phosphorus and gold elemental PIXE images were obtained of the region scanned by the beam. Fig. 1 shows an example of these images. A high phosphorus signal shows where a cell is, and the corresponding region on the gold map indicates how much gold is in each cell. From the PIXE image the position of each cell is identified, from which the EBS spectra (and PIXE spectra too, although these are not used here) for that cell are reconstructed off-line. Fig. 1c shows the selected regions identifying all the cells in a scan. See the ESI† for more details.
The EBS spectra of a total of 332 individual cells, with at least 55 cells for each condition were analysed, counting the number of each atomic species in each cell, including the light elements C, N, O, P & S as well as Au. Since we know the size of the GNPs, this is equivalent to counting the GNPs per cell. PIXE is used to image the cells and isolate their spectra, with OMDAQ2007 software21 for imaging, and the DataFurnace (NDF) code22 for interpreting the EBS spectra. NDFv9.4h23 was used to accurately fit the EBS spectra from individual cells and quantify by mass (in picograms) the elements within each cell. Briefly, each EBS spectrum is accurately fitted using the proper non-Rutherford scattering cross-sections24 and taking account of the inhomogeneity of the cell thickness, allowing the polypropylene foil to be unambiguously identified. Quantification is through the composition of the polypropylene foil, and the spectrometric determination of proton energy loss in the foil, where the energy loss function is known at around 4%.25 The ESI† shows IBA spectra with further details.
Firstly, it is interesting to directly compare the mean, median and spread of the mass of gold in the TAT-coated and FBS-coated GNPs and how this relates to the number of GNPs in the cells. Fig. 2 shows that the mass of gold in the average (mean) cell incubated with TAT-coated GNPs is about six times that of those incubated with FBS-coated GNPs, whereas the difference in mass uptake between the sizes (30 nm and 50 nm) is not significant. Note, the sizes here refer the initial core sizes of the GNPs as given by the manufacturer, and not the final size of the GNP-conjugate, which can be tens of nanometers larger when conjugated.18,19 When the mass of gold is converted into numbers of GNPs it is clear that there are many more “30” nm GNPs compared to “50” nm GNPs imported, with this difference accentuated in the TAT-coated samples. In other words, the number of GNPs which have a core of 30 nm are imported to a greater extent than those which have a core of 50 nm, regardless of whether they are coated by TAT or FBS.
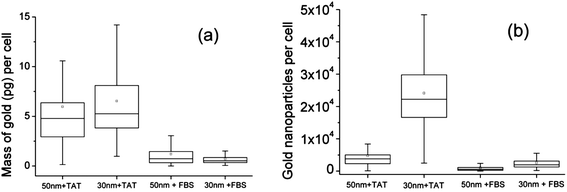 | ||
| Fig. 2 (a) Box plots of cell uptake of gold (pg) depending on TAT-coated or FBS-coated 30 nm or 50 nm. The boxes are the 25th & 75th percentiles and medians, the whiskers are the 5th and 95th percentiles and the squares are the means. (b) Comparison of the mean number of GNPs per cell, after converting the mass of gold into numbers of GNPs. In both graphs, the means for the TAT-coated GNPs are significantly different from the FBS-coated GNPs using ANOVA (P < 0.05). | ||
To explain why GNP-TAT is imported to a greater extent than GNP-FBS, regardless of the initial core size of the nanoparticle, it is important to understand the mechanism by which the GNPs enter the cells. Chithrani et al.2 investigated how GNP-conjugates are imported, and concluded that there is an optimum binding ratio between GNPs-conjugates and endocytosis receptors; some GNP-conjugates are imported to a greater extent than others, simply because there are more receptors on the cell surface for that particular conjugate molecule.
To explain why one initial core size of GNP is imported to a greater extent than another; Chithrani found that a certain “activation energy” must be overcome before a lysosome is formed to invaginate and engulf gold nanoparticles. GNPs-conjugates can be too small or large for optimum peturbation of the cell membrane, lysosome formation and invagination. These parameters have also been mathematically modelled.26 Although from the results shown in the present paper it cannot be shown exactly what is the optimum size of uptake, it is clear that some sizes are imported better than others and this is dependent on the type of molecule the nanoparticle is conjugated to.
Chithrani et al.2 also measured the kinetics of GNP uptake hour by hour and found after an initially high uptake, the rate plateaus after about 5.5 hours, presumably when receptor saturation is reached. We found that a Hill equation27 fitted their data very well (see Fig. S6a†) and we used this equation to also fit our data (see Fig. S6b†). Thus we are able to define the rate of uptake of each of our four conditions (30 nm + FBS, 30 nm + TAT, 50 nm + FBS, 50 nm + TAT). We have used this as one of the parameters in a Monte Carlo simulation of individual cells importing GNPs over time (see below).
As shown in Fig. 2, large variations of gold content are observed for each tested condition highlighting a wide cell-to-cell variability in the GNP uptake. This is also easily observable in Fig. 3 which shows gold mass as a function of cell mass, with both gold and carbon measured absolutely in picograms. Interestingly, there is a large scatter in both the mass of the cell and the gold content in each cell, which perhaps is to be expected with asynchronous growing cells. To make sense of this scatter, we modelled the data with parameters drawn from the literature, and from our own data. Firstly, as mentioned above, we fitted Chithrani et al.'s data with a Hill equation to explain the uptake rate of GNPs over time (see ESI† for more on this fit).
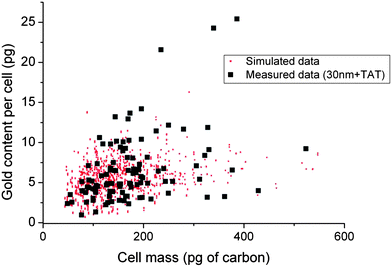 | ||
| Fig. 3 GNP content per cell compared with Monte Carlo simulation. Points are simulated using Hill model parameters for both cell size and uptake rates fitted from the data (see Table S4 and discussion in the ESI†). The three outlier data points with >20 pg GNP per cell are probably superposed cells. | ||
We then simulated uptake of GNPs in virtual “cells” over 48 hours using two parameters; a log-normally distributed GNP uptake rate with a sigma of 0.2, and a log-normally distributed cell size with a sigma of 0.3. This functional form of the latter is demonstrated by Bettega et al.28 from diameter measurements of populations of cells. The cells divide at 20 hours, binomially distributing the GNPs between daughters with a mean of 0.7, according to Summers et al.9 The most probable value of a cell's “birth” mass is modelled from our data as 100 pg, a value corroborated by Son et al.29 The Monte Carlo simulation produces data points which fit the measured data remarkably well, as Fig. 3 shows. Overall, Fig. 3 demonstrates that the uptake of GNPs on a cell by cell basis, can be modelled using simple parameters.
To further investigate this cell-to-cell variability in the gold content of cells, a histogram of the numbers of nanoparticles in each cell treated with TAT-coated 30 nm GNPs can be seen in Fig. 4a. Here the most probable number of GNPs per cell is 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, but the range is very large at between 250 and 50000 GNPs per cell. The data are fitted with a log-normal distribution giving an adjusted R2 value of 0.94 (indicating a good fit between the model and the data: fitting parameters are shown with discussion in Table S3†). Fig. 4a shows that a few cells have 2000 or less GNPs in them while the majority of cells have 20000 GNPs or more. A small minority of cells can have more than double the mode number of GNPs. This is an important finding to factor into dosimetry calculations when using GNPs therapeutically, either as drug vectors or in radiotherapy as some cells could receive much more or much less dose than the average cell.
000, but the range is very large at between 250 and 50000 GNPs per cell. The data are fitted with a log-normal distribution giving an adjusted R2 value of 0.94 (indicating a good fit between the model and the data: fitting parameters are shown with discussion in Table S3†). Fig. 4a shows that a few cells have 2000 or less GNPs in them while the majority of cells have 20000 GNPs or more. A small minority of cells can have more than double the mode number of GNPs. This is an important finding to factor into dosimetry calculations when using GNPs therapeutically, either as drug vectors or in radiotherapy as some cells could receive much more or much less dose than the average cell.
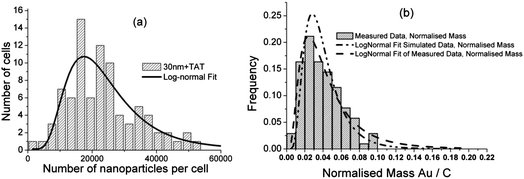 | ||
| Fig. 4 (a) Histogram of the number of 30 nm + TAT gold nanoparticles per cell. There is considerable variation from cell to cell with a mode of about 20000 per cell. The data are fitted with a log-normal distribution. (b) Data of (a) replotted as normalised mass: the ratio of the mass of gold in the cell and its size (as the mass of carbon). This also follows a log-normal distribution showing that cells of the same size can have large differences in GNP uptake rate. Simulated distributions are also log-normal with parameters in agreement from the measured data (see text: details of all fits are shown in Table S3†). | ||
To answer the question as to whether the heaviest cells also have the highest density of GNPs (that is, whether the uptake rate is correlated with growth rate), Fig. 4b shows the distribution of gold in cells, where the plot is normalised to the size of the cell. This is achieved by dividing the mass of gold in a cell by the mass of that cell. The frequency histogram is displayed for the 30 nm + TAT data, and fitted with a log-normal distribution (fitting parameters shown in Table S4†). It shows that some cells import GNPs up to ten times faster than the mean rate, independent of the cell size. Hence, two cells that are exactly the same size can have greatly differing GNP uptake rates. Interestingly, these results are consistent with the log-normal distribution of the number of receptors (e.g. VEGF) found recently on individual cells within a population by Imoukhuede & Popel.30 When the GNP-uptake of the cells is modelled (see below), we find that the log-normal fit to the model is in agreement with the observed data. Of note, when a histogram was made at earlier timepoints below 5 hours using data generated by the simulation, similar log-normal distributions were found to the 48 hours timepoint (data not shown). Hence it is likely that a heterogenous gold content in the cell population is reached remarkably quickly, making this more relevant to in vivo situations. Blood concentration curves in rats showed that after 4 hours, 1 day and 7 days, 317 μg g−1, 29 μg g−1, and 0.02 μg g−1 gold nanoshells remain respectively, after intravenous injection with 10 μg g−1 (for a review of this topic see Khlebtsov and Dykman31).
Overall, Fig. 4 shows the actual cell-to-cell variation in both the number of GNPs per cell and, independently, in the GNP density of the cells. Heavier (older) cells have more GNPs, but cells with a higher initial uptake rate have a higher GNP density. We have modelled both of these effects, but the most interesting one is that the GNP uptake rate is log-normally distributed and has model parameters comparable to those obtained by fitting Chithrani et al.'s data2 (see ESI for more details and also Fig. S4 & S5† for comparable data for the FBS and TAT coated 50 nm and 30 nm GNPs).
It is plausible that the distribution in uptake of GNPs can be explained by a log-normally distributed number of receptors on cell surfaces. By chance, a single cell could have up to ten times as many receptors as its same-sized sibling, and so will import many more GNPs. Indeed, much work has investigated cell-to-cell variability and the consequences for pharmacology (for a review see Niepal et al.32). Such log-normally distributed levels of proteome expression from cell-to-cell are likely to be an important but poorly appreciated factor in drug responsiveness.33 For instance, drug-resistant cancer cells, rather than being a static genetically distinct subfraction, may represent population outliers resulting from proteomic heterogeneity. There may be various origins of this variability, including molecule expression, local cell density or cell size.10 A good example is cell-to-cell variability of SV40 virus infection, where the probability of infection can be largely attributed to nucleus size but where local cell density can also make a contribution.33
In conclusion, we have for the first time directly measured the GNP uptake per cell together with the mass of each cell in asynchronous populations under different conditions, and demonstrated a log-normal distribution of both the GNP uptake and the cell size. We have interpreted these data by Monte Carlo simulation on the Hill model, which is the simplest relevant model available. Moreover we have shown by modelling, that the parameters extracted from our data are consistent with the literature. We discuss the pervasiveness of the log-normal distribution in biological processes in terms of the underlying biological mechanisms. We have also shown that the conjugation of a TAT-peptide to a gold nanoparticle increases the uptake by about six times in comparison to GNP-FBS complexes. Here it is assumed that the prevalence and binding affinity of TAT-receptors on the cell membrane surface to TAT molecules is much greater then those of FBS-receptors to FBS molecules, and overall governs the uptake rate of the GNPs. We conclude that when drug dosimetry calculations are made using GNPs as delivery vectors, care should be taken to include the effects of cell-to cell variability of GNP uptake.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Notes
The authors declare no competing financial interests.Acknowledgements
JCGJ thanks Brian Jones for helpful advice and Anne-Catherine Wéra for editing the manuscript. We thank John Peacock (Kingston University, U.K.) for giving us the RT112 cell line. This work has been supported by the European Community as an Integrating Activity ≪Support of Public and Industrial Research Using Ion Beam Technology (SPIRIT)≫ under EC contract no. 227012 and by the UK EPSRC under grant EP/C009592/1.References
- P. Juzenas, W. Chen, Y.-P. Sun, M. A. N. Coelho, R. Genera-lov, N. Generalova and I. L. Christensen, Adv. Drug Delivery Rev., 2008, 60, 1600–1614 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- J. A. Kim, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62–68 CrossRef CAS PubMed.
- X. Cai, H.-H. Chen, C.-L. Wang, S.-T. Chen, S.-F. Lai, C.-C. Chien, Y.-Y. Chen, I. M. Kempson, Y. Hwu, C. S. Yang and G. Margaritondo, Anal. Bioanal. Chem., 2011, 40, 1809–1816 Search PubMed.
- H.-H. Chen, C.-C. Chien, C. Petibois, C.-L. Wang, Y. S. Chu, S.-F. Lai, T.-E. Hua, Y.-Y. Chen, X. Cai, I. M. Kempson, Y. Hwu and G. Margaritondo, J. Nanobiotechnol., 2011, 9, 14–20 CrossRef CAS PubMed.
- Y. Hao, X. Yang, S. Song, M. Huang, C. He, M. Cui and J. Chen, Nanotechnology, 2012, 3, 045103 CrossRef PubMed.
- N. Ogrinc, P. Pelicon, P. Vavpetič, M. Kelemen, N. Grlj, L. Jeromel, S. Tomić, M. Čolić and A. Beran, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 306, 121–124 CrossRef CAS.
- S. Corezzi, L. Urbanelli, P. Cloetens, C. Emiliani, L. Helfen, S. Bohic, F. Elisei and D. Fioretto, Anal. Biochem., 2009, 388, 33–39 CrossRef CAS PubMed.
- H. D. Summers, P. Rees, M. D. Holton, M. R. Brown, S. C. Chappell, P. J. Smith and R. J. Errington, Nat. Nanotechnol., 2011, 6, 170–174 CrossRef CAS PubMed.
- B. Snijder and L. Pelkmans, Nat. Rev. Mol. Cell Biol., 2011, 12, 119–125 CrossRef CAS PubMed.
- C. Jeynes, R. P. Webb and A. Lohstroh, Rev. Accel. Sci. Technol., 2011, 4, 41–82 CrossRef.
- J. P. Landsberg, B. McDonald and F. Watt, Nature, 1992, 360, 65–68 CrossRef CAS PubMed.
- V. Valkovic, D. Miljanic, R. M. Wheeler, R. B. Liebert, T. Zabel and G. C. Phillips, Nature, 1973, 243, 543–544 CrossRef CAS PubMed.
- J. C. G. Jeynes, M. J. Bailey, H. Coley, K. J. Kirkby and C. Jeynes, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 2168–2171 CrossRef CAS.
- E. F. Garman and G. W. Grime, Prog. Biophys. Mol. Biol., 2005, 89(173), 205 Search PubMed.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- G. Ruan, A. Agrawal, A. I. Marcus and S. Nie, J. Am. Chem. Soc., 2007, 129, 14759–14766 CrossRef CAS PubMed.
- C. P. Patel, D. A. Giljohann, D. S. Seferos and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17222–17226 CrossRef PubMed.
- D.-H. Tsai, F. W. DelRio, A. M. Keene, K. M. Tyner, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2011, 27, 2464–2477 CrossRef CAS PubMed.
- A. Simon, C. Jeynes, R. P. Webb and R. M. Gwilliam, et al., Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 219, 405–409 CrossRef.
- G. W. Grime and M. Dawson, Nucl. Instrum. Methods Phys. Res., Sect. B, 1995, 104, 107–113 CrossRef CAS , http://www.microbeams.co.uk/download.html.
- N. P. Barradas, C. Jeynes and R. P. Webb, Appl. Phys. Lett., 1997, 71, 291–293 CrossRef CAS.
- N. P. Barradas and C. Jeynes, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 1875–1879 CrossRef CAS , http://www.surreyibc.ac.uk/ndf.
- A. F. Gurbich, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1703 CrossRef CAS.
- Ziegler, F. James, M. D. Ziegler and J. P. Biersack, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1818–1823 CrossRef CAS , http://www.srim.org.
- A. Chaudhuri, G. Battaglia and R. Golestanian, Phys. Biol., 2011, 8, 046002 CrossRef PubMed.
- S. Goutelle, M. Maurin, F. Rougier, X. Barbaut, L. Bourguignon, M. Ducher and P. Maire, Fundam. Clin. Pharmacol., 2008, 633–648 CrossRef CAS PubMed.
- D. Bettaga, P. Calzolari, S. M. Doglia, L. Tallone and A. M. Villa, Int. J. Radiat. Biol., 1998, 74, 397–403 CrossRef.
- S. Son, A. Tzur, Y. weng, P. Jorgensen, J. Kim, M. W. Kirschner and S. R. Manalis, Nat. Methods, 2012, 9, 910–912 CrossRef CAS PubMed.
- P. I. Imoukhuede and A. S. Popel, Exp. Cell Res., 2011, 17, 955 CrossRef PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2010, 40, 1647–1671 RSC.
- M. Niepel, S. L. Spencer and P. K. Sorger, Curr. Opin. Chem. Biol., 2009, 13, 556–561 CrossRef CAS PubMed.
- B. Snijder, R. Sacher, P. Ramo, E.-M. Damm, P. Liberali and L. Pelkmans, Nature, 2011, 461, 520–523 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3an01406a |
| This journal is © The Royal Society of Chemistry 2013 |