 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extruded polymer films pigmented with a heterogeneous ion-pair based lumophore for O2 sensing
Andrew Mills* and Ashleigh Graham
School of Chemistry and Chemical Engineering, Queen's University, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: andrew.mills@qub.ac.uk; Fax: +44 (0)28 9097 6524; Tel: +44 (0)28 9097 4339
First published on 5th September 2013
Abstract
A novel approach to polymeric Ru(II)–diimine luminescent O2 sensors is described. The Ru(II)–diimine, tris(4,7-diphenyl-1,10-phenanthroline)ruthenium(II) dichloride ([Ru(dpp)3]2+), is first ion-paired to the surface of heterogeneous TiO2 particles, rendered negatively charged due to the alkali nature of the aqueous solution, to produce an O2 sensitive pigment with a strikingly high oxygen sensitivity (i.e. PO2 (S = 1/2) = 0.002 atm, where PO2 (S = 1/2) is defined as the amount of oxygen required to reduce the initial, oxygen free luminescence by 50%), and a rapid response to oxygen. The pigment is extruded in low density polyethylene (LDPE) to produce a thin (60 μm), flexible, O2 sensing plastic film, with an O2 sensitivity (PO2 (S = 1/2) = 0.84 atm) comparable to the more traditional homogeneous lumophore ion-pair based O2 sensor ink films reported in the literature.
1. Introduction
The detection and quantification of oxygen is of great importance in many industries and practical applications, not least environmental monitoring, medical applications, and more recently, food packaging.1–3 Research into O2 detection has increasingly moved away from the more traditional Clark electrode4 and GC analysis, which is limited by its expense and bulkiness, towards optical detection. Most optical sensors for oxygen are based on luminescence quenching, and within the area of such luminescence-based O2 sensors, two main dye groups have been studied as the O2 quenchable lumophores, namely: platinum and palladium metal porphyrins, and ruthenium α-diimines. The latter are favoured because of their large Stokes shift, high quantum yield of luminescence, and high photostability,5 although the former have generally higher sensitivities (especially the Pd porphyrins).6 These complexes are quenched by molecular oxygen and so, upon exposure to O2 the lifetime and luminescence intensity of the lumophore decreases, i.e.| D + hv →D* | (1a) |
| D* + O2 →D + O*2 | (1b) |
In homogeneous media, such as aqueous solution, this quenching is found to obey the linear Stern–Volmer equation, eqn (2), where I0 (and τ0) and I (and τ) are the luminescence intensities (and lifetimes) in the absence and presence of oxygen, respectively, and KSV is the Stern–Volmer constant.
| I0/I = τ0/τ − (1 = KSV[O2]) | (2) |
Usually, when these Ru(II)–diimine complexes are used in O2 sensors, they are immobilized as a homogeneous ion-pair with a lipophilic anion, such as tetraphenyl borate, in a hydrophobic encapsulating medium, such as a polymer.7 More often than not, upon encapsulation, the quenching behaviour of these complexes no longer obeys the linear Stern–Volmer relationship, but instead exhibits a negative deviation from linearity and a new kinetic model is required to fit the quenching data. Probably the most commonly accepted and widely used model to fit such data is the two-site model,8,9 reported by Demas and co-workers, in which it is assumed that the lumophore exists in two different microdomains within the encapsulating medium, with each microdomain associated with a different quenching response by the lumophore contained therein. The modified Stern–Volmer equation for this system is given in eqn (3), where f01 and f02 are the fractions of the dye molecules in the two different microdomains, the sum of which is unity, and KSV1 and KSV2 are the Stern–Volmer constants of the lumophore in these two microdomains. Although the model is based on two different O2-quenching microdomains, it is recognised as a likely gross oversimplification of the true nature of the system.8 However, eqn (3) does provide a good fit to most data sets generated by lumophores in polymer based O2 sensors.
| I/I0 = f01/(1 + KSV1[O2]) + f02/(1 + KSV2[O2]) | (3) |
One of the earliest O2 sensors based on [Ru(dpp)3]2+, was reported by Wolfbeis et al., in 1986,10 and incorporates the lumophore into silica gel beads (5 microns diameter with 30 nm pores), which were then dried, homogenously mixed with a silicone prepolymer, cured for 12 h at 40 °C, and the volume inside the silica beads – now in a cured silicone membrane – then filled with water by dipping the sensor membrane in boiled water. It is possible in this work that the silica initially binds the [Ru(dpp)3]2+ electrostatically, although the authors note the silica has ‘a low ion-exchanging capacity’ and the need for a film hydration step and to store the film in an aqueous or moist environment, suggests that the dye might be instead simply adsorbed as the dichloride salt onto the internal surfaces of the silica beads. Whatever the binding, these films suffer problems of low luminescence and a tendency for the lumophore to undergo self-quenching of luminescence when its concentration exceeds a critical value.11
Given these difficulties, other means of encapsulating [Ru(dpp)3]2+ into a lipophilic polymer, such as silicone, were investigated subsequently, and now most O2 sensors utilise this lumophore in a form in which it is electrostatically bound as an ion-pair to a lipophilic anion, such as perchlorate, dodecyl sulphate (DS−), tetramethylsilypropansulfonate (TSPS−) or tetraphenyl borate (Ph4B−). These are homogeneous ion-pair lumophores, many examples of which are given in Table 1 (ref. 7 and 10–16) and, not surprisingly, these ion-pair, lipophilic lumophores are a major feature of most commercial O2 optical sensors (e.g. OxySense17) and pressure sensitive paints.18
| Ion-pair | Encapsulating polymer | f01 | KSV1 (atm−1) | KSV2 (atm−1) | PO2 (S = 1/2) (atm) | Ref. |
|---|---|---|---|---|---|---|
| [Ru(dpp)3Cl2] in silica beads | Silicone, E43 | 0.83 | 9.13 | 0.17 | 0.16 | 10 |
| [Ru(dpp)3ClO4)2] | Silicone, RTV-118 | 0.88 | 53.65 | 4.67 | 0.02 | 12 |
| [Ru(dpp)3(DS)2] | Silicone, E4 | 0.35 | 11.55 | 0.55 | 0.73 | 11 |
| [Ru(dpp)3(TSPS)2] | Silicone, E4 | 0.28 | 15.69 | 0.60 | 0.83 | 11 |
| [Ru(dpp)3(Ph4B)2] | Cellulose acetate | 0.7 | 20.44 | 1.76 | 0.09 | 13 |
| [Ru(dpp)3(ClO4)2] | Silicone, RTV-118 | 0.98 | 29.25 | 1.22 | 0.04 | 14 |
| [Ru(dpp)3(ClO)4)2] | Polystyrene | 0.88 | 2.02 | 0.05 | 0.64 | 15 |
| [Ru(dpp)3(ClO)4)2] | PVC | 0.54 | 8.66 | 3.29 | 0.18 | 16 |
| [Ru(dpp)3(Ph4B)2] | PMMA | 0.98 | 23.53 | 0.001 | 0.04 | 7 |
Two of the examples in Table 1 utilise the one component, acetic acid releasing, prepolymer, E4 from Wacker (Burhausen, Germany) to encapsulate the [Ru(dpp)3]2+ homogeneous ion-pair. This prepolymer, as with many silicone prepolymers, contains a hydrophobic silica gel as a filler.11 The latter is prepared from hydrophilic silica via a reaction of the surface silanol groups with an organosilane, to generate Si–R surface groups, so rendering the material hydrophobic. Klimant and Wolfbeis note that with their numerous homogeneous [Ru(dpp)3]2+ ion-pair sensors based on E4, ‘the lumophore doesn't accumulate at the filler/silicone interface’, presumably based on the observation that the ‘quenching constants are similar for sensor membranes based on silicones containing either silica gel as filler or no filler’.9 Thus, in O2 sensors containing hydrophobic silica as a filler, there is no evidence that the lumophore is in anything but its homogeneous ion-pair form.
Most oxygen sensors based on the ruthenium diimine lumophores, with and without filler, exhibit a negative deviation from linearity in the Stern–Volmer plots of the observed luminescence data as a function of O2 concentration. As a consequence the data is often fitted to eqn (3), values for which (i.e. f01, KSV1 and KSV2) are given in Table 1 for the various [Ru(dpp)3]2+-based O2 sensors. Although formally sensitivity is defined as signal change/concentration, in systems in which a non-linear response curve is generated this formal definition is not considered particularly useful.19 Thus, with regard to O2 sensors, others20 have used the ratio, R = I0/I100, as a measure of sensitivity, where I100 is the luminescence signal in 100% O2. Here, as elsewhere,7 we use the parameter, PO2 (S = 1/2) as a rough measure of sensitivity of O2 sensors, which is defined as: the level of oxygen required for a 50% reduction in luminescence intensity. A brief examination of Table 1 reveals the [Ru(dpp)3]2+/silicone rubber O2-sensor of Bacon and Demas,12 with a PO2 (S = 1/2) = 0.02 atm, to be one of the most sensitive of all the O2 sensitive inks based on [Ru(dpp)3]2+ that have been reported to date.
In contrast to most of the examples in Table 1, in this paper we present a novel method of luminescent, Ru(II)–diimine-based O2 sensor fabrication, in which the cationic, lumophoric dye, [Ru(dpp)3]2+, is ion-paired to the surface of heterogeneous TiO2 nanoparticles in aqueous solution, which have been rendered anionic through the exploitation of the pzc (point of zero charge) of the TiO2 (∼pH 6). After filtering, the result is a pigment, which can then be extruded in LDPE to produce a thin (60 μm), highly flexible, luminescence-based O2 sensitive plastic film. Such heterogeneous O2 sensors overcome the problems associated with the more traditional, homogeneous ion-pair lumophore O2-sensitive inks, such as film curling12 and the need for a solid, largely inflexible support (such as glass or Mylar).11,12,15,16
2. Experimental
2.1 Materials and instrumentation
The [Ru(dpp)3]2+, in its dichloride salt form, was purchased from GFS Chemicals, and used as received. The P25 TiO2 powder was a gift from Evonik Industries, and comprised particles with a primary size of 21 nm, in turn comprised of an intimate mixture of 75% anatase and 25% rutile crystalline phases.21 Titania was chosen as the inorganic support, over silica (pzc ∼ 3) due to its higher surface density of ionisable protons (12.5 cf. 5.9 sites per nm2 based on surface crystal structure calculations).22 The low density polyethylene, LDPE, powder (for masterbatch work) had a melt flow index (MFI) of 20 was supplied by PW Hall UK and the LDPE for film preparation had an MFI of 4 (lupolen 3020 K) and was supplied as pellets by Ultrapolymers UK.LDPE has been used as an O2 lumophore encapsulating material before.23 In this, and other examples,12 the O2-sensitive lumophore (ref. 23 used fluoranthene) is incorporated into the polymer film by soaking the film in a volatile organic solvent, such as cyclohexane, in which the lumophore is dissolved. The solvent causes the polymer film to swell and adsorb some of the solution and rapid drying then leaves some of the dye entrapped in the polymer. Since this approach requires the lumophore to be lipophilic, it follows that such indicators are examples of homogeneous O2 sensing lumophoric systems. One problem with such systems is the tendency for the lumophore to crystallise over time.11
All gases were purchased from BOC gases and blended using a Cole Parmer gas blender. All fluorescence work was carried out using a PerkinElmer LS45 Fluorescence Spectrometer. The pigmented polymer film was extruded using a Rondol Microlab Twin Screw extruder.
2.2 Preparation of intelligent pigments
The cationic nature of the [Ru(dpp)3]2+ dye and the point of zero charge (pzc) of the TiO2 (pH 6.6) were exploited in order to ion-pair the dye to the surface of the titania particles. Thus, at pH 11, the TiO2 forms surface![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–O− groups, (the latter is a common representation24 of such surface groups, in this case with the Ti forming 3 bonds to three different atoms in the bulk and one to a surface oxygen) to which the cationic dye can bind via ion-pairing, i.e.
Ti–O− groups, (the latter is a common representation24 of such surface groups, in this case with the Ti forming 3 bonds to three different atoms in the bulk and one to a surface oxygen) to which the cationic dye can bind via ion-pairing, i.e.| 2Ti–O− + [Ru(dpp)3]2+ → [(Ti–O−)2(Ru(dpp)3] | (4) |
Hence, typically, to 100 ml of a 10−4 M solution of the dye in 10−3 M NaOH (i.e. at pH 11) were added 5 g of P25 TiO2, the mixture was stirred for 2 hours, filtered, air dried, and the resulting solid washed with three 10 ml aliquots of a 10−3 M NaOH aqueous solution. The pale yellow/orange powdered solid product was allowed to air dry for a few hours before being transferred to an oven (50 °C) for 1 hour, ground with a mortar and pestle, and sieved through a 250 μm sieve. The final, fine, evenly coated, yellow powder luminesced brightly under UV light in the absence of O2.
2.3 Preparation of O2-sensitive plastic films
The O2-sensitive plastic films were fabricated by the extrusion of the TiO2–[Ru(dpp)3]2+ pigment powder in low density polyethylene (LDPE), with a final pigment loading of 5 wt%, using a Rondol Microlab 10 mm twin screw extruder (barrel L/D 25/1). The pigment was first dispersed through LDPE powder (melt flow index, MFI, 20) to produce a masterbatch of pigmented pellets with 10 wt% pigmentation, using the extruder to create masterbatch pellets at processing temperatures ramped gradually from 90 °C at the feed zone, to 140 °C at the die, using a feed hopper rate of 41 rpm, extruder screw speed of 80 rpm and a pelletizer speed of 0.5 m min−1. These pellets were then diluted by 50% w/w with virgin LDPE pellets (MFI 4) and extruded to produce a cast film at the following processing conditions: temperatures starting at 90 °C (at the feed zone) then increasing to: 110–125–135 °C (across the barrel) and finally 140 °C (at the die), at a feed hopper rate of 41 rpm, extruder screw speed of 100 rpm, and take-off speed of 1.5 m min−1. The average, central thickness of the O2-sensitive LDPE film was measured by micrometry to be 60 μm. The TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film was a pale yellow colour and luminesced brightly under UV light even in air, as illustrated in Fig. 1.![Photographs of the TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film in air under room light, and under UV light.](/image/article/2013/AN/c3an01141k/c3an01141k-f1.gif) | ||
| Fig. 1 Photographs of the TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film in air under room light, and under UV light. | ||
2.4 Characterization
For luminescence intensity measurements, the pigment and plastic film were both mounted on a microscope slide using double-sided tape, which in turn was cut to fit the diagonal of a typical 1 cm quartz fluorescence cell. In order to maximise the measured luminescence intensity of these solid-state polymer film samples, the slide was placed in the cuvette, and positioned so that the edge of the sensor films was directly in line with the emission detector of the fluorimeter and so square on to the excitation beam, allowing the luminescence, which was mostly gathered by total internal reflection, and exits from the edge of the film, to be more easily measured.3. Results and discussion
3.1 TiO2–[Ru(dpp)3]2+ pigment as an O2 indicator
Upon excitation at 430 nm, the emission maximum of the TiO2–[Ru(dpp)3]2+ pigment (adhered to a microscope slide using double-sided tape) was observed at 615 nm. Quenching of this luminescence was monitored as a function of partial pressure of O2, PO2, and the resulting recorded emission spectra are shown in Fig. 2, with the corresponding Stern–Volmer plot shown as the inset diagram. As expected, a negative deviation from linearity was observed in this plot, so eqn (3) was used to model the data, and the resulting calculated optimum fit parameters are presented in Table 2.![Emission spectra of [Ru(dpp)3]2+–TiO2 pigment upon exposure to (from top to bottom) 0, 0.01, 0.05, 0.3, 0.6 and 1 atm O2; inset is Stern–Volmer plot ○ = experimental data, solid line is 2-site model fit to data.](/image/article/2013/AN/c3an01141k/c3an01141k-f2.gif) | ||
| Fig. 2 Emission spectra of [Ru(dpp)3]2+–TiO2 pigment upon exposure to (from top to bottom) 0, 0.01, 0.05, 0.3, 0.6 and 1 atm O2; inset is Stern–Volmer plot ○ = experimental data, solid line is 2-site model fit to data. | ||
| Indicator type | f01 | KSV1 (atm−1) | KSV2 (atm−1) | PO2 (S = 1/2) (atm) |
|---|---|---|---|---|
| TiO2 pigment | 0.959 ± 0.004 | 459.7 ± 44.2 | 3.6 ± 0.4 | 0.002 |
| LDPE film | 0.24 ± 0.01 | 30.7 ± 3.5 | 0.65 ± 0.02 | 0.84 |
These quenching parameters reveal the pigment to be extremely sensitive to oxygen, more so than the homogeneous, ion-pair, ink-based O2 sensors reported in Table 1; indeed, the calculated PO2 (S = 1/2) (0.002 atm) for the pigment (see Table 2) is ten times less than that of the Bacon and Demas12 silicone-based sensor (see Table 1). This is not too surprising given the O2-sensitive lumophoric dye is bound on the surface of the TiO2 pigment particles and so much more easily quenched by oxygen than when the homogeneous ion-paired dye is embedded in the polymer of a dried ink film, since in the latter case the oxygen must first dissolve and then diffuse through the polymer in order to quench the homogeneously dispersed lumophore.
In a separate experiment, the luminescence intensity response of the titania–[Ru(dpp)3]2+ pigment towards repeated alternating streams of 0 atm and 1 atm O2, and its response and recovery times, were measured by measuring the luminescence intensity as a function of time at 615 nm, and switching the gas stream between pure Ar and O2 (Fig. 3). From this data, it was possible to determine the 50% response and recovery times, t↓50 and t↑50, defined as the time taken for the luminescence intensity to decrease/recover by 50% on switching from Ar → O2, and back again, which were 2 s and 25 s, respectively.
![Repeat response and recovery of TiO2–[Ru(dpp)3]2+ pigment to alternating streams of 0 and 1 atm O2.](/image/article/2013/AN/c3an01141k/c3an01141k-f3.gif) | ||
| Fig. 3 Repeat response and recovery of TiO2–[Ru(dpp)3]2+ pigment to alternating streams of 0 and 1 atm O2. | ||
The extremely rapid change in luminescence of the [Ru(dpp)3]2+ bound to the surface of the TiO2 particle pigment upon exposure to O2 (≤2 s) is also likely due to the large surface area of the pigment (ca. 50 m2 g−1) which provides much greater access for the O2 to the quenchable, surface-bound lumophore. Note that in this work, the relative humidity sensitivity of the pigment was not evaluated and may alter the PO2 (S = ½) value reported in Table 2.
3.2 TiO2–[Ru(dpp)3]2+ – pigmented LDPE film as an O2 indicator
![Emission spectra of [Ru(dpp)3]2+ LDPE film upon exposure to (from top to bottom) 0, 0.1, 0.3, 0.6 and 1 atm O2; inset is Stern–Volmer plot ○ = experimental data, solid line is 2-site model fit to data.](/image/article/2013/AN/c3an01141k/c3an01141k-f4.gif) | ||
| Fig. 4 Emission spectra of [Ru(dpp)3]2+ LDPE film upon exposure to (from top to bottom) 0, 0.1, 0.3, 0.6 and 1 atm O2; inset is Stern–Volmer plot ○ = experimental data, solid line is 2-site model fit to data. | ||
A study of the variation in luminescence intensity upon switching the gas stream alternately from O2 to Ar was carried out and the results are illustrated in Fig. 5, from which values for t↓50 and t↑50 were calculated to be 6 s and 14 s, respectively. These results show that the heterogeneous lumophore ion-pair O2 sensitive film has a much (ca. 420 times) reduced O2 sensitivity compared to the pigment alone most probably due to the low O2 permeability in LDPE (2.3 × 1010 cm3 cm cm−2 s−1 cm Hg−1 (ref. 25)). However, despite this, the extruded TiO2–[Ru(dpp)3]2+ pigmented LDPE film reported here is of comparable O2 sensitivity to the [Ru(dpp)3]2+–silicone sensors previously reported and listed in Table 2, which is possibly not too surprising given the similarity in O2 permeability, i.e. PM (silicone) = 6.2 × 1010 cm3 cm cm−2 s−1 cm Hg−1.26
![Repeat response and recovery of TiO2–[Ru(dpp)3]2+ pigmented LDPE film to alternating streams of 0 and 1 atm O2.](/image/article/2013/AN/c3an01141k/c3an01141k-f5.gif) | ||
| Fig. 5 Repeat response and recovery of TiO2–[Ru(dpp)3]2+ pigmented LDPE film to alternating streams of 0 and 1 atm O2. | ||
Finally, the luminescence responses of the TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film to 0, 0.21 and 1 atm O2 in the presence (100%) and absence (0%) of relative humidity (RH) were measured, and the results indicated little, if any, change in the O2 sensitivity of the indicator to changes in RH. This is most likely due to the low permeability of water in polyethylene (0.05 × 1010 cm3 cm cm−2 s−1 cm Hg−1), which is in striking contrast to silicone rubber (51.8 × 1010 cm3 cm cm−2 s−1 cm Hg−1).27
3.2.2 Luminescence lifetime studies
Quenching analysis of the extruded film was additionally carried out via lifetime measurements, and typical nanosecond flash photolysis luminescence decay curves of the lumophore in the extruded film in the presence and absence of O2 are shown in Fig. 6.![Typical decay curves of the TiO2–[Ru(dpp)3]2+ pigmented LDPE film in the absence and presence of O2.](/image/article/2013/AN/c3an01141k/c3an01141k-f6.gif) | ||
| Fig. 6 Typical decay curves of the TiO2–[Ru(dpp)3]2+ pigmented LDPE film in the absence and presence of O2. | ||
As expected, these decays were not described by simple first order kinetics, as expected for homogeneous solutions, but rather a multi-exponential decay was observed. Such decays fit eqn (5), as proposed by Demas,28 for a two quenching domain, heterogeneous system:
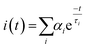 | (5) |
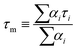 | (6) |
These weighted lifetimes can be compared directly to the intensity values gained from the fluorimetry work, via a corresponding Stern–Volmer (lifetime) plot, the results of which are illustrated in Fig. 7. The calculated quenching parameters derived from fitting the data in Fig. 7 to eqn (3) are in extremely good agreement with those calculated from the intensity data illustrated in Fig. 4, notably a value for PO2 (S = 1/2) of 0.78 atm can be calculated from the lifetime study that compares well with that from the luminescence intensity study (see Fig. 4), of 0.84 atm.
![Lifetime Stern–Volmer plot of TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film: ○ experimental data, solid line is two-site model fit to the data; f01 = 0.29 ± 0.02, KSV1 = 26.4 ± 7.3 atm−1, KSV2 = 0.59 ± 0.05 atm−1, PO2 (S = 1/2) = 0.78 atm.](/image/article/2013/AN/c3an01141k/c3an01141k-f7.gif) | ||
| Fig. 7 Lifetime Stern–Volmer plot of TiO2–[Ru(dpp)3]2+ pigmented extruded LDPE film: ○ experimental data, solid line is two-site model fit to the data; f01 = 0.29 ± 0.02, KSV1 = 26.4 ± 7.3 atm−1, KSV2 = 0.59 ± 0.05 atm−1, PO2 (S = 1/2) = 0.78 atm. | ||
4. Conclusions
Most previous work on O2 sensors involving Ru(II) diimine complexes as the lumophores have combined the cationic lumophores with a lipophilic anion, rendering the homogeneous dye-anion ion-pair lipophilic, and so soluble in a solvent containing a polymer such as polystyrene or silicone. In contrast, in this work –[Ru(dpp)3]2+ is ion-paired to the surface of TiO2 nanoparticles to produce a heterogeneous O2 sensing pigment, with a strikingly high O2 sensitivity, most likely due to the ready access afforded to the ambient O2 to the quenchable luminescent dye, [Ru(dpp)3]2+. This pigment was extruded in LDPE to form a thin, flexible, plastic film which was readily quenched by O2. A major drawback of using a homogeneous ion-pair polymer ink is the need for a largely inflexible, support on which the ink must be cast and dried prior to O2 sensing.11,12,15,16 In contrast, extrusion of polymer films containing the O2 sensitive lumophore (in this case in pigment form) produces a fully flexible and self-supporting, ready to use, O2 sensitive film. Other reported problems with the traditional O2 sensors include: dye leaching in solvents which cause the polymer to swell, such as acetone,12 and water,11 film curling,12 water condensation on the film surface,12 and a reduction of sensitivity at high RH.11 The LDPE film produced in this work shows none of these undesirable characteristics and from the results of this work, dye-pigment pairing followed by extrusion into a polymer appears a promising route to make O2 sensitive, flexible, polymer films.References
- B. Lei, B. Lei, H. Zhang, S. Lu, Z. Zheng, W. Li and Y. Wang, Adv. Funct. Mater., 2006, 16, 1883 CrossRef CAS.
- C. S. Chu, Appl. Opt., 2011, 50, E145 CrossRef CAS.
- A. N. Watkins, R. Weriner, J. D. Jordan, W. Xu, J. N. Demas and F. U. Bright, Appl. Spectrosc., 1998, 52, 751 Search PubMed.
- L. C. Clark, R. Wolf, D. Granger and Z. Taylor, J. Appl. Physiol., 1953, 6, 189 CAS.
- A. Mills, Platinum Met. Rev., 1997, 41, 115 CAS.
- A. Mills and A. Lepre, Anal. Chem., 1997, 69, 4653 CrossRef CAS.
- A. Mills and M. Thomas, Analyst, 1997, 122, 63 RSC.
- J. N. Demas and B. A. DeGraff, in Topics in Fluorescence Spectroscopy, ed. J. R. Lakowicz, Plenum Press, New York and London, 1994, vol. 4, p. 71 Search PubMed.
- J. N. Demas, B. A. DeGraff and W. Xu, Anal. Chem., 1995, 67, 1377 CrossRef CAS.
- O. S. Wolfbeis, M. J. P. Leiner and H. E. Posch, Mikrochim. Acta, 1986, 90, 359 CrossRef.
- I. Klimant and O. Wolfbeis, Anal. Chem., 1995, 67, 3160 CrossRef CAS.
- J. R. Bacon and J. N. Demas, Anal. Chem., 1987, 59, 2780 CrossRef CAS.
- A. Mills and F. C. Williams, Thin Solid Films, 1997, 306, 163 CrossRef CAS.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 5337 Search PubMed.
- P. Hartmann, M. J. P. Leiner and M. E. Lippitsch, Anal. Chem., 1995, 67, 88 CrossRef CAS.
- C. Preininger, I. Kilmant and O. S. Wolfbeis, Anal. Chem., 1994, 66, 1841 CrossRef CAS.
- http://www.oxysense.com, accessed June 2013.
- J. W. Gregory, K. Asai, M. Kameda, T. Liu and J. P. Sullivan, Journal of Aerospace Engineering, 2008, 222, 249 CAS.
- R. Erkins and P. Edwards, Clin. Chem., 1997, 43, 1824 Search PubMed.
- B. D. MacCraith, C. McDonagh, G. O'Keefe, E. T. Keyes, J. G. Vos, B. O'Kelly and J. F. McGlip, Analyst, 1993, 118, 385 RSC.
- T. Ohno, K. Surukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82 CrossRef CAS.
- R. O. James and G. A. Parks, Characterization of aqueous colloids by their electrical double-layer and intrinsic chemical properties, in Surface and Colloidal Science, ed. E. Matijevic, Plenum Press, New York, ch. 2, 1982 Search PubMed.
- I. Bergman, Nature, 1968, 218, 396 CrossRef CAS.
- T. Hiemstra and W. H. Van Riemsdijk, J. Colloid Interface Sci., 2006, 301, 1 CrossRef CAS PubMed.
- H. Zhang and A. Cloud, The Permeability Characteristics of Silicone Rubber, in Global Advances in Materials and Process Engineering proceedings, November 6–9, 2006, Dallas, pp. 72–75 Search PubMed.
- S. Marias, J. M. Saiter, C. Devallencourt, Q. T. Nguyen and M. Métayer, Polym. Test., 2002, 21, 425 CrossRef.
- Shin-Etsu, Characteristic properties of silicone rubber compounds, Tokyo, Japan, 2005 Search PubMed.
- E. R. Carraway, J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63, 332 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |