Role of innate and adaptive immunity during drug-induced liver injury
C. David
Williams
and
Hartmut
Jaeschke
*
Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, Kansas City, KS, USA. E-mail: hjaeschke@kumc.edu; Fax: +1 913 588 7501; Tel: +1 913 588 7969
First published on 28th August 2012
Abstract
Drug-induced liver injury (DILI) is a major human health concern and is the most frequent cause of FDA boxed warnings and the removal of drugs from the market. Idiosyncratic DILI (IDILI) is highly variable in its time to onset and no one clear hypothesis exists to explain the mechanism. The general belief is most cases of IDILI involve some immune mediated component; however no animal models can recapitulate human IDILI. Despite numerous drugs that have IDILI potential, the most common cause of DILI is acetaminophen (APAP) overdose. APAP in animal models and in patients is a dose-dependent hepatotoxicant resulting in severe centrilobular necrosis and a robust inflammatory response. In this review we will compare the existing hypotheses for potential causes of IDILI and discuss the potential roles of immune involvement in DILI. Additionally we will focus on what we have learned from the mechanisms of APAP toxicity (protein adduction, mitochondrial dysfunction, oxidant stress, DNA damage, release of damage associated molecular patterns (DAMPs)) and useful interventions to alleviate APAP-toxicity (reduced protein binding, scavenging of reactive oxygen, induction of autophagy). Mechanistically APAP-induced liver injury appears to be fundamentally different from IDILI, however, there are potential critical events shared between APAP-induced liver injury and IDILI. The strategies and methods currently being used to study APAP-induced liver injury are described in this review. This improved insight into mechanisms of APAP-induced injury with initiation, propagation and inflammation may also help to better understand IDILI.
 Clarence David Williams | Dr Clarence David Williams received his doctorate degree in toxicology from the University of Kansas Medical Center and currently works in the laboratory of Dr Jaeschke. Dave has previously studied, in an industry setting, the mechanisms of immunogenicity to biologic therapeutics. Currently his work focuses on liver injury and in particular the role of inflammation in the progression and resolution of injury. The primary model that Dave focuses on is acetaminophen-induced hepatotoxicity in both rodents and humans. |
 Hartmut Jaeschke | Dr Hartmut Jaeschke is currently Professor and Chairman of the Department of Pharmacology, Toxicology and Therapeutics at the University of Kansas Medical Center in Kansas City. He received a MSc and PhD degree in biochemistry and toxicology from the University of Tübingen, Germany. Since 1988 he held faculty positions at Baylor College of Medicine in Hoston, TX, The University of Arkansas for Medical Sciences in Little Rock and The University of Arizona in Tucson and was a scientist at the Upjohn Company. Dr Jaeschke has published more than 270 original manuscripts and invited reviews and book chapters on mechanisms of liver pathophysiology and drug hepatotoxicity. |
Introduction
The liver is generally considered a target of drug toxicity because of its first-pass exposure to orally administered drugs and its high capacity for xenobiotic metabolism. Phase I (oxidation-reduction) and phase II (conjugation) reactions occur within hepatocytes to detoxify and facilitate the removal of xenobiotics, however in this process toxic metabolites can be generated and accumulate within hepatocytes. Additionally, many phase III (transporter) reactions lead to the accumulation of xenobiotics and their metabolites within the liver. These metabolic capacities, in combination with the liver's portal blood supply, and unique immune system make it vulnerable to drug toxicity.Despite the relative infrequency of idiosyncratic drug-induced liver injury (IDILI) it poses a very serious health concern with potentially fatal consequences. In a retrospective study from 1998 to 2007 it was reported that of all causes of acute liver failure, APAP was the predominant cause (46%) and DILI from other drugs accounted for 11%.1 Additionally, the spontaneous patient survival is quite different between etiologies; APAP showed a 65% spontaneous survival rate while the spontaneous survival rate from other drugs was only 29%.1 In many cases, IDILI cannot be detected in preclinical testing or clinical trials because the occurrence is so infrequent that the study number is insufficient to detect it (Table 1), however guidelines have been implemented to minimize these risks. If a drug is suspected of IDILI the decision making process if the drug should be discontinued is generally determined by the frequency of patients that fall into “Hy's Law” (Table 2). If a patient meets the three criteria there is a 10–50% chance of patient mortality.2
| Severity | Clinical manifestation | Frequency rate |
|---|---|---|
| a The frequency of IDILI is variable due to failures in reporting and potential conflicting causes of liver injury (i.e. autoimmune hepatitis, viral hepatitis and others), however the individual susceptibility for altered liver function due to a drug is shown. Content adapted with permission from Robert J. Fontana, MD, University of Michigan. | ||
| Mild | <3-fold ULN ALT | 1/10 to 1/1000 |
| Moderate | Impaired hepatic function (>3-fold ULN ALT) | 1/100 to 1/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Severe | Acute liver failure or death | 1/10000 to 1/1000000 |
| Hepatocellular Injury | 3-fold ULN for ALT or AST |
| Hepatic Impairment | 2-fold ULN for total bilirubin |
| Exclusion of other causes | No evidence of viral hepatitis, confounding drugs, acute liver disease or cholestasis (should not present elevated serum ALP) |
Drugs with the highest IDILI concern are antineoplastic agents, NSAIDs, antivirals, antidepressants, and antimicrobials,3 and in the US, antimicrobials account for 46% of IDILI.4 In a majority of cases (73%) a single prescription was implicated as the cause of IDILI.5 Clinically, the only effective treatment for DILI is to stop the administration of the drug and supportive care (with the exception of N-acetyl-cysteine for APAP overdose and potentially carnitine for valproic acid).6
There are several reasons to assume IDILI is immune mediated. The most striking is the time to onset; depending on the drug it could take months to over one year prior to initiation of IDILI which would indicate an adaptive immune response. Additionally elevated transaminase activities can be observed with some drugs up to one month following discontinuation of drug.7 Some IDILI also involves the generation of antidrug antibodies or autoantibodies.8,9
In most cases IDILI is regarded as dose-independent, however this is most likely an incorrect statement. IDILI is very rarely seen at doses of <10 mg per day and more than three fourth of IDILI cases occur when the drug is given at >50 mg per day.10 The notion that IDILI is dose independent probably arises from the fact that the overwhelming majority of patients taking the drug are non-responders in regard to toxicity.8 In this review we will compare and contrast what is known about mechanisms of APAP-induced liver injury and IDILI.
Why is the liver a target of drug toxicity?
Drug disposition alone cannot explain IDILI because there is no association with the accumulation of excess drug or metabolite that would result in concentrations that are toxic to the liver.11 This is in contrast to what is seen in APAP overdose. It is interesting however that deletion of certain drug detoxification enzymes cause an increased risk for IDILI. Patient with glutathione S-transferase M1 and T1 null mutations were 2.7-fold more likely to develop IDILI; these odds ratios increased to 3.5-fold for antibacterial and 5.6-fold for NSAIDs.12 Also of interest in this study was the predominance of women with this double null genotype as there is generally a female predominance of IDILI cases.12 IDILI is classified by its bizarre and, as to date, unexplainable toxicity unlike APAP which is explainable based on its chemical structure and generation of excess reactive metabolite.Liver toxicity is the leading cause of removing drugs from the market and issuance of boxed warnings.11 There is no one single genetic, environmental or other factor that can be the sole cause of IDILI. This is illustrated in the case of flucloxacillin. A genome wide association study showed patients with the HLA-B*5701 genotype were at a much higher risk of developing IDILI to flucloxacillin with an incredibly high odds ratio (OR) of 80.6.13 This genotype was in no means the sole determinant of DILI, however. Despite this very strong association, only 1 out of every 500 to 1000 patients treated with flucloxacillin who have the HLA-B*5701 genotype develop IDILI. The HLA-B*5701 genotype is also associated with abacavir hypersensitivity.10 This is a clear illustration of the multifactorial nature of IDILI and certain genotypes and environmental queues are risk factors rather than causative. This study also demonstrates a direct link to risk of IDILI with the adaptive immune system as HLA-B is an MHC class I (MHC-I) molecule that presents self-antigen to CD8+ T cells. For additional detail of MHC-related risk factors for IDILI we recommend a current critical review by Daly and Day.14
The liver is considered a site of immune tolerance, and perhaps in IDILI this tolerance is lost. Tolerance in the liver has been demonstrated in several ways.15 Systemic donor-specific T cell tolerance can be achieved after orthotopic liver transplantation. Introduction of antigen via portal vein can promote tolerance. Liver transplantations can be performed between patients with MHC mismatches. The liver can function as a site for clonal deletion of T cells and has been referred to as a “sink” for activated T cells.16,17
Perhaps this tolerance is due in part to altered leukocyte distribution relative to other organs, blood and secondary lymphoid tissue. In the human liver the ratio of CD4+/CD8+ T cells is 1:3.5 but 2:1 in peripheral blood.18 Additionally the liver has a much higher number of cells that blur the line between innate and adaptive immune response. This includes the ‘unconventional’ γδ T cells with frequency in the liver five-times that found in peripheral blood,18 natural killer (NK) T cells (CD3+CD56+) which account from 5% to 25% of liver lymphocytes but are rarely seen in blood19 and NK cells which account for approximately one third of human liver lymphocytes.20 Additionally, between species the frequencies of these cell types can differ dramatically with invariant NK T cells being relatively rare in the human liver.21
In the liver the accumulation of leukocytes occurs in a manner substantially different from other organs mediated at least in part by the blood supply and architecture of the liver. The sinusoidal vasculature is small in diameter, low pressure and fenestrated, and leukocyte accumulation is not dependent on selectins or integrins.22,23 It was reported that leukocyte accumulation into the liver in the endotoxin model was dependent on CD44-hyaluronan interaction,24 however this does not appear to be the mechanism of neutrophil accumulation during APAP overdose (Williams and Jaeschke, unpublished data). Most leukocytes that leave the vasculature and enter into the parenchyma do so via the sinusoids rather than postcapillary venules.25,26
Liver immunity and its role during APAP overdose
The most common model to study drug-induced hepatotoxicity is APAP. APAP overdose results in centrilobular necrosis that is not exacerbated by the recruitment of inflammatory cells.27 Despite extensive evidence of the benign role of inflammation in regard to injury several groups still report that inflammatory cells can increase injury, and this will be discussed in more detail in later sections and has previously been reviewed in detail.27 If biopsy is performed in IDILI patients the mode of cell death appears to necrosis with inflammatory infiltrates,11 however the mechanisms of T cell mediated death is generally apoptosis. From these data it is very difficult to determine the mode of cell death in IDILI because it is possible that at the time of biopsy the injury has already degraded into secondary necrosis. Clearly there are striking differences between IDILI and APAP toxicity. APAP toxicity is dose dependent with a very rapid onset of injury (within hours). Therefore the mechanisms of toxicity and cell death are most likely different between idiosyncratic hepatotoxic drugs and APAP, but understanding of intracellular events of APAP toxicity (i.e. formation of reactive metabolite, covalent binding of cellular proteins, mitochondrial dysfunction, release of mitochondrial components, nuclear DNA damage) could lead to more clues regarding susceptibility or outcome of IDILI.The initiation of APAP toxicity is dependent on the metabolic conversion of APAP to N-acetyl-p-benzoquinone imine (NAPQI) in hepatocytes predominantly by CYP2E1 and to a lesser extent by CYP1A2, CYP2D6 and potentially other P450 enzymes. The reactive metabolite then adducts cellular proteins; of particular interest is the adduction of mitochondrial proteins.28 This mitochondrial binding propagates injury by initiating an oxidant stress that leads to mitochondrial failure, the formation of ROS and the mitochondrial release of endonucleases.29 These endonucleases translocate to the nucleus and damage DNA.30 All of these events ultimately lead to cellular oncotic necrosis.31
NK and NK T cells
Despite the liver's role in immune tolerance, it is also critical during inflammation and capable of very quick and robust inflammatory response. Of particular interest in this regard are the liver NK and NKT cells that have the capacity to become activated very quickly and produce large amounts of cytokines, in particular IFN-γ. Examples of this massive inflammatory response can be seen in hepatitis A and E infections where these viral vectors are delivered to the liver via the portal vein.32 Interestingly, depletion of Kupffer cells (KCs) results in decreased NK cell numbers in the liver.21NK and NK T cells were suspected to cause increased injury during APAP overdose via IFN-γ production and enhanced neutrophil recruitment.33 It was later demonstrated that the DMSO vehicle in these studies using C57BL/6 mice artificially recruited and activated these cells types.34 It is known that even between different strains of mice the surface marker expression of NK cells is variable.21 It is therefore reasonable to conclude that function or activation could be different between strains as well, and it was shown that elimination of NK and NKT cells reduced APAP-induced injury in IL-13 deficient mice.35 While these cell types in C57BL/6 mice do not cause additional injury in APAP overdose they are still suspected as potential participants in other forms of DILI primarily because these cells function as innate immune cells but perform these functions in ways similar to the adaptive immune system. It was shown in a case report of two patients with IDILI-induced hepatic failure that NK and NKT cell quantity and expression of the costimulatory factor CD28 was different than control patients.36 Additionally, of potential importance is the Fas/Fas ligand interaction that can be mediated by these cells on hepatocytes.27 Generally, hepatocytes are resistant to perforin/granzyme-induced cell death which means T cell mediated killing could involve Fas/FasL interaction or TNF-α.37
Neutrophils
Neutrophils can occasionally be seen in normal, healthy liver, however upon inflammatory insult these cells are rapidly recruited into the liver and the number of total hepatic neutrophils can be increased several orders of magnitude.38 If proper stimuli are present, these cells will extravasate from the vasculature into the hepatic parenchyma and cause injury.39 Without these signals the neutrophils will remain in the sinusoids, undergo apoptosis and be cleared by KCs. Normally the half-life of a neutrophil is 6–12 h but during active inflammation this can be increased to 48 h.40It has been extensively demonstrated that neutrophils do not participate in APAP induced injury,27 however, some controversy still exists. A neutropenia-inducing antibody results in protection against APAP toxicity but only if given 24 h prior to APAP,41–43 but not if given after APAP overdose despite functional inactivation of neutrophils.44–46 Also, neutrophils show no enhanced activation status during the APAP-induced injury phase,47 priming and activating neutrophils during APAP overdose by IL-1β or endotoxin does not increase injury,47,48 CD18-47 and ICAM-1-deficient mice44 are not protected, an anti-CD 18 antibody did not affect injury,49 and mice that have inhibited NADPH oxidase or lack NADPH oxidase (phagocytic respiratory burst) have no difference in oxidant stress or APAP-induced injury.44,50 Often times neutrophils are thought to be the cause of enhanced injury because in genetic or pharmacologic interventions mice with reduced injury will have lower hepatic neutrophil counts or reduced myeloperoxidase staining,43,51 however, this is most like due to the fact that mice with less injury, release less DAMPs, and therefore recruit fewer neutrophils. In simple terms, blocking upstream events (injury) will reduce downstream events (inflammation). A caveat of most immunological interventions is these studies often times do not assess drug metabolism, protein binding or other intracellular events. Without these control experiments results are difficult if not impossible to interpret and a variety of off-target effects could occur. This should be a particular concern when the data contradict many published findings.
Most theories and models of IDILI do not have neutrophils being a major contributor to injury with a few exceptions. If neutrophils are participating in IDILI then it would most likely occur as a secondary effect to damage caused by an adaptive immune response. One exception is the case of halothane. Halothane was used clinically as an inhaled anesthetic, but patients upon reexposure had an increased risk of potentially fatal hepatotoxicity.9 Halothane is converted to the reactive metabolite trifluoroacetic acid which adducts hepatic protein and initiates liver injury.9 It was shown that depletion of neutrophils in this model could attenuate injury.52 It was later demonstrated that NK T cells were critical for the recruitment of neutrophils in this model.53
Kupffer cells (KCs)
Administration of LPS or other inflammatory insults results in massive cytokine formation and accumulation of leukocytes in the liver. KCs are instrumental in the removal of gut-derived endotoxin and the phagocytosis and killing of bacteria. Within minutes of the inflammatory stimuli KCs produce numerous cytokines, chemokines and subsequently stimulate hepatocytes (predominantly through IL-6) to produce acute phase proteins which include pentraxins, protease inhibitors, coagulation factors, complement components and others.37 Interestingly, inactivation of KCs using gadolinium chloride (GdCl3) prevents portal vein tolerance.54 This was determined by the administration of alloantigen (spleen cells from different mouse strains) with or without GdCl3 treatment and measurement of delayed hypersensitivity reaction by foot swelling.54 In another model of T cell mediated delayed hypersensitivity it was shown that KCs were involved in the induction of tolerance for protein adducts or haptens.55 This was determined by sensitization of mice to 2,4-dinitrochlorobenzene (DCNB) with or without KC depletion via liposome/clodronate. Additionally this tolerance could be induced upon adoptive transfer of KCs from tolerized mice.55 KCs are also important in attenuating this inflammatory response by producing high levels of IL-10.37During APAP overdose distinct macrophage populations can be observed during the injury phase and injury resolution. These different populations have unique phenotypes which can be determined by surface markers. Kupffer cells are often referred to as M1 (classically activated) macrophages are typically thought of as pro-inflammatory and drive cells toward a Th1 phenotype through high IL-12 production; traditionally these cells are activated by IFN-γ or LPS. M2 (alternatively activated) are typically anti-inflammatory, activated by IL-4 or IL-13, highly phagocytic and promote tissue repair.56 During injury resolution monocytes are recruited into the liver and become M2-biased. Depletion of KCs prior to APAP overdose actually results in increased injury which is mediated by the loss of IL-10 production.57,58 Also of importance during APAP overdose is IL-6 which is also hepatoprotective and generally considered pro-regenerative.59 IDILI most likely involves the loss of immune tolerance. Alteration of the cytokine production profile could cause of a shift away from immune tolerance which would most likely begin with the resident tissue macrophages of the liver.
Dendritic cells
As stated previously, KCs produce fairly high levels of IL-10 which is capable of altering the function of the liver's other antigen presenting cell type, dendritic cells (DCs). The liver contains both plasmacytoid and myeloid DCs which are most commonly found within portal tracts.32 High levels of IL-10 tend to drive DCs away from effector pathways, and DCs of the liver are generally immature and express low levels of co-stimulatory molecules, which tend to produce regulatory T cells.37It was reported that dendritic cell depletion results in enhanced APAP-induced injury.60 DC depletion was induced in CD11c.DTR mice via diphtheria toxin prior to APAP; this resulted in enhanced inflammatory cytokine and chemokine profiles and increased liver injury. Conversely, expansion of DC populations via Fms-related tyrosine kinase 3 ligand (Flt3L) decreased APAP-induced injury. Additionally, neutrophil or NK cell depletion in addition to DC depletion did not modulate the injury further confirming these innate immune cells do not contribute to APAP-induced injury.60 The immature phenotype of DCs in the liver almost certainly participates in the maintenance of tolerance and loss of tolerance could induce IDILI. It is not clear if loss of tolerance would be mediated by DCs directly or modulation of the microenvironment (i.e. exposure to cytokines and stimulatory factors).
Nalp3 inflammasome
The NACHT, LRR and PYD domains containing protein 3 (Nalp3) inflammasome is assembled after the initiation of sterile inflammation and is responsible for the maturation of IL-1β and IL-18 through activation of caspase-1.61 It was reported that mice deficient for each component of the Nalp3 are protected from APAP-induced injury62 and ATP released during early injury signals through the P2X7 purinergic receptor to activate the Nalp3 inflammasome.63 These studies were repeated and no protection was observed in mice deficient for each component of the Nalp3 inflammasome.64 Additionally, inhibiting caspase-1 prevented IL-1β maturation without altering liver injury, and pharmacologic addition of recombinant IL-1β during APAP overdose did not modulate liver injury.48 In addition, the purinergic receptor antagonist can directly act on hepatocytes and inhibit protein adduct formation, oxidant stress and c-Jun N-terminal kinase (JNK) activation (Williams and Jaeschke, unpublished data). These data suggest that this compound alters the mechanisms of intracellular cell death, which occur upstream and independently of the Nalp3 inflammasome. From these data it is highly unlikely that the Nalp3 inflammasome contributes to APAP-induced injury but its role in IDILI is yet to be determined. This is just one of many examples where data from immunological or pharmacological interventions are misinterpreted because effects of these agents on intracellular signaling events are not considered.Major theories of IDILI
There are a number of hypotheses regarding the cause of IDILI. Most likely no one hypothesis alone can explain the mechanism of all cases of IDILI and several theories might occur sequentially or concurrently to explain the mechanism. As described previously IDILI most likely involves an adaptive immune response which is fundamentally different from what is observed in APAP-induced injury. The accepted dogma of adaptive immune activation requires presentation of antigen loaded on an MHC molecule to the T cell receptor (TCR) of a T cell. Additionally, a second costimulatory signal is required which could be a B7 molecule (CD80 or CD86) on antigen presenting cell (APC) interacting with CD28 on the T cell. When both of these signals occur an immune response is mounted.65 Without this second costimulatory signal the T cell becomes anergic for the maintenance of immune tolerance. This central dogma of immunology gives rise to the “danger hypothesis”.65 APCs present antigen to CD4+ T cells via MHC class II (MHC-II); all other cells present antigen to CD8+ T cells via MHC class I (MHC-I). Classically this means exogenous antigens are presented via the MHC-II and endogenous antigens are presented via MHC-I. There are exceptions to this rule however with the most predominant being that of “cross-presentation”.66 In this process antigens from the extracellular environment can be processed, loaded into MHC-I and presented to CD8+ T cells.“Danger hypothesis in IDILI”
Due to the environment and diet we are constantly exposed to countless exogenous substances through multiple routes of delivery yet mount immune response to very few of these things. For this reason the “danger hypothesis” was proposed.8,9 It is believed that the immune system responds to “danger” (i.e. cell injury) rather than specifically to “non-self”.64 In this theory necrotic cell death is referred to as “bad death” and only bad death triggers immune response mediated by the co-stimulatory factors described previously. Additionally, this theory proposes that only professional APCs (i.e. DCs) can prime naïve T cells where subsequent activation can be performed by other APCs (macrophages and B cells). Effector T cells that are presented antigen without the required co-stimulatory factors will become senescent. Initially, it is thought that cells dying by apoptosis are non-inflammatory and therefore tolerogenic and necrotic cells are pro-inflammatory and therefore immunogenic, however it has been shown that this is not always the case. Antigens from apoptotic cells can be cross-presented to CD8+ T cells and prime an immune response, and cells that die at the peak of an immune response can be immunogenic but cells dying as inflammation wanes can induce tolerance.67 It was also shown that DCs that phagocytosed necrotic cells debris presented antigen to both CD4+ and CD8+ T cells, but DCs that phagocytosed apoptotic cells only presented antigen to CD8+ T cells.68The first step of the danger hypothesis involves some initial cell injury or at the very least some cell stress to cause the initial danger signal. Potentially these danger signals could be cytokines released by innate immune cells or DAMPs released by the injured cells. High mobility group box-1 (HMGB-1) protein currently is one of the most commonly studied and measured DAMPs. APAP causes the release of HMGB-1.64,69 In agreement with one of Matzinger's original hypotheses, oxidation of HMGB-1 which occurs in apoptotic but not necrotic cells was critical to neutralizing its stimulatory activity and blocking oxidation sites prevented tolerance.70 Potentially the oxidation state of HMGB-1 could be of prognostic value during drug-induced liver injury.71
“Hapten hypothesis”
A hapten is a small molecule (typically incapable of initiating an immune response) that covalently binds to a larger molecule (cellular protein) and becomes immunogenic. This theory of IDILI involves the concept of a drug or more-likely the drug's reactive metabolite binding to a cellular protein and triggering the initiation of an immune response. This is a rational theory because of the high expression of CYP450's (drug metabolizing enzymes) and drug transporters found in the liver, therefore most of these reactive metabolites are present within the liver as is the case with the conversion of APAP to the reactive metabolite NAPQI. Adduction of cellular proteins by the reactive metabolite then can initiate an adaptive immune response following antigen processing and MHC presentation; this new immunological entity is referred to as a hapten. The haptenized protein can elicit a T cell response to the drug, the protein itself or a mixed response can occur depending on the antigen presentation.9,72 If the drug metabolite adducts a cellular protein and results in cell death then these aberrant antigens during an inflammatory response (increase costimulatory factors) could be presented by APCs and trigger a CD4+ response, so this theory generally involves the danger hypothesis to propagate the injury.Evidence for the pathogenic role of haptens can be seen with drugs like tienilic acid, dihydralazine, halothane, phenytoin, carbamazepine and others.9 In these cases, common targets of haptenation are the enzymes responsible for metabolism of the drug as well as other liver-specific proteins.73 It has been demonstrated that these antibodies or autoantibodies can react with P450 enzymes (CYP2D6, CYP2C9, CYP1A2, and others) and UDP glucuronyltransferases.74,75 It is unclear if these antibodies exert a direct toxicity. However, it does clearly demonstrate that immune cell activation has occurred, and in the case of autoantibodies immune tolerance has been compromised.8,9
An argument against the hapten hypothesis is some IDILI drugs do not form reactive metabolites to covalently bind proteins.8,76 Additionally, it should be noted, that covalent binding should be regarded as a bioactivation event and not one of overt toxicity, and conversely not all drugs must be metabolized to trigger an adverse drug reaction.77 Additionally, the efficacy of some drugs is dependent on covalent binding like aspirin and proton-pump inhibitors.78
Not all drugs have to form a reactive metabolite to cause IDILI and generation of reactive metabolite does not mean an immune response will be generated even in the presence of substantial injury and inflammation which is the case with APAP. It was hypothesized that APAP toxicity creates a tolerance to APAP protein adducts.79 In a mouse model of APAP overdose lymphocyte apoptosis could be seen in the spleen, thymus and liver-draining lymph nodes, and APAP overdose also suppressed the hypersensitivity response of DNCB.79 This theory might be a mechanism of how progression of IDILI is avoided in most people with minor or subclinical drug toxicity.
“Failure to adapt hypothesis”
Another concept with IDILI is the notion of a lack of adaptation to a toxic insult in which injury caused by the drug or metabolite triggers a response or repair mechanism. This injury could be initiated in the mitochondria (as discussed later), in the endoplasmic reticulum as unfolded protein response or other cellular stressors.Occasionally, patients on a drug with known IDILI potential will present with serum ALT >3 times upper limit of normal (ULN) but return to baseline levels even when continued on the drug.9 This concept is known as adaption. Why will some patients return to normal and other progress to severe liver injury? The response mechanisms could involve phase I, II or III metabolic processes; the induction of anti-oxidant responses, enhanced autophagy, enhanced cell proliferation or other protective measures. On the other hand, this could be indicative of the development of immune tolerance. It is possible the loss of tolerance could be mediated by KCs. This could occur by generation of reactive metabolite within the KCs9 or cross-presentation of antigen. The mode of this initial cell death could also be important. It has been shown that caspases or other cellular proteases upon activation may create new antigenic epitopes within the cell.67
“Pharmacologic interaction (P-I) hypothesis”
This theory involves the direct binding of drug to TCR and T cell activation upon MHC interaction with an APC, or the drug non-covalently interacts with MHC and then presents it to a responsive T cell.80 This concept can explain why drugs that do not form reactive metabolites can initiate IDILI, however this hypothesis is fundamentally different from the danger hypothesis because T cell activation in this hypothesis does not require co-stimulatory factors. This theory was first used to describe the IDILI potential of sulfamethoxazole. In this study T cell clones could be activated by drug even after the fixation of APCs indicating antigen processing was not required.81 While this theory appears to be plausible for some drugs it does not apply for all IDILI drugs. It has been shown that the P-I hypothesis can be applied to other drugs like carbamazepine, lamotrigine, lidocaine and others;80 however, these drugs also can produce reactive metabolites, so perhaps P-I hypothesis occurs in concert with another model of IDILI.“Mitochondria hypothesis” (SOD2+/− model)
This hypothesis involves a subclinical mitochondrial dysfunction that gradually accumulates until a critical threshold is met at which time liver injury rapidly occurs.82 Mitochondria lack DNA repair mechanisms, and cumulative damage over time might induce toxicity. Typically speaking DNA damage is associated with hard electrophiles which have a high positive charge density; these electrophiles are also more likely to react with amino acid residues like lysine. Soft electrophiles (i.e. NAPQI) generally do not attack nucleic acids and interact with amino acids like cysteine and are predominantly detoxified by GSH.77,83 However during mitochondrial dysfunction ROS generated within the mitochondria is the most likely cause of mitochondrial failure. Mitochondrial DNA damage is a late event (after drug metabolism) which can be prevented by scavenging ROS in the mitochondria.84The inner membrane of mitochondria contains high levels of cardiolipin which has many unsaturated bonds potentially making in vulnerable to lipid peroxidation.82 This would be an organelle-specific toxicity potentially leading to impaired mitochondrial respiration and damage as previously described. It is unknown if lipid peroxidation (on a cellular level) is a relevant mechanism of cell death in IDILI, but it is not a relevant general mechanism during APAP overdose.85,86
Sod2-heterozyous mice are more susceptible to mitochondrial injury, and SOD is critical for the detoxification of superoxide to prevent peroxynitrite formation.87–89 It was shown that in Sod2+/− mice troglitazone caused delayed hepatic injury.90 This injury is consistent with a delay in toxicity seen in patients, however these findings could not be repeated in a subsequent study using a very similar model.87 Additionally, long-term administration of flutamide resulted in oxidant stress, mitochondrial dysfunction and hepatic injury in Sod2+/− mice.91 Clinically it has been shown that patients with SOD2 Val16Ala mutation are prone to mixed cholestatic injury (OR = 2.3) and patients homozygous for this mutation with this type of injury are more susceptible to drugs with known mitochondrial hazards (OR = 3.6).92
If a mitochondrial threshold effect does occur and causes toxicity this will not explain why patients with idiosyncratic toxicities are susceptible to toxicity upon re-challenge. Most likely the mitochondria would be an initiating event in IDILI that would then proceed to an immune response to propagate the injury. Thus, the Sod2+/− model could be useful to evaluate the potential of drugs to cause a mitochondrial stress but does not mimic the entire pathophysiology of IDILI.
“Inflammagen model”
This model uses drugs with IDILI potential and co-treats animals with LPS. The assumption behind this approach is that the hepatotoxicity of idiosyncratic drugs is not detectable because of interfering events (death of animals). In addition an inflammagen like LPS causes a leftward shift of the dose-response curve and thus makes the toxicity detectable. The goal of this model is to induce hepatotoxicity in a large percentage of treated animals which will allow for better statistical analysis and provide an economical way to test for IDILI.93 The inflammagen model has been used for drugs like amiodarone, chlorpromazine, diclofenac, trovafloxacin, sulindac and others.93 In these models the dosing of drug and LPS was highly variable; occasionally LPS was given prior to drug, sometimes post-drug, sometimes drug was given multiple times.93 In these studies the liver injury ranged from moderate to severe liver necrosis with a relatively rapid onset.However, there are concerns regarding the relevance of this model. First, the generally used dose of endotoxin (44 × 106 EU kg−1 or ∼4 mg kg−1) results in plasma levels approximately five orders of magnitude higher than the maximum serum concentration measured in septic patients with confirmed Gram-negative infection.94 And, sepsis has not been identified to precede or accompany the onset of IDILI. Second, the mechanism of liver injury in the inflammagen model appears to always involve TNF-α, neutrophils and the coagulation cascade93 independent of the drug. Thus it is likely that the model mimics LPS-induced liver injury where a subtoxic dose of LPS causes neutrophil-mediated liver injury when additional stress (exposure to drug) is applied.
Support of these concerns comes from the application of the inflammagen model to APAP toxicity. Pretreatment with a high dose of LPS before a subtoxic dose of APAP caused severe but delayed liver injury.95 The interpretation was that LPS caused a leftward shift of toxicity implying the same mechanism of toxicity just with a lower dose of APAP in the presence of LPS pretreatment.95 However the time course of toxicity with LPS suggests a different mechanism. Pretreatment with LPS will cause the recruitment of primed neutrophils into the liver vasculature.25 The subtoxic dose of APAP still causes GSH depletion and protein adduct formation (McGill and Jaeschke, unpublished data), which in the centrilobular area is likely a sufficient stress signal for the neutrophils to extravaste and attack39 resulting in a neutrophil-mediated liver injury. Thus, a high dose of APAP and low dose of APAP with LPS pretreatment have fundamentally different mechanisms of liver injury.
Role of autophagy in DILI
As mentioned previously autophagy could play a major role in the adaptation process to prevent the progression of IDILI. It is becoming more apparent that autophagy is critical for various functions in both the innate and adaptive immune response. Current investigations involve the modulation of autophagy in vaccine efficacy and chronic inflammatory disease. Dysfunction or altered regulation of autophagy can lead to loss of immune tolerance, which is most likely a critical component to the induction and progression of IDILI. Autophagy has a multitude of immune functions.96,97 These functions include digestion and removal of intracellular pathogens, promotion of Th1-Th2 polarization during intracellular infection, modulation of TLR response and inflammation via pathogen associated molecular pattern (PAMP) delivery, presentation of endogenous peptides to MHC-II molecules, regulation of T cell homeostasis, and immune tolerance. Autophagy can result in the “non-classical” MHC class II presentation of autophagosomal peptides98 and self-antigen presentation on MHC-II by DCs might be responsible for CD4+ mediated peripheral tolerance and immature dendritic cells (like those observed in the liver) have a high rate of autophagy and are considered pro-tolerogenic.96 Following pattern recognition receptor signaling, autophagy is enhanced thereby promoting non-classical MHC-II presentation.97 Interestingly autophagy has been shown to be critical for type I interferon production, however, loss of autophagy can also up-regulate several pro-inflammatory cytokines and generation of ROS in macrophages.99 Autophagy is also critical in T cell homeostasis and activation. It has been shown that autophagy is induced during T-cell activation and it has been reported that it is required for clonal expansion.100,101 In this regard, autophagy promotes T cell expansion. Conversely, if autophagy is blocked the immune synapse (MHC-TCR interaction) can become hyperstablized. Ultimately this leads to enhanced T cell activation.102In addition to the potential implications of autophagy within the immune system autophagy within the liver could be, equally, if not more important. Autophagy of damaged mitochondria or other cellular organelles could be critical to limit oxidant stress or ER stress as was previously described in the “failure to adapt” section. The role of autophagy and DILI has only very recently been evaluated.
Just this year several papers have shown the role of autophagy during APAP overdose however caution must be used when using these models as we will discuss in more detail. The primary hypothesis regarding the role of autophagy after APAP is that of hepatoprotection which is not surprising since autophagy is normally considered a cell survival pathway. Following overdose, APAP adducts cellular proteins potentially making some of them non-functional; of particular interest is the adduction of mitochondrial proteins ultimately resulting in mitochondrial dysfunction. As some mitochondria begin to fail an oxidant stress is generated, mitochondrial membrane potential is lost, and mitochondria swell then rupture releasing endonucleases; the combination of these events ultimately lead to cell death by oncotic necrosis.29,86,89 During this process there must be some critical threshold for mitochondrial loss that determines cell fate; the most centrilobular hepatocytes which have the highest P450 enzyme activities and lowest GSH levels presumably have the highest reactive metabolite burden and therefore are the most susceptible. Depending on the dose, the centrilobular injury can expand and potentially invade midzonal or even periportal areas, however the injury always begins with the most centrilobular hepatocytes. The hepatocytes at the threshold between necrotic and healthy tissue are the cells where autophagy plays a role. These cells remove damaged mitochondria and adducted proteins thereby preventing lethal oxidant stress and the release of endonucleases allowing cell survival.
The first paper investigating the role of autophagy during APAP overdose demonstrated that APAP does in fact activate autophagy and this activation is protective.103 This was determined by using both inhibitors (3-methyladenine or chloroquine) and activators (rapamycin) of autophagy in vivo and in primary cultured mouse hepatocytes. The predominant mechanism of protection appears to be mitophagy (autophagy of mitochondria) which is then capable of reducing APAP-induced injury.
Most recently it was shown that mice deficient for Atg5 in the liver were actually resistant to APAP toxicity.104 This finding is contradictory to the dogma that autophagy is protective, but it was determined that the protection from APAP toxicity was due to the aberrant phenotype seen in these mice. Atg5 was deleted from hepatocytes under the albumin-Cre promoter. The Atg5 liver knockout mice develop mild liver injury resulting in increased apoptosis and compensatory proliferation. Additionally the liver becomes preconditioned and Nrf2 activation increases the expression of target genes which increases basal GSH levels, and therefore these mice are protected by compensatory effects rather than by loss of Atg5. Clinically the induction of Nrf-2 could be indicative of a “danger signal” because it is activated by oxidant stress or the induction of autophagy.
Recently, another study has demonstrated that loss of autophagy can promote APAP toxicity. In this study Atg7 conditional knockout animals were generated under Mx1-Cre by treating mice with poly I:C.105 The poly I:C treatment is capable of modulating the metabolic activation of APAP, and this study shows a mechanism of cell death in the Atg7-deficient mice quite different from control mice, however. The Atg7-deficient mice have caspase processing and apoptosis. It has previously been reported that the poly I:C treatment in Atg7-Mx1-Cre mice results in hepatomegaly and liver injury.106 The abnormal phenotype of these mice is a major concern, therefore, care must be used when interpreting data from these types of models.107
The role of autophagy and DILI is now beginning to be established. Potentially, autophagy might be critical in the clearance of adducted proteins, removal of damaged organelles or modulation of immune tolerance (Fig. 1).
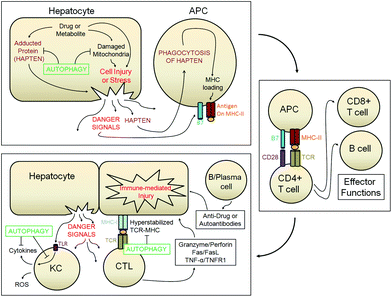 | ||
| Fig. 1 There are multiple hypotheses regarding the initiation and propagation of IDILI. This illustration depicts how several of these events could occur and potential key events that could involve autophagy. Autophagy could clear adducted or nonfunctional proteins or could remove damaged organelles. If these processes are insufficient drug or metabolite could cause cell stress or death within the hepatocyte. This could then trigger an immune response and activation. This activation could then trigger various downstream effector functions in additional immune cells. Propagation of injury can then occur through these adaptive or innate immune responses. Autophagy could potentially attenuate these responses through the modulation of TLR/ligand interaction or destabilization of the immune synapse. These roles of autophagy during IDILI are speculative however convincing evidence of the participation of autophagy during APAP overdose has been shown. The figure is a modification of the concepts previously described.72 | ||
Conclusions
During APAP overdose there are several critical stages which can be defined as initiation and propagation. The initiating event is reactive metabolite formation resulting in protein adduction. The injury is then propagated by mitochondrial dysfunction and ROS production. In APAP overdose, both of these events occur independently of immune cell infiltration27 and depletion of resident immune cells (KCs and DCs) actually causes increased injury.57,60,108 A different situation most likely occurs in IDILI, but this toxicity probably involves initiation and propagation stages as well (Fig. 1). The initiation stage likely involves the generation of reactive metabolites or the parent drug impairs some cell function (mitochondrial respiration or autophagy); this could lead to formation of a hapten or trigger mild liver injury. The propagation stage most likely involves an immune component which then greatly enhances the liver injury. In theory the generation of reactive metabolites or location of parent drug will determine the type of immune response by the antigenic presentation via MHC-I or MHC-II. If the reactive metabolite is generated within the cell and then presented via MHC-I this will lead to CD8+ T cell recognition; however in absence of co-stimulatory factors this should result in immune tolerance. If the reactive metabolite is released and then phagocytosed by an APC, these antigens will be presented to CD4+ T cells via MHC-II. These helper T cells can then lead to B cell or CTL activation.72,109Clearly, there will be no easy answer to explain the cause of IDILI. As evidenced by genetic polymorphisms there are clearly risk factors involved with the development of IDILI but genetics alone do not explain the cause. No better example illustrates this point than the HLA-B*5701 genotype with flucloxacillin, which greatly increases the risk but is by no means the determinant of DILI.14 There are multiple factors involved like dose of drug, gender, age, alcohol or concomitant drug use, underlying disease state, environmental exposures, and potentially other as yet unidentified risk factors.110,111 Additional risk could be attributed to enhanced production or impaired detoxification of oxidant stress, or altered mitochondrial respiration due to inherited or acquired mitochondrial dysfunction.112 Because of these complexities most likely not all IDILI will share the same mechanism of toxicity. Therefore, uncovering new risk factors for patients and trying to develop animal models of IDILI is critical to reduce this human health concern.
References
- W. M. Lee, R. H. Squires Jr., S. L. Nyberg, E. Doo and J. H. Hoofnagle, Hepatology, 2008, 47, 1401–1415 CrossRef.
- H. J. Zimmerman, Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver, Lippincott Williams & Wilkins, Philadelphia, 2nd edn, 1999 Search PubMed.
- M. Chen, V. Vijay, Q. Shi, Z. Liu, H. Fang and W. Tong, Drug Discovery Today, 2011, 16, 697–703 CrossRef.
- R. J. Fontana, L. B. Seeff, R. J. Andrade, E. Bjornsson, C. P. Day, J. Serrano and J. H. Hoofnagle, Hepatology, 2010, 52, 730–742 CrossRef.
- N. Chalasani, R. J. Fontana, H. L. Bonkovsky, P. B. Watkins, T. Davern, J. Serrano, H. Yang and J. Rochon, Gastroenterology, 2008, 135, 1924–1934 CrossRef , 1934 e1921–1924.
- CDER, Drug-induced Liver Injury: Premarketing Clinical Evaluation, 2009. http://www.fda.gov/downloads/Drugs?GuidanceComplianceRegulatoryInformation/Guidances/UCM174090.pdf.
- S. Verma and N. Kaplowitz, Gut, 2009, 58, 1555–1564 CrossRef CAS.
- J. Uetrecht, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 513–539 CrossRef CAS.
- J. Uetrecht, Semin. Liver Dis., 2009, 29, 383–392 CrossRef CAS.
- X. Zhang, F. Liu, X. Chen, X. Zhu and J. Uetrecht, Drug Metab. Pharmacokinet., 2011, 26, 47–59 CrossRef CAS.
- N. Kaplowitz, Nat. Rev. Drug Discovery, 2005, 4, 489–499 CrossRef CAS.
- M. I. Lucena, R. J. Andrade, C. Martinez, E. Ulzurrun, E. Garcia-Martin, Y. Borraz, M. C. Fernandez, M. Romero-Gomez, A. Castiella, R. Planas, J. Costa, S. Anzola and J. A. Agundez, Hepatology, 2008, 48, 588–596 CrossRef.
- A. K. Daly, P. T. Donaldson, P. Bhatnagar, Y. Shen, I. Pe'er, A. Floratos, M. J. Daly, D. B. Goldstein, S. John, M. R. Nelson, J. Graham, B. K. Park, J. F. Dillon, W. Bernal, H. J. Cordell, M. Pirmohamed, G. P. Aithal and C. P. Day, Nat. Genet., 2009, 41, 816–819 CrossRef CAS.
- A. K. Daly and C. P. Day, Drug Metab. Rev., 2012, 44, 116–126 CrossRef CAS.
- I. N. Crispe, M. Giannandrea, I. Klein, B. John, B. Sampson and S. Wuensch, Immunol. Rev., 2006, 213, 101–118 CrossRef.
- B. John and I. N. Crispe, J. Immunol, 2004, 172, 5222–5229 CAS.
- W. Z. Mehal, A. E. Juedes and I. N. Crispe, J. Immunol., 1999, 163, 3202–3210 CAS.
- S. Norris, C. Collins, D. G. Doherty, F. Smith, G. McEntee, O. Traynor, N. Nolan, J. Hegarty and C. O'Farrelly, J. Hepatol., 1998, 28, 84–90 CrossRef CAS.
- S. Norris, D. G. Doherty, C. Collins, G. McEntee, O. Traynor, J. E. Hegarty and C. O'Farrelly, Hum. Immunol., 1999, 60, 20–31 CrossRef CAS.
- D. G. Doherty and C. O'Farrelly, Immunol. Rev., 2000, 174, 5–20 CrossRef CAS.
- B. Gao, S. Radaeva and O. Park, J. Leukocyte. Biol., 2009, 86, 513–528 CAS.
- H. Jaeschke, Am. J. Physiol., 1997, 273, G602–G611 CAS.
- W. Y. Lee and P. Kubes, J. Hepatol., 2008, 48, 504–512 CrossRef CAS.
- B. McDonald, E. F. McAvoy, F. Lam, V. Gill, C. de la Motte, R. C. Savani and P. Kubes, J. Exp. Med., 2008, 205, 915–927 CrossRef CAS.
- J. G. Chosay, N. A. Essani, C. J. Dunn and H. Jaeschke, Am. J. Physiol., 1997, 272, G1195–G1200 CAS.
- D. H. Adams, C. Ju, S. K. Ramaiah, J. Uetrecht and H. Jaeschke, Toxicol. Sci., 2010, 115, 307–321 CrossRef CAS.
- H. Jaeschke, C. D. Williams, A. Ramachandran and M. L. Bajt, Liver Int., 2012, 32, 8–20 CrossRef CAS.
- M. A. Tirmenstein and S. D. Nelson, J. Biol. Chem., 1989, 264, 9814–9819 CAS.
- H. Jaeschke and M. L. Bajt, Toxicol. Sci., 2006, 89, 31–41 CrossRef CAS.
- M. L. Bajt, C. Cover, J. J. Lemasters and H. Jaeschke, Toxicol. Sci., 2006, 94, 217–225 CrossRef CAS.
- J. S. Gujral, T. R. Knight, A. Farhood, M. L. Bajt and H. Jaeschke, Toxicol. Sci., 2002, 67, 322–328 CrossRef CAS.
- D. H. Adams, B. Eksteen and S. M. Curbishley, Gut, 2008, 57, 838–848 CrossRef CAS.
- Z. X. Liu, S. Govindarajan and N. Kaplowitz, Gastroenterology, 2004, 127, 1760–1774 CrossRef CAS.
- M. J. Masson, L. D. Carpenter, M. L. Graf and L. R. Pohl, Hepatology, 2008, 48, 889–897 CrossRef CAS.
- S. B. Yee, M. Bourdi, M. J. Masson and L. R. Pohl, Chem. Res. Toxicol., 2007, 20, 734–744 CrossRef CAS.
- R. Miyakawa, T. Ichida, S. Yamagiwa, C. Miyaji, H. Watanabe, Y. Sato, H. Yokoyama, C. Tsukada, Y. Ishimoto, S. Sugahara, X. H. Yang, T. Abo and H. Asakura, J. Gastroenterol. Hepatol., 2005, 20, 1126–1130 CrossRef.
- B. Eksteen, S. C. Afford, S. J. Wigmore, A. P. Holt and D. H. Adams, Semin. Liver Dis., 2007, 27, 351–366 CrossRef CAS.
- M. L. Bajt, A. Farhood and H. Jaeschke, Am. J. Physiol. Gastrointest. Liver Physiol., 2001, 281, G1188–G1195 CAS.
- H. Jaeschke, Am. J. Physiol.: Gastrointest. Liver Physiol., 2006, 290, G1083–G1088 CrossRef CAS.
- S. Yamashiro, H. Kamohara, J. M. Wang, D. Yang, W. H. Gong and T. Yoshimura, J. Leukocyte Biol., 2001, 69, 698–704 CAS.
- Y. Ishida, T. Kondo, A. Kimura, K. Tsuneyama, T. Takayasu and N. Mukaida, Eur. J. Immunol., 2006, 36, 1028–1038 CrossRef CAS.
- Z. X. Liu, D. Han, B. Gunawan and N. Kaplowitz, Hepatology, 2006, 43, 1220–1230 CrossRef CAS.
- P. E. Marques, S. S. Amaral, D. A. Pires, L. L. Nogueira, F. M. Soriani, B. H. Freire Lima, G. A. Oliveira Lopes, R. C. Russo, T. V. Avila, J. G. Melgaco, A. G. Oliveira, M. A. Pinto, C. X. Lima, A. M. de Paula, D. C. Cara, M. F. Leite, M. M. Teixeira and G. B. Menezes, Hepatology, 2012 DOI:10.1002/hep.25801.
- C. Cover, J. Liu, A. Farhood, E. Malle, M. P. Waalkes, M. L. Bajt and H. Jaeschke, Toxicol. Appl. Pharmacol., 2006, 216, 98–107 CrossRef CAS.
- H. Jaeschke, Hepatology, 2008, 48, 699–701 CrossRef.
- H. Jaeschke, M. R. McGill and C. D. Williams, Hepatology, 2012 DOI:10.1002/hep.25877.
- C. D. Williams, M. L. Bajt, A. Farhood and H. Jaeschke, Liver Int., 2010, 30, 1280–1292 CrossRef CAS.
- C. D. Williams, A. Farhood and H. Jaeschke, Toxicol. Appl. Pharmacol., 2010, 247, 169–178 CrossRef CAS.
- J. A. Lawson, A. Farhood, R. D. Hopper, M. L. Bajt and H. Jaeschke, Toxicol. Sci., 2000, 54, 509–516 CrossRef CAS.
- L. P. James, S. S. McCullough, T. R. Knight, H. Jaeschke and J. A. Hinson, Free Radical Res., 2003, 37, 1289–1297 CrossRef CAS.
- X. Wang, R. Sun, H. Wei and Z. Tian, Hepatology, 2012 DOI:10.1002/hep.25982.
- Q. You, L. Cheng, T. P. Reilly, D. Wegmann and C. Ju, Hepatology, 2006, 44, 1421–1431 CrossRef CAS.
- L. Cheng, Q. You, H. Yin, M. P. Holt and C. Ju, Biochem. Pharmacol., 2010, 80, 255–261 CrossRef CAS.
- M. P. Callery, T. Kamei and M. W. Flye, Transplantation, 1989, 47, 1092–1094 CrossRef CAS.
- C. Ju and L. R. Pohl, Toxicology, 2005, 209, 109–112 CrossRef CAS.
- M. P. Holt, L. Cheng and C. Ju, J. Leukocyte Biol., 2008, 84, 1410–1421 CrossRef CAS.
- C. Ju, T. P. Reilly, M. Bourdi, M. F. Radonovich, J. N. Brady, J. W. George and L. R. Pohl, Chem. Res. Toxicol., 2002, 15, 1504–1513 CrossRef CAS.
- M. Bourdi, Y. Masubuchi, T. P. Reilly, H. R. Amouzadeh, J. L. Martin, J. W. George, A. G. Shah and L. R. Pohl, Hepatology, 2002, 35, 289–298 CrossRef CAS.
- Y. Masubuchi, M. Bourdi, T. P. Reilly, M. L. Graf, J. W. George and L. R. Pohl, Biochem. Biophys. Res. Commun., 2003, 304, 207–212 CrossRef CAS.
- M. K. Connolly, D. Ayo, A. Malhotra, M. Hackman, A. S. Bedrosian, J. Ibrahim, N. E. Cieza-Rubio, A. H. Nguyen, J. R. Henning, M. Dorvil-Castro, H. L. Pachter and G. Miller, Hepatology, 2011, 54, 959–968 CrossRef CAS.
- J. E. Sims and D. E. Smith, Nat. Rev. Immunol., 2010, 10, 89–102 CrossRef CAS.
- A. B. Imaeda, A. Watanabe, M. A. Sohail, S. Mahmood, M. Mohamadnejad, F. S. Sutterwala, R. A. Flavell and W. Z. Mehal, J. Clin. Invest., 2009, 119, 305–314 CAS.
- R. Hoque, M. A. Sohail, S. Salhanick, A. F. Malik, A. Ghani, S. C. Robson and W. Z. Mehal, Am. J. Physiol.: Gastrointest. Liver Physiol., 2012, 302, G1171–G1179 CrossRef CAS.
- C. D. Williams, D. J. Antoine, P. J. Shaw, C. Benson, A. Farhood, D. P. Williams, T. D. Kanneganti, B. K. Park and H. Jaeschke, Toxicol. Appl. Pharmacol., 2011, 252, 289–297 CrossRef CAS.
- P. Matzinger, Annu. Rev. Immunol., 1994, 12, 991–1045 CrossRef CAS.
- K. L. Rock and L. Shen, Immunol. Rev., 2005, 207, 166–183 CrossRef CAS.
- D. R. Green, T. Ferguson, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2009, 9, 353–363 CrossRef CAS.
- T. S. Griffith, H. Kazama, R. L. VanOosten, J. K. Earle, Jr., J. M. Herndon, D. R. Green and T. A. Ferguson, J. Immunol., 2007, 178, 2679–2687 CAS.
- D. J. Antoine, D. P. Williams, A. Kipar, R. E. Jenkins, S. L. Regan, J. G. Sathish, N. R. Kitteringham and B. K. Park, Toxicol. Sci., 2009, 112, 521–531 CrossRef CAS.
- H. Kazama, J. E. Ricci, J. M. Herndon, G. Hoppe, D. R. Green and T. A. Ferguson, Immunity, 2008, 29, 21–32 CrossRef CAS.
- D. J. Antoine, R. E. Jenkins, J. W. Dear, D. P. Williams, M. R. McGill, M. R. Sharpe, D. G. Craig, K. J. Simpson, H. Jaeschke and B. K. Park, J. Hepatol., 2012, 56, 1070–1079 CrossRef CAS.
- J. P. Uetrecht, Chem. Res. Toxicol., 1999, 12, 387–395 CrossRef CAS.
- Z. X. Liu and N. Kaplowitz, Clin. Liver Dis., 2002, 6, 755–774 CrossRef.
- M. P. Manns and P. Obermayer-Straub, Hepatology, 1997, 26, 1054–1066 CrossRef CAS.
- T. Mizutani, M. Shinoda, Y. Tanaka, T. Kuno, A. Hattori, T. Usui, N. Kuno and T. Osaka, Drug Metab. Rev., 2005, 37, 235–252 CAS.
- J. Li and J. P. Uetrecht, Handb. Exp. Pharmacol., 2010, 493–509 CrossRef.
- B. K. Park, A. Boobis, S. Clarke, C. E. Goldring, D. Jones, J. G. Kenna, C. Lambert, H. G. Laverty, D. J. Naisbitt, S. Nelson, D. A. Nicoll-Griffith, R. S. Obach, P. Routledge, D. A. Smith, D. J. Tweedie, N. Vermeulen, D. P. Williams, I. D. Wilson and T. A. Baillie, Nat. Rev. Drug Discovery, 2011, 10, 292–306 CrossRef CAS.
- D. P. Williams, Toxicology, 2006, 226, 1–11 CrossRef CAS.
- M. J. Masson, R. A. Peterson, C. J. Chung, M. L. Graf, L. D. Carpenter, J. L. Ambroso, D. L. Krull, J. Sciarrotta and L. R. Pohl, Chem. Res. Toxicol., 2007, 20, 20–26 CrossRef CAS.
- J. Adam, W. J. Pichler and D. Yerly, Br. J. Clin. Pharmacol., 2011, 71, 701–707 CrossRef CAS.
- B. Schnyder, D. Mauri-Hellweg, M. Zanni, F. Bettens and W. J. Pichler, J. Clin. Invest., 1997, 100, 136–141 CrossRef CAS.
- U. A. Boelsterli and P. L. Lim, Toxicol. Appl. Pharmacol., 2007, 220, 92–107 CrossRef CAS.
- A. Srivastava, J. L. Maggs, D. J. Antoine, D. P. Williams, D. A. Smith and B. K. Park, Handb. Exp. Pharmacol., 2010, 165–194 CrossRef CAS.
- C. Cover, A. Mansouri, T. R. Knight, M. L. Bajt, J. J. Lemasters, D. Pessayre and H. Jaeschke, J. Pharmacol. Exp. Ther., 2005, 315, 879–887 CrossRef CAS.
- T. R. Knight, M. W. Fariss, A. Farhood and H. Jaeschke, Toxicol. Sci., 2003, 76, 229–236 CrossRef CAS.
- H. Jaeschke, M. R. McGill, C. D. Williams and A. Ramachandran, Life Sci., 2011, 88, 737–745 CrossRef CAS.
- K. Fujimoto, K. Kumagai, K. Ito, S. Arakawa, Y. Ando, S. Oda, T. Yamoto and S. Manabe, Toxicol. Pathol., 2009, 37, 193–200 CrossRef CAS.
- A. Ramachandran, M. Lebofsky, S. A. Weinman and H. Jaeschke, Toxicol. Appl. Pharmacol., 2011, 251, 226–233 CrossRef CAS.
- H. Jaeschke, M. R. McGill and A. Ramachandran, Drug Metab. Rev., 2012, 44, 88–106 CrossRef CAS.
- M. M. Ong, C. Latchoumycandane and U. A. Boelsterli, Toxicol. Sci., 2007, 97, 205–213 CrossRef CAS.
- R. Kashimshetty, V. G. Desai, V. M. Kale, T. Lee, C. L. Moland, W. S. Branham, L. S. New, E. C. Chan, H. Younis and U. A. Boelsterli, Toxicol. Appl. Pharmacol., 2009, 238, 150–159 CrossRef CAS.
- M. I. Lucena, E. Garcia-Martin, R. J. Andrade, C. Martinez, C. Stephens, J. D. Ruiz, E. Ulzurrun, M. C. Fernandez, M. Romero-Gomez, A. Castiella, R. Planas, J. A. Duran, A. M. De Dios, C. Guarner, G. Soriano, Y. Borraz and J. A. Agundez, Hepatology, 2010, 52, 303–312 CrossRef CAS.
- R. A. Roth and P. E. Ganey, Crit. Rev. Toxicol., 2011, 41, 723–739 CrossRef CAS.
- J. C. Marshall, P. M. Walker, D. M. Foster, D. Harris, M. Ribeiro, J. Paice, A. D. Romaschin and A. N. Derzko, Crit. Care, 2002, 6, 342–348 CrossRef.
- J. F. Maddox, C. J. Amuzie, M. Li, S. W. Newport, E. Sparkenbaugh, C. F. Cuff, J. J. Pestka, G. H. Cantor, R. A. Roth and P. E. Ganey, J. Toxicol. Environ. Health, Part A, 2010, 73, 58–73 CrossRef CAS.
- D. Schmid and C. Munz, Immunity, 2007, 27, 11–21 CrossRef CAS.
- V. Deretic, Curr. Opin. Immunol., 2009, 21, 53–62 CrossRef CAS.
- C. Paludan, D. Schmid, M. Landthaler, M. Vockerodt, D. Kube, T. Tuschl and C. Munz, Science, 2005, 307, 593–596 CrossRef CAS.
- H. W. Virgin and B. Levine, Nat. Immunol., 2009, 10, 461–470 CrossRef CAS.
- H. H. Pua, I. Dzhagalov, M. Chuck, N. Mizushima and Y. W. He, J. Exp. Med., 2007, 204, 25–31 CrossRef CAS.
- C. M. Walsh and A. L. Edinger, Immunol. Rev., 2010, 236, 95–109 CrossRef CAS.
- M. E. Wildenberg, A. C. Vos, S. C. Wolfkamp, M. Duijvestein, A. P. Verhaar, A. A. Te Velde, G. R. van den Brink and D. W. Hommes, Gastroenterology, 2012, 142, 1493–1503 CrossRef CAS.
- H. M. Ni, A. Bockus, N. Boggess, H. Jaeschke and W. X. Ding, Hepatology, 2012, 55, 222–232 CrossRef CAS.
- H. M. Ni, N. Boggess, M. R. McGill, M. Lebofsky, P. Borude, U. Apte, H. Jaeschke and W. X. Ding, Toxicol. Sci., 2012, 127, 438–450 CrossRef CAS.
- Y. Igusa, S. Yamashina, K. Izumi, Y. Inami, H. Fukada, M. Komatsu, K. Tanaka, K. Ikejima and S. Watanabe, J. Gastroenterol., 2012, 47, 433–443 CrossRef CAS.
- M. Komatsu, H. Kurokawa, S. Waguri, K. Taguchi, A. Kobayashi, Y. Ichimura, Y. S. Sou, I. Ueno, A. Sakamoto, K. I. Tong, M. Kim, Y. Nishito, S. Iemura, T. Natsume, T. Ueno, E. Kominami, H. Motohashi, K. Tanaka and M. Yamamoto, Nat. Cell. Biol., 2010, 12, 213–223 CAS.
- H. Jaeschke and W. X. Ding, J. Gastroenterol., 2012, 47, 845–846 CrossRef CAS.
- C. Ju and T. Reilly, Drug Metab. Rev., 2012, 44, 107–115 CrossRef CAS.
- B. K. Park, M. Pirmohamed and N. R. Kitteringham, Chem. Res. Toxicol., 1998, 11, 969–988 CrossRef CAS.
- R. J. Andrade, J. A. Agundez, M. I. Lucena, C. Martinez, R. Cueto and E. Garcia-Martin, Curr. Drug Metab., 2009, 10, 956–970 CrossRef CAS.
- N. Chalasani and E. Bjornsson, Gastroenterology, 2010, 138, 2246–2259 CrossRef CAS.
- S. Russmann, A. Jetter and G. A. Kullak-Ublick, Hepatology, 2010, 52, 748–761 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |