Evolution of molecular machines: from solution to soft matter interface
Katsuhiko
Ariga
*ab,
Taizo
Mori
ab and
Jonathan P.
Hill
ab
aWorld Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, 305-0044, Japan. E-mail: ARIGA.Katsuhiko@nims.go.jp; Fax: +81-29-860-4832; Tel: +81-29-860-4597
bJapan Science and Technology Agency (JST), CREST, 1-1 Namiki, Tsukuba, 305-0044, Japan
First published on 21st November 2011
Abstract
In the molecular machine concept, single molecules and/or small molecular assemblies operate as independent machines in the ultimate miniaturization of functional systems. For the development of molecular machines, changing the medium of operation from solution to solid surface satisfies several demands for machine functions including allowing direct observation, enabling connection with devices and facilitation of sequential actions. However, the field of molecular machines on solid surfaces is still immature when compared with the sophisticated molecular mechanical systems observed in Nature. Required further developments of molecular machines include improved dynamic operations so that investigations of molecular machine functionality at dynamic interfacial media have become important. In this paper, the development of research on molecular machines in solution and at solid interfaces is described together with an introduction of recent challenges in the operation of molecular machines at soft matter interfaces.
 Katsuhiko Ariga | Katsuhiko Ariga is principal investigator of World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS). He also works as a director of the Supermolecules Group at NIMS and professor of Tokyo University of Science. His major interests are the fabrication of novel nanostructures based on molecular recognition and self-assembly, including Langmuir–Blodgett films, layer-by-layer films, and mesoporous materials. |
 Taizo Mori | Taizo Mori is a postdoctoral researcher in the Supermolecules Group at the National Institute for Materials Science (NIMS). He graduated from the Department of Polymer Chemistry at Kyoto University and obtained his doctorate in 2008. His research interests include supramolecular science, and synthesis of conjugated polymer in liquid crystal and chirality control of molecules. |
 Jonathan P. Hill | Jonathan P. Hill is a senior researcher in the Supermolecules Group at the National Institute for Materials Science (NIMS). Prior to working in NIMS, he occupied postdoctoral positions in Tokyo, University of Karlsruhe (Germany), and Osaka. His research interests include supramolecular science, chemistry of the tetrapyrroles, molecular self-assembly in the bulk state and at interfaces, and the sensing, optical and electronic properties of molecules. |
Introduction
Current technologies are focused on miniaturization of functional systems with molecular machines as one of the ultimate goals. In the concept of molecular machines, single molecules and/or small molecular assemblies operate as independent machines.1 Although research on molecular machines is still at a challenging stage and correspondingly distant from practical application, there has recently been rapid progress. Advanced specimens might be synthetic organisms in which numerous machine-like objects, such as enzymes, operate in sophisticated harmony. Nature has already developed such fine systems over billions of evolutional processes.As illustrated in Fig. 1, evolution of living creatures and development of molecular machines share one important feature. Improvements of their structures and functions are motivated by changes in their surroundings. Pioneering research on molecular machines includes molecular rotors in solutions where chemical and physical stimuli enabled specifically designed molecules to rotate in particular directions.2 These machine-like molecules have also been positioned on solid surfaces due to demands for direct observability of the machine motions. This variation in medium of operation is also important for establishing connections between molecular machine functions and device outputs and it is anticipated that second generation molecular machines immobilized on solid surfaces will present a new hot topic in this research field.
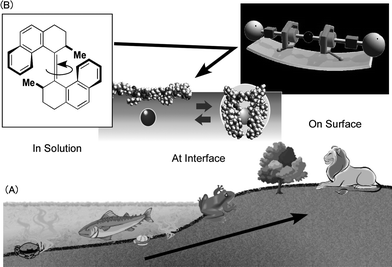 | ||
| Fig. 1 Evolution processes for (A) living creatures and (B) molecular machines. | ||
Immobilization of molecular machines may cause certain disadvantages including restriction of mechanical activity. Unlike artificial molecular machines, molecular machines in Nature such as proteins and their assemblies operate at dynamic interfaces such as surfaces of cell membranes. Therefore, bringing our molecular machines to a soft matter interface could cause the next evolution in molecular machine science.3 In this paper, these stages of molecular machine evolution are highlighted. First, we describe progress of molecular machine research in solution state and at solid interfaces, and then we introduce recent challenges in the operation of molecular machines at soft matter interfaces.
Molecular machines in solutions
The concept of molecular machines was realized using selected molecules in solution. Their machine-like motions could be analyzed spectroscopically at certain concentrations although analysis at the single molecule level is near impossible. Motions of molecular machines must be easily controllable and random operations should not occur. A pioneering example of rotational molecular motion is shown in Fig. 2A.2a Sequential application of light and thermal stimuli causes directional rotation in overcrowded alkene molecules. Axial chirality with two stereogenic centers and photoinduced isomerization are essential for the unidirectional rotation. This can be a good analogy of a biomolecular motor because this nanometre-sized molecular motor is driven by external energy sources similarly to a ten-nanometre-sized ATP-synthase motor driven by proton flow (see Fig. 2A). Other pioneering examples of molecular machines are molecular shuttles where translational motion of molecules along a molecular axis is realized (Fig. 2B).4 A viologen-based cyclophane ring (an electron acceptor) is threaded by a molecular wire containing two kinds of biphenyl groups (or ‘stations’). The viologen ring positions itself near the benzidine moiety on the wire due to its stronger electron-donating ability. Oxidation of the benzidine unit to a cation radical form induces the viologen ring to move to the biphenol part of the wire due to electrostatic repulsion. Molecular machines whose components can undergo externally stimulated predictable movements between molecular stations are called molecular shuttles.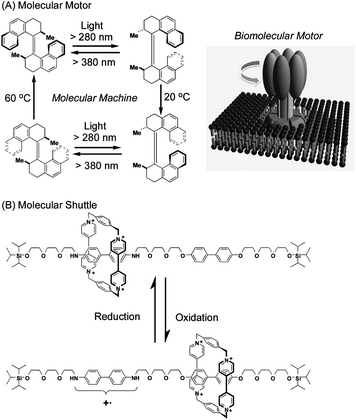 | ||
| Fig. 2 Examples of molecular machines in solution: (A) molecular motor (analogous to biomolecular motor) and (B) molecular shuttle. | ||
The research area of molecular shuttles and molecular machines in solution is now very well established and many variations on this theme are available.5 For example, Leigh and coworkers synthesized molecular shuttles with inorganic ring components.6 Heterometallic rings containing seven or more trivalent Cr(III) ions and one or two divalent metal ions (typically Ni(II), Co(II), Fe(II) or Cu(II)) bridged by multiple fluoride and alkyl/aryl carboxylate anions were threaded by a dumb-bell-shaped organic molecule with dialkylammonium groups. Shuttling of inorganic ring components was expected to be accompanied by the variation of the electronic, magnetic and paramagnetic characteristics, which might prove to be useful as qubits for quantum computers. Jiang, Huc, and coworkers proposed a new concept for dynamically assembling molecular shuttles.7 In their design, helical molecular tapes slowly wind around a rod-like molecule and rapidly slide along it. The former winding process requires helix unfolding and refolding. The rod, which is terminated with bulky ends, forms a complex via a slow helix unfolding–refolding process, thereby allowing the helix to rapidly shuttle along the guest without dissociating. This example introduces dynamic assembly into molecular shuttle methodology. Detailed analysis on the shuttle motion has also become an important research target. Panman et al. investigated a nanosecond shuttling mechanism, which involved reversing the relative binding affinities of two stations, by monitoring changes in the CO-stretching bands of the two stations and the shuttling macrocycle using an infrared probing pulse.8 The shuttling process commences with thermally driven escape from the initial station, followed by rapid motion along the track ending either at the initial or final station. The rapid motion approximates a biased one-dimensional random walk. The ring component eventually arrives at the more thermodynamically stable station. Increasing the length of the shuttle's track made its time of arrival increasingly unpredictable.
Molecular machines at solid surfaces
After pioneering research on molecular machines in solutions had been proposed, several workers began applying this concept using solid surfaces rather than solution states.9 For instance, the Nano-car presented by Tour and coworkers is an outstanding example.10 They synthesized car-like molecules with fullerene moieties as rolling wheels connected to a rigid molecular frame as the car body. Incorporation of four fullerene units provided four-wheeled nano-cars while nano-tricycles with three fullerenes in one molecule could also be prepared. Motion (or ‘driving’) could be directly observed by scanning tunneling microscopy (STM) and confirmed translational motion for the nano-car and rotational motion for the nano-tricycle.Another motivation for fixing molecular machines at a solid surface is the collectivisation of molecular movements in order to output to artificial devices. Fig. 3 shows one such system where motions of molecular shuttles could be converted to macroscopic mechanical output of a solid object. In the approach by Stoddart and coworkers, numerous molecular shuttle units were immobilized on the surface of a cantilever of an atomic force microscope (AFM).11 Shifting the position of the shuttle unit along the molecular axis resulted in molecular-level torsions, which accumulated tension at the AFM cantilever surface causing bending of the much larger cantilever. A mean molecular force of 14 to 21 pN caused a cantilever deformation of 34 to 50 nm. Recently, Feringa and coworkers investigated performance of their molecular motor attached to a surface.12 The molecular motors were designed containing azide or alkyne groups for attachment to alkyne- or azide-modified surfaces. Immobilisation of the molecular motor at a surface sometimes reduces the rate of the thermal isomerization process. The rate of thermal isomerization was especially dependent on the surface coverage of the motors. On the other hand, the kinetics of thermal isomerization were not affected by the valency of attachment. These facts suggest that differences in the molecular motor performance are related to interactions between the surface-bound motors.
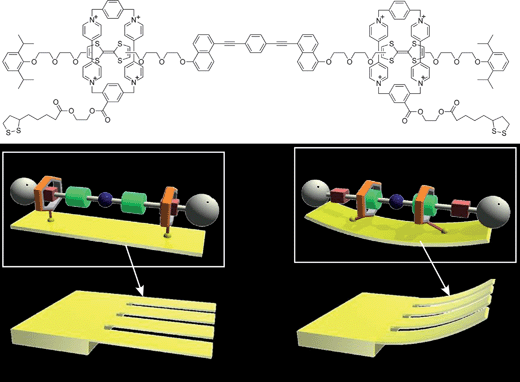 | ||
| Fig. 3 Molecular machine immobilized on a surface. In this example, molecular shuttle motion induces bending of the molecules' substrate (an AFM cantilever). | ||
Localization of molecular machines on a surface should have advantages in sequential operations compared with those randomly dispersed in solution. Several pioneering examples of sequential molecular actions have been realized based on the DNA origami concept.13 In this concept, specific nanoscale patterns can be surface immobilized upon DNA folding directed by complimentary base-pairing within specific DNA sequences. DNA nano-objects as DNA walkers can be driven along specific DNA patterns through sequential interaction between oligomeric DNA sequences. For example, Lund et al. proposed the use of a DNA spider molecule, which is comprised of a streptavidin molecule as an inert body and three deoxyribozymes as catalytic legs for robot-like behaviour under a precisely defined environment.14 The legs bind and cleave oligodeoxynucleotide substrates with a single ribose moiety into two shorter products that have lower affinities for the leg. The different substrate and product affinities control the spider's interactions with a patterned DNA on the surface, resulting in sequences of actions such as start, follow, turn and stop. Seeman and coworkers reported advanced actions of the similar DNA walker.15 They demonstrated a nanoscale assembly through addition of cassettes containing three independently controlled two-state DNA machines to the DNA walker system. The DNA walker generates the target product by moving along the track and collecting cargo from those devices that are switched on.
Molecular machines' soft matter interfaces
Immobilization of molecular machine components at an interfacial environment has certain advantages in the promotion of sequential machine-like actions. Similar sequential events can be frequently observed in biological processes such as photosynthesis, respiration, and signal transduction. These sophisticated functions are accomplished by cooperative actions of several machine-like components including receptors and proteins. As machine functions become more complex, the dynamic nature of the components seems to be more crucial. These biological machinery functions usually operate on semi-fluid biomembranes. Therefore, placing functional units at a dynamic soft matter interface appears to be a logical step in the evolution of molecular machines. Currently, development of molecular machines at solid surfaces and their molecular level observation has been intensively investigated. Observation of molecular machine operations at soft matter interfaces probably represents the next phase of investigation and some emerging examples are highlighted below.The use of soft matter as interfacial material could be advantageous for incorporation of biological machines such as enzymes. Kikuchi and coworkers immobilized synthetic receptor molecules and lactate dehydrogenase (LDH) on the surface of a dynamic lipid bilayer.16 Activity of LDH could be controlled by added metallic cations such as Cu(II). Association and dissociation of Cu(II) could be used to switch off and switch on the LDH activity. The affinity of the former receptor to Cu(II) is adjustable by applying light irradiation or a Schiff's base inducing reagent. Therefore, switching of enzymatic activities of LDH on the lipid membrane could be regulated by the presence of two factors, light and particular chemical species. Combined application of these factors made it possible to switch on the enzyme. Similar control of enzymatic activity was realized using soft and dynamic layer-by-layer (LbL) films as reported by Voegel and coworkers.17 They embedded alkaline phosphatase (ALP) in an LbL assembly within a second polyelectrolyte layer acting as a mechanically sensitive nanobarrier. Simple stretching of the LbL films induced exposure of ALP to the external interface so that biocatalysis was permitted and hydrolysis of fluorescein diphosphate molecules to fluorescein and phosphate ions commenced. This catalysis could be switched on/off through mechanical processes of soft matter.
The latter example also suggests that placing molecular machines at a dynamic interface could introduce possibilities of molecular machine operation by external macroscopic mechanical stimuli. This concept could be important for connecting macroscopic motions with molecular operations at the nanoscopic scale. Dynamically deformable interfaces have dimension-dependent properties. The direction of thickness can be defined as the Z-axis, which remains always within the nanometre length regime. In contrast, X- and Y-axes can have macroscopically visible dimensions. Coupling between stimuli in the X–Y plane with molecular motion in the Z-direction results in connection of macroscopic mechanical stimuli with molecular machine functions. A useful model environment is a Langmuir monolayer at the air–water interface where the monolayer structure is deformable over tens of centimetres and molecular interactions are limited within the interfacial thickness.18 A pioneering example is shown in Fig. 4A.19 Molecular machines embedded at an air–water interface are capable of selective capture and release of target guest molecules dissolved in the water subphase through open-cavity conversion of the machine. This molecular machine, a steroid cyclophane, can adopt ‘cavity’ and ‘open’ conformations upon monolayer compression and expansion by macroscopic mechanical forces, respectively. Guest capture upon cavity formation of the machine and guest release upon opening of the cavity are repeatable by mechanical compression and expansion of the Langmuir monolayer. The obtained results demonstrate that the dynamic interfacial environment makes it possible to control molecular machines by macroscopic mechanical motion.
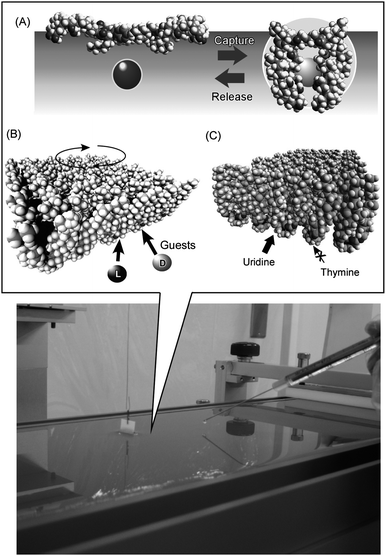 | ||
| Fig. 4 Molecular machines at the air–water interface with functions of (A) capture and release of the target molecule, (B) chiral recognition, and (C) discrimination of nucleic acid bases. | ||
This concept can be applied for mechanical optimization of receptor structure. So far, one of the important machine-like molecular functions, molecule selection, has been mostly considered using stable static formations such as X-ray crystal structures. However, the flexible nature of molecular receptors introduces the potential to adopt structures optimized for guest selection. In order to explore this possibility, molecular machines capable of molecular recognition have been embedded on dynamic interfaces of soft matter, and their best-performance structures were mechanically optimized as shown in Fig. 4B.20 In this example, variation in the twisting behaviour of a molecular machine, N-substituted cyclen containing four cholesteric side arms, resulted in chiral discrimination induced by the pairwise packing of adjacent chiral centers. Inversion of the magnitude of the binding constant between L- and D-enantiomers in the case of a subphase guest (valine) was observed. Another example shown in Fig. 4C uses cholesterol-substituted triazacyclononane as a molecular machine for discrimination of uracil from thymine.21 Mechanical optimization of the molecular machine structures for the best discrimination resulted in ca. 64 times higher binding constant for uracil than for thymine. It is notable that these nucleic acid bases cannot even be distinguished by DNA and RNA. Only specialized enzymes can perform this discrimination operation in nature. It is surprising that such a simple organic compound can perform functions corresponding to natural molecular machines. However, this feature emphasises the potential importance of the dynamic interface for operation of molecular machines.
Future perspectives
In this paper, we have briefly summarized the development of molecular machine research based on their medium of operation. Interestingly, variation of this medium bears some conceptual analogy with the shifts that have occurred during evolution of life processes. During development of molecular machines, changing from solution to a solid surface satisfies several demands of machine function including allowing direct observation, enabling connection with devices and facilitation of sequential actions. Varying the molecules environment has permitted evolution of design, structures and functions of molecular machines, perhaps in analogy with natural evolution processes. However, knowledge of molecular machines on solid surfaces remains immature compared to the sophisticated mechanical systems observed in Nature. Required developments of molecular machines include greater dynamic functionality similar to that contained in many complex cell-membrane-bound systems. The highly dynamic nature of media for molecular machines leads to several advantages including combination of multiple molecular machine functions and connection between macroscopic stimuli and molecular functions. Research on molecular machine functions at dynamic interfacial media will become an important subject because such media both satisfy conditions of surface confinement and promote dynamic motions. The examples used for illustration in this paper all use a Langmuir monolayer as the medium, which is a good medium for basic demonstration and characterization but is far from allowing practical applications partly because it lacks versatility in materials' selection. Therefore, molecular machines at dynamic interfaces should be investigated at interfaces composed of other soft matters such as polymer membranes or gels. Such research is anticipated to accelerate the evolution of molecular machines.Acknowledgements
This work was in part supported by World Premier International Research Center (WPI) Initiative on Materials Nanoarchitectonics, MEXT, Japan and JST, CREST. We thank all the people in the world who support recovery in Japan after the earthquakes in March 2011.Notes and references
- (a) A. Harada, Acc. Chem. Res., 2001, 34, 456 CrossRef CAS; (b) K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377 CrossRef CAS; (c) S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344 CrossRef CAS; (d) Y. B. Zheng, Y.-W. Yang, L. Jensen, L. Fang, B. K. Juluri, A. H. Flood, P. S. Weiss, J. F. Stoddart and T. J. Huang, Nano Lett., 2009, 9, 819 CrossRef CAS; (e) Y. B. Zheng, Q. Hao, Y.-W. Yang and T. J. Huang, J. Nanophotonics, 2010, 4, 042501 CrossRef; (f) Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124 CrossRef CAS; (g) K. Ariga, S. Ishihara, H. Izawa, H. Xia and J. P. Hill, Phys. Chem. Chem. Phys., 2011, 13, 4802 RSC; (h) Y.-W. Yang, Med. Chem. Commun., 2011, 2, 1033 RSC.
- (a) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS; (b) T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef CAS; (c) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174 CrossRef CAS; (d) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS.
- (a) K. Ariga, T. Nakanishi and J. P. Hill, Soft Matter, 2006, 2, 465 RSC; (b) K. Ariga, T. Mori and J. P. Hill, Chem. Sci., 2011, 2, 195 RSC.
- R. A. Bissell, E. Cordova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133 CrossRef CAS.
- (a) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC; (b) F. Durola, J.-P. Sauvage and O. S. Wenger, Coord. Chem. Rev., 2010, 254, 1748 CrossRef CAS.
- C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314 CrossRef CAS.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172 CrossRef CAS.
- M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, A. M. Brouwer, W. J. Buma, D. A. Leigh and S. Woutersen, Science, 2010, 328, 1255 CrossRef CAS.
- (a) E. Coronado, P. Gaviña and S. Tatay, Chem. Soc. Rev., 2009, 38, 1674 RSC; (b) D. Y. Zhong, T. Blomker, K. Wedeking, L. F. Chi, G. Erker and H. Fuchs, Nano Lett., 2009, 9, 4387 CrossRef CAS; (c) C. Manzano, W. H. Soe, H. S. Wong, F. Ample, A. Gourdon, N. Chandrasekhar and C. Joachim, Nat. Mater., 2009, 8, 576 CrossRef CAS; (d) A. Kausar, H. Nagano, T. Ogata, T. Nonaka and S. Kurihara, Angew. Chem., Int. Ed., 2009, 48, 2144 CrossRef CAS; (e) K. Miyake, M. Fukuta, M. Asakawa, Y. Hori, T. Ikeda and T. Shimizu, J. Am. Chem. Soc., 2009, 131, 17808 CrossRef CAS; (f) M. Yu, N. Kalashnyk, W. Xu, R. Barattin, Y. Benjalal, E. Laegsgaard, I. Stensgaard, M. Hliwa, X. Bouju, A. Gourdon, C. Joachim, F. Besenbacher and T. R. Linderoth, ACS Nano, 2010, 4, 4097 CrossRef CAS; (g) J. J. Davis, G. A. Orlowski, H. Rahman and P. D. Beer, Chem. Commun., 2010, 46, 54 RSC; (h) A. Coskun, P. J. Wesson, R. Klajn, A. Trabolsi, L. Fang, M. A. Olson, S. K. Dey, B. A. Grzybowski and J. F. Stoddart, J. Am. Chem. Soc., 2010, 132, 4310 CrossRef CAS; (i) J. Otsuki, Y. Komatsu, D. Kobayashi, M. Asakawa and K. Miyake, J. Am. Chem. Soc., 2010, 132, 6870 CrossRef CAS; (j) T. Takami, T. Ye, B. K. Pathem, D. P. Arnold, K. Sugiura, Y. Z. Bian, J. Z. Jiang and P. S. Weiss, J. Am. Chem. Soc., 2010, 132, 16460 CrossRef CAS; (k) D. Ecija, W. Auwarter, S. Vijayaraghavan, K. Seufert, F. Bischoff, K. Tashiro and J. V. Barth, Angew. Chem., Int. Ed., 2011, 50, 3872 CrossRef CAS; (l) E. H. Witlicki, C. Johnsen, S. W. Hansen, D. W. Silverstein, V. J. Bottomley, J. O. Jeppesen, E. W. Wong, L. Jensen and A. H. Flood, J. Am. Chem. Soc., 2011, 133, 7288 CrossRef CAS; (m) F. Xia, X. L. Zuo, R. Q. Yang, R. J. White, Y. Xiao, D. Kang, X. O. Gong, A. A. Lubin, A. Vallee-Belisle, J. D. Yuen, B. Y. B. Hsu and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 8557 CrossRef CAS; (n) F. Barka-Bouaifel, J. Niedziolka-Jonsson, X. Castel, O. Saison, A. Akjouj, Y. Pennec, B. Djafari-Rouhani, P. Woisel, J. Lyskawa, L. Sambe, G. Cooke, N. Bezzi, R. Boukherroub and S. Szunerits, J. Mater. Chem., 2011, 21, 3006 RSC; (o) Y. Xiang, X. Q. Qian, Y. Chen, Y. Y. Zhang, Y. Q. Chai and R. Yuan, Chem. Commun., 2011, 47, 2080 RSC; (p) M. M. Boyle, R. A. Smaldone, A. C. Whalley, M. W. Ambrogio, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2011, 2, 204 RSC.
- (a) Y. Shirai, A. J. Osgood, Y. Zhao, K. F. Kelly and J. M. Tour, Nano Lett., 2005, 5, 2330 CrossRef CAS; (b) Y. Shirai, J.-F. Morin, T. Sasaki, J. M. Guerrero and J. M. Tour, Chem. Soc. Rev., 2006, 35, 1043 RSC; (c) G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473 CrossRef CAS; (d) C. J. Villagomez, T. Sasaki, J. M. Tour and L. Grill, J. Am. Chem. Soc., 2010, 132, 16848 CrossRef CAS.
- (a) B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y. W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291 CrossRef CAS; (b) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745 CrossRef CAS.
- G. T. Carroll, G. London, T. F. Landaluce, P. Rudolf and B. L. Feringa, ACS Nano, 2011, 5, 622 CrossRef CAS.
- (a) Y. Ke, S. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180 CrossRef CAS; (b) E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73 CrossRef CAS.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206 CrossRef CAS.
- H. Gu, J. Chao, S.-J. Xiao and N. C. Seeman, Nature, 2010, 465, 202 CrossRef CAS.
- (a) J. Kikuchi, K. Ariga, T. Miyazaki and K. Ikeda, Chem. Lett., 1999, 253 CrossRef CAS; (b) J. Kikuchi, K. Ariga and T. Ikeda, Chem. Commun., 1999, 547 RSC; (c) J. Kikuchi, K. Ariga, Y. Sasaki and K. Ikeda, J. Mol. Catal. B: Enzym., 2001, 11, 977 CrossRef CAS; (d) K. Fukuda, Y. Sasaki, K. Ariga and J. Kikuchi, J. Mol. Catal. B: Enzym., 2001, 11, 971 CrossRef CAS.
- (a) D. Mertz, J. Hemmerlé, J. Mutterer, S. Ollivier, J.-C. Voegel, P. Schaaf and P. Lavalle, Nano Lett., 2007, 7, 657 CrossRef CAS; (b) D. Mertz, C. Vogt, J. Hemmerlé, J. Mutterer, V. Ball, J.-C. Voegel, P. Schaaf and P. Lavalle, Nat. Mater., 2009, 8, 731 CrossRef CAS.
- (a) S. Acharya, J. P. Hill and K. Ariga, Adv. Mater., 2009, 21, 2959 CrossRef CAS; (b) S. Acharya, A. Shundo, J. P. Hill and K. Ariga, J. Nanosci. Nanotechnol., 2009, 9, 3 CrossRef; (c) K. Ariga, M. V. Lee, T. Mori, X.-Y. Yu and J. P. Hill, Adv. Colloid Interface Sci., 2010, 154, 20 CrossRef CAS; (d) K. Sakakibara, J. P. Hill and K. Ariga, Small, 2011, 7, 1288 CrossRef CAS.
- (a) K. Ariga, Y. Terasaka, D. Sakai, H. Tsuji and J. Kikuchi, J. Am. Chem. Soc., 2000, 122, 7835 CrossRef CAS; (b) K. Ariga, T. Nakanishi, Y. Terasaka, H. Tsuji, D. Sakai and J. Kikuchi, Langmuir, 2005, 21, 976 CrossRef CAS; (c) K. Ariga, T. Nakanishi, J. P. Hill, Y. Terasaka, D. Sakai and J. Kikuchi, Soft Matter, 2005, 1, 132 RSC; (d) K. Ariga, T. Nakanishi, Y. Terasaka and J. Kikuchi, J. Porous Mater., 2006, 13, 427 CrossRef CAS.
- (a) T. Michinobu, S. Shinoda, T. Nakanishi, J. P. Hill, K. Fujii, T. N. Player, H. Tsukube and K. Ariga, J. Am. Chem. Soc., 2006, 128, 14478 CrossRef CAS; (b) K. Ariga, T. Michinobu, T. Nakanishi and J. P. Hill, Curr. Opin. Colloid Interface Sci., 2008, 13, 23 CrossRef CAS; (c) T. Michinobu, S. Shinoda, T. Nakanishi, J. P. Hill, K. Fujii, T. N. Player, H. Tsukube and K. Ariga, Phys. Chem. Chem. Phys., 2011, 13, 4895 RSC.
- (a) T. Mori, K. Okamoto, H. Endo, J. P. Hill, S. Shinoda, M. Matsukura, H. Tsukube, Y. Suzuki, Y. Kanekiyo and K. Ariga, J. Am. Chem. Soc., 2010, 132, 12868 CrossRef CAS; (b) T. Mori, K. Okamoto, H. Endo, K. Sakakibara, J. P. Hill, S. Shinoda, M. Matsukura, H. Tsukube, Y. Suzuki, Y. Kanekiyo and K. Ariga, Nanoscale Res. Lett., 2011, 6, 304 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |