Adsorption behaviour of sulfur containing polymers to gold surfaces using QCM-D†
Stacy
Slavin
a,
Alexander H.
Soeriyadi
b,
Lenny
Voorhaar
a,
Michael R.
Whittaker
b,
C. Remzi
Becer
a,
Cyrille
Boyer
b,
Thomas P.
Davis
b and
David M.
Haddleton
*a
aUniversity of Warwick, Department of Chemistry, Gibbet Hill Road, Coventry, CV4 7AL, United Kingdom. E-mail: d.m.haddleton@warwick.ac.uk; Fax: +44 (24765); Tel: +44 (24765) 23256
bCentre for Advanced Macromolecular Design (CAMD), School of Chemical Sciences and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 13th October 2011
Abstract
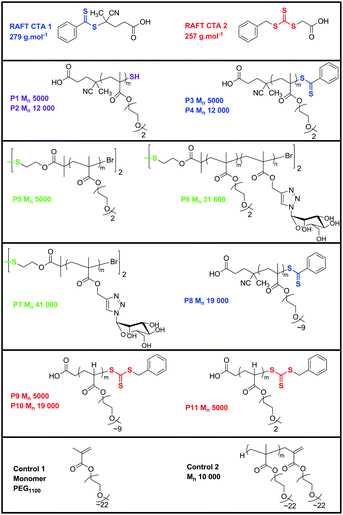
We investigate the influences that functional group, polymer molecular weight and polymer molecular architecture have on the adsorption behaviour of some sulfur containing oligo(ethylene glycol) polymers to gold. QCM-D and XPS was used in this investigation revealing that disulfide functional groups bind with more mass deposited than dithio, trithio or thiols. This was observed with small di(ethylene glycol) polymers and with higher mass polymers. The effect of the sulfo-groups was not as apparent with higher mass polymers. Longer PEG pendent chains resulted in lower binding overall on the gold surface in comparison to shorter DEG chains caused by shielding of sulfur by the longer pendent chains. Thiols undergo two steps during the adsorption process while all other sulfur species adsorb in one step. XPS revealed the dissociation of disulfide bonds when binding to gold. These findings are important when forming stable polymer films on gold efficiently, with uses in applications from bio-fouling to polymer-lipid bilayers.
Introduction
The high affinity between organosulfur compounds and a gold surface can result in self-assembled mono-layers and provides an anchoring unit onto the gold. This provides a stable covalent bond between the gold substrate and the organosulfur compound. A flow of appropriate molecules over this surface which are known to react with these compounds allows binding interactions to be studied and can be quantified using a quartz crystal microbalance with dissipation monitoring (QCM-D).There are many examples of small organosulfur compounds which coordinate to metal surfaces, including gold, platinum, silver.1–15 The study of the adsorption of these interactions is fundamental to the understanding of parameters, both structural and chemical, that influence the molecular assembly onto the surface. Previous work has shown that the electron density of the adsorbing sulphur, and the intermolecular interactions between assembled molecules, both play key roles in determining adsorption behaviour.16QCM-D has also been applied to investigate sulfo-containing macromolecules.17–24
Sulfur is inherently present in polymers synthesised via radical addition-fragmentation chain transfer (RAFT) polymerisation25 as they contain dithiobenzoates, trithiocarbonates, xanthates and dithiocarbamates as end-groups originating from the chain transfer agent.26–30 In transition metal mediated living radical polymerisations, such as atom transfer radical polymerisation (ATRP), disulfide bridges and dithiomaleimides can be incorporated into initiators31 or by post-modification of the end-group.32 Finally, the insertion of a disulfide bond can also be achieved through judicious choice of functional monomer, for example, using pyridine disulfide functionality.33–39
With the various classes of sulfur containing polymers available, we were inspired to investigate their interactions with gold in consideration of their functional group, polymer molecular weight and molecular architecture. QCM-D is a powerful tool, and emerging technique, for studying this type of interaction as the measurements are made in real-time and provide information of the film mass along with how the chains interact in solution. A detailed overview describing QCM-D can be found elsewhere.40 Finally, X-ray photoelectron spectroscopy (XPS) was used to determine the nature of the interactions between polymer end group and the gold surface.
Materials and methods
Polymer synthesis
The initiator, 2,2-azobisisobutyronitrile (AIBN) was recrystallised twice from methanol prior to use. The RAFT agents 4-cyano-4-(phenylcarbonothiolthio)pentanoic acid (CPADB) and 3-benzylsulfanylthiocarbonylsulfanyl)-propionic acid (BSPA) were synthesised as previously described.47 The initiator 4,4′-azobis(4-cyanovaleric acid) (ACVA) was used as received and the catalytic chain transfer (CCT) agent bis(methanol) complex, (CH3OH)2Co-(dmgBF2)2 (bis(boron difluorodimethylglyoximate)cobalt(II), CoBF), was synthesised according to the method used previously.48 The chain transfer agent (CTA) transfer efficiency was checked by CCT polymerisation of methyl methacrylate (MMA) which yielded a Cs value of 25 000.
RAFT polymer synthesis
Polymerisations were carried out using RAFT polymerisation employing AIBN as an initiator and CPADB and BSPA as RAFT agents. A typical polymerisation was carried out with the monomer DEGMEMA (3.0 g, 0.016 moles) mixed with AIBN (6.4 × 10−3 g, 3.9 × 10−5 moles) and CPADB (0.11 g, 3.8 × 10−4 moles) in a round bottom flask. The mixture was then thoroughly deoxygenated by purging with nitrogen gas for 20 min and then placed in an oil bath at 70 °C. The polymerisation was then stopped at a designated time and the polymer purified by precipitation into cold petroleum ether (40–60 °C). All polymers were then characterised viaGPC and NMR spectroscopy.Reduction of the polymer to free thiol
Polymers that contained RAFT agent were reduced to free thiol by aminolysis in tghe presence of n-hexyl amine (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) molar ratio to the RAFT polymer and catalytic amount of dimethylphenyl phosphine (DMPP) to prevent the formation of a disulfide (1% mol. ratio).
:1) molar ratio to the RAFT polymer and catalytic amount of dimethylphenyl phosphine (DMPP) to prevent the formation of a disulfide (1% mol. ratio).
Disulfide containing polymer synthesis
The synthesis of polymers containing a disulfide bond was based on published procedures.31 Briefly, a Schlenk tube was charged with the RAFT agent BSPA (1.0 mmol), monomer (50 mmol), N-(ethyl)-2-pyridylmethanimine (ethyl ligand) as ligand (2.0 mmol) and toluene as solvent (same volume as monomer). Two different methods were used to remove oxygen from the solution; three freeze-pump-thaw cycles and via bubbling the solution with nitrogen for at least 20 min. A second Schlenk tube was charged with CuBr (1.0 mmol) and a magnetic stirrer bar, oxygen was removed by evacuating and subsequently filling the tube with nitrogen three times. The solution was transferred to the second tube by cannulation, heated to 90 °C and kept under a nitrogen atmosphere for the duration of the reaction. Samples were taken periodically using a degassed syringe. The polymerisation was stopped by cooling and exposing it to air. The mixture was diluted with CH2Cl2 (approximately same volume as mixture) and passed through a basic alumina column. The polymer was precipitated into either hexanes, cooled with dry ice/acetone, (p(DEGMEMA)), a 5:1 vol/vol methanol/water mixture (p(TMSMA)) or water at 70 °C (p(DEGMEMA)-p(TMSMA) 8-arm diblock). The solid was isolated by filtration and dried in a vacuum oven overnight. The kinetic plot for the polymerisation of DEGMEMA shows a relatively good linear conversion up to approximately 90%.
Mannose azide clicking to deprotected p(TMSMA) polymers
A solution containing p(TMSMA) (100 mg, 0.81 mmol propargyl groups), mannose azide (213 mg, 1.05 mmol) and CuBr (116 mg, 0.81 mmol) in DMSO (10 mL) was bubbled with nitrogen for 20 min. Ethyl ligand (216 mg, 1.61 mmol) was added and the solution was bubbled with nitrogen for several more minutes. The solution was subsequently stirred at ambient temperature for two days. The mixture was purified by dialysis (MWCO: 1,000 g mol−1) against distilled water for two days, while changing the water at least four times. It was then concentrated under reduced pressure and freeze-dried overnight.Control polymer p(PEGMEMA1100)
Methanol (30 mL) and demineralised water (46 mL) were placed into a round bottomed flask with a magnetic stirrer bar, sealed with a suba-seal, parafilm and purged with nitrogen for 2 h prior to the experiment starting. Poly(ethylene glycol) methyl ether methacrylate (PEGMEMA)1100 monomer (20 g, 0.018 mol, 1100 g mol−1), initiator 4,4′-azobis(4-cyano-valeric acid) (ACVA) (0.025 g, 9 × 10−5 mol, 280.28 g mol−1) and CoBF as chain transfer agent (0.45 mg, 9 × 10−7 mol, 503 g. mol−1) were placed into a round bottom flask, sealed with a suba-seal, parafilm and purged with nitrogen for 2 h. The solvent mixture was cannulated into the round bottom flask containing ACVA, CoBF and monomer, and the solution was heated to 80 °C in an oil bath with a fuzzy-logic temperature controller. After four hours the reaction was removed from the oil bath and air was bubbled through the reaction solution. The remaining monomer and solvent in the reaction were removed using dialysis tubing with a MWCO: 1,000 g mol−1.QCM-D
The Q-Sense E4 System: QE 401 Electronics Unit, QCP 401 Chamber Platform, QFM 401 Flow Module with Ismatec IPC-N Pump. Au coated quartz sensor (Frequency: 4.95 MHz ± 50 kHz; Cut: AT; Diameter: 14 mm; Thickness: 0.3 mm; Finish: Optically polished, surface roughness of electrode less than 3 nm (RMS); Electrode layer: 10–300 nm). All measurements were carried out in at least duplicate, at 20 °C, harmonics from f(3) and D(3) through to f(11) and D(11) were recorded to follow the polymer adsorption (10 mg mL−1) to the surface. f(5) was used to calculate mass adsorbed via Sauerbrey's equation and all harmonics were modelled using Voigt model. In both cases the last data point from f(5) was used. Before use, all QCM-D sensors were cleaned using piranha solution, rinsed with water, ethanol and dried under nitrogen gas.X-ray photoelectron spectrometer (XPS)
A Kratos Axis ULTRA XPS incorporating a 165 mm hemispherical electron energy analyser was used. The incident radiation was monochromatic A1 X-rays (1486.6 eV) at 225 W (15 kV, 15 mA). Survey (wide) scans were taken at analyser pass energy of 160 eV and multiplex (narrow) higher resolution scans at 20 eV. Survey scans were carried out over 1200–0 eV binding energy range with 1.0 eV steps and a dwell time of 100 ms. Narrow higher resolution scans were run with 0.2 eV steps and 250 ms dwell time. Base pressure in the analysis chamber was 1.0 × 10−9 Torr and during sample analysis 1.0 × 10−8 Torr and the data analysed by XPS peak 4.1.Results and discussion
A series of specifically designed sulfur functional oligo(ethylene glycol) polymers were synthesised and their adsorption onto a gold surface examined. The mass adsorbed, an estimated surface coverage and apparent thickness of each polymer film was calculated using the mass obtained from the Sauerbrey relation (Tables 1 and 2, eqn (1)). The effects of a range of chemical and structural variables including, sulfur functional groups studied including thiol, disulfide, dithiocarbonate and trithiocarbonate, pendent chain length and polymer molecular weight, were determined by QCM-D. QCM-D was used to study the adsorption profile of each polymer with each solution tested several times. The adsorbed mass was calculated using two methods: Sauerbrey's relation (eqn (1)) and Voigt model (eqn (2)).
|
| Entry | Monomer used | Sulfur Moiety | Mn/g mol−1 | Mass/ng cm2 | Surface coverage/nmol cm2 | Thickness/pm | |
|---|---|---|---|---|---|---|---|
| Sauerbrey | Model | ||||||
| RAFT CPADB | n/a |

|
279 | 263 | 565 | 0.94 | 0.25 |
| RAFT BSPA | n/a |

|
257 | 88 | 114 | 0.34 | 0.08 |
| P1 | DEGMEMA |
|
5000 | 140 | 173 | 0.03 | 0.13 |
| P2 | 12 000 | 505 | 589 | 0.04 | 0.48 | ||
| P3 | DEGMEMA |

|
5000 | 225 | 258 | 0.01 | 0.21 |
| P4 | 12 000 | 497 | 579 | 0.04 | 0.47 | ||
| P5 | DEGMEMA |
|
5000 | 788 | 745 | 0.16 | 0.75 |
| P6 | DEGMEMA/MANNOSE | 31 600 | 523 | 593 | 0.02 | 0.50 | |
| P7 | MANNOSE | 41 000 | 675 | 734 | 0.02 | 0.64 | |
| P8 | PEGMEMA |

|
19 000 | 125 | 157 | 0.01 | 0.12 |
| Control 1 | PEGMEMA | n/a | Monomer PEG1100 | −22 | 35 | — | — |
| Control 2 | PEGMEMA | n/a | 10 000 | −7 | 25 | — | — |
| P9 | PEGA |

|
5000 | 227 | 268 | 0.05 | 0.22 |
| P10 | 19 000 | 300 | 341 | 0.06 | 0.29 | ||
| P11 | DEGA |

|
5000 | — | — | — | — |
Sauerbrey states that the frequency shift of a quartz crystal resonator is directly proportional to the added mass.49 Moving from air to liquid, Kanazawaet al.50 stated that the frequency shift of a quartz crystal microbalance is proportional to the square root of the liquid's density-viscosity product. The resonance frequency of the sensor depends on the total oscillating mass, so the method for calculating the mass of adsorbed layers depends on the type of thin film. Rigid films use the Sauerbrey relation which can be simplified to:
| Δm = −kΔf | (1) |
| G* = G′ + iG′′ = μ1 + i2πfη1 | (2) |
| heff = Δm/ρeff | (3) |
The mass calculated via the Sauerbrey relation is expected to be an underestimation of the actual mass on the surface when the film formed has viscoelastic properties. When this is the case the application of the Voigt model is expected to give a closer to actual polymer mass adsorbed on the gold surface. Control studies were carried out with both PEGMEMA monomer (Control 1, ESI) and PEGMEMA polymer (Control 2, Fig. 1) which contain no sulfur atoms and in both cases, no binding to gold was observed.
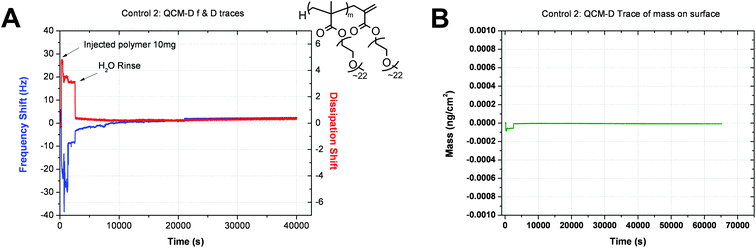 | ||
| Fig. 1 QCM-D traces showing (A) the shift in frequency and dissipation over time and (B) Sauerbrey mass adsorption profile for non-functional PEGMEMA polymer. | ||
It has been previously demonstrated that RAFT end groups can directly bind to the gold surface without modification.51–54 However, in the literature there is no consistent investigation on the relative affinity of these RAFT agents. Two RAFT chain transfer agents were studied to examine which had the better adsorption profile (Fig. 2). RAFT CTA 1, CPADB, containing a dithio functional group was found to have a higher bound mass after rinsing than RAFT CTA 2, BSPA, which contained a trithio functional group. It was observed that a change in dissipation was apparent during both experiments, indicating that both of the RAFT CTA's formed viscoelastic films on the gold surface interacting with the flowing solutions.
 | ||
| Fig. 2 QCM-D traces showing the shift in frequency and dissipation with time for (A) RAFT CTA 1 and (B) RAFT CTA 2 as they are passed over a gold surface. The corresponding frequency vs. dissipation trace showing the adsorption profile is shown in plot (C). | ||
A trace comparing f against D removes time explicitly, this allows interpretation of the relationship between mass adsorbed per unit mass on gold with changes in rigidity; i.e. changes in polymer conformations as the layer builds up. If the plot of f against D is linear this indicates no conformational changes occur during the adsorption process.55–57Fig. 2 shows a linear relationship exists between f and D for CTA 2. Slight curvature is observed in the CTA 1 trace suggesting conformational change on the surface. An indication of density can also be studied using f vs. D plots. A high f value and low D value signifies highly dense films. Both show similar steep gradients which correlates to extended structures on the surface. The frequency and dissipation traces for the four Mn ∼ 5000 polymers (Fig. 3) show binding to gold and after rinsing with water, the polymers remained bound. Interestingly, the dissipation is different between the polymers. P1 forms a rigid film after all unbound material has been removed, whereas polymers P3, P5 and P9 display a somewhat viscoelastic film. Yet during the adsorption process P1, P3 and P9 have much steeper gradients than P5 (Fig. 3E). This demonstrates faster initial adsorption kinetics and either a more extended structure or loose binding between interacting molecules. The overall change in dissipation is relatively small suggesting that the resulting film has little interaction with the solution. Initial asymmetry between the f and D plots shows a change in conformation at the surface for P1, P3 and P9. Longer time-frame experiments show a steady state is reached with no deviation in dissipation. P1, P3 and P5 are all DEGMEMA polymers with relatively low lower critical solution temperatures (LCST) (∼26 °C). These experiments were conducted below their LCST's and thus it is expected that the LCST transition will not affect dissipation.
 | ||
| Fig. 3 QCM-D traces showing the shift in frequency and dissipation against time for Mn ∼ 5000 polymers 1 (A), 3 (B), 5 (C) and 9 (D). The corresponding frequency vs. dissipation traces showing the adsorption profile are shown in plot (E). | ||
Adsorptions of P1, P3, P5 and P9 are compared in Fig. 4 (Sauerbrey mass traces in ESI). This polymer set has a variety of sulfur functionality, while all have a constant molecular weight (Mn ∼ 5000 g mol−1). The following trend in final mass bound (after unbound residue has been removed by a water rinse) to gold was observed: disulfide > dithio ∼ trithio > free thiol, suggesting that disulfide functional groups in polymers have a higher affinity to gold or are able to create more densely packed surface coverage. Since all of the polymers are the same molecular weight, the influence of Mn in this case was found to be negligible. An attempt was made to measure p(DEGMEA) (P11) with trithiocarbonyl end group however the LCST of this polymer was very low and the instruments baseline stability at such a low temperature was not sufficient to attain a stable measurement. Instead a longer poly(ethylene glycol) repeat unit (P9) was used.
 | ||
| Fig. 4 Comparison of Mn ∼ 5000 polymers: P1, P3, P5 and P9 mass adsorption to gold. | ||
A rationalisation for the differences observed in the sulfur polymers adsorption to gold can be attributed to steric effects on the sulfur atoms. Weak disulfide bonds are known to dissociate, resulting in the chemisorptions of thiolates on gold surfaces with low surface coverage.58,59 Whilst Fenter et al. proposed that dimerisation occurs when long chain thiols are adsorbed at high coverage resulting in molecular adsorption of disulfides.60 These studies focused on simple alkyl thiols for SAM's applications, while in this study we investigated higher molecular weight polymers. Clearly how these polymer chains order themselves at the gold surface is an important consideration; with polymer chain properties such as rigidity/stiffness and its hydrodynamic volume in solution influencing their adsorption and packing. The interaction between water and PEG is defined by reduced mobility, hydrogen bonding from water to the polar oxygen atom in PEG and anisotropic motion as studied by Arnold et al. and is most likely to cause disruption as the sulfo-groups try to adsorb on gold.61 The interaction between the polymer backbone and the surface can be neglected since control experiments revealed non-sulfo polymers did not interact with the gold surface.
A similar trend in the mass adsorption profiles was observed when all of the larger Mn polymers were compared: with disulfide polymers adsorbing more than free thiol-, dithio- and trithio-polymers. Two polymer sets were investigated: DEGMEMA (P2, P4 and P6) and PEGMEMA polymers (P7, P8 and P10). The frequency and dissipation traces for the DEGMEMA polymers are shown in Fig. 5. All polymers remain bound to the gold surface and stay on the surface as a viscoelastic film, although the dissipation shift for P6, a copolymer of DEGMEMA and a mannose repeating unit, is tailing towards zero. It would be expected that non-covalent interactions between the side chains and steric hindrance caused by the bulkier sugar side chains would have an impact on the disulfide ability to bind to the gold surface. This can be compared to P7, a homopolymer of the mannose repeat unit, which shows has better binding, as expected. P2 undergoes a two step adsorption onto the surface while the other polymers only show one step adsorption profile. The f vs. D plots (Fig. 5D) of the larger polymers show different kinetics to those seen in Fig. 3. None of the traces are linear, suggesting that they all undergo conformational change during adsorption. While it was easy to see this for P2 in Fig. 5A it was not as obvious in Fig. 5B and C. The shallowest gradient in Fig. 5D is difficult to determine between P4 and P6, both traces indicate that the interaction between the polymer chains is strong.
 | ||
| Fig. 5 QCM-D traces showing the shift in frequency and dissipation against time for larger Mn DEGMEMA polymers: 2 (A), 4 (B) and 6 (C). The corresponding f vs. D trace showing the adsorption profile is shown in (D). | ||
Comparison of adsorbed polymer 2, 4 and 6 on gold (Fig. 6) shows similar binding between all polymers (Mn ∼ 20 000 g mol−1). P6, with the disulfide moiety as middle group, presents a slightly better grafting (mass adsorption) than the thiol and dithioester terminated polymers. However, the effect of sulfur groups binding is less pronounced for higher molecular weight polymers.
 | ||
| Fig. 6 Comparison of larger Mn polymers: P2, P4 and P6 mass adsorption to gold. | ||
The larger Mn PEGMEMA polymer set (P8 and P10) show lower binding to gold after rinsing, demonstrated by a lower overall shift in frequency of ∼15–20 Hz for P8 and P10 compared to ∼30 Hz for P2, 4 and 6. At first P8 and P10 show a greater dissipation shift during adsorption however the overall change in dissipation for P8 is similar to that of P2, 4 and 6 indicating a viscoelastic film is formed (Fig. 7A, B and 8). The dissipation shift for P10 is close to zero after rinsing. In the adsorption profile trace for P8 and P10 (Fig. 7C), the gradient of both lines is steeper than the respective lines for P2, 4 and 6 (Fig. 5D) and more akin to those observed in Fig. 3E suggesting an extended polymeric structure during the adsorption process.
 | ||
| Fig. 7 QCM-D traces showing the shift in frequency and dissipation against time for larger Mn PEGMEMA polymers: 8 (A) and 10 (B). The corresponding f vs. D trace showing the adsorption profile is shown in (C). | ||
 | ||
| Fig. 8 Comparison of larger Mn PEGMEMA polymers: P8 and P10 mass adsorption to gold. | ||
The experiments investigating the DEG containing polymer indicate that it is the disulfides in oligo(ethylene glycol) polymers that react better than dithio, trithio and thiol species with the gold surface. The disulfides are in the centre of the polymer chains whereas all other thio species are at the end of the polymer chains. Sulfur atoms as neighbours provide better bonding than sulfur species which contain a carbon spacer between the sulfur atoms or single thiol units. The distribution of charge is spread more equally, leading to better binding.
The chain length of the oligo(ethylene glycol) side chains was also found to have an impact on the polymer adsorption to gold. This can be rationalised as the shorter side chains of the DEGMEMA polymers in solution cause less steric hindrance during the adsorption process compared to longer flexible PEG chains. This concept is illustrated in the results obtained for P4 (DEGMEMA) and P8 (PEGMEMA) polymers which both contain dithio end groups where P4 has better binding that P8. Walker et al. discuss Zolk et al.'s paper on oligo ethylene oxide (OEO) segments who suggested these segments are more disordered in water compared to their dry state since they become extensively hydrated in the OEO segment relevant to their conformation (helical) in air.62,63 They explain that in situ analysis of the film structure is necessary, as differences are observed between mass calculated viaQCM-D in the hydrated state, and XPS in the dry state. Walker et al. demonstrated that disulfide SAMs equilibrate faster than thiols, and although thiols adsorb slower they eventually result in a more densely packed polymer layer at the gold surface.
In our present study, the rate at which the polymers bound to the surface was similar for all the disulfide, dithio and trithio species studied. However, the thiol polymer plots showing adsorption on gold revealed two adsorption steps (P1 and P2, Fig. 3A and 5A respectively). The first is similar to that found for the polymers and is characterised by a steep adsorption gradient, after which all the species plateau and reach a steady state. In the case of the thiol species, however, a second shallow slope is evident, with more adsorption taking place before a plateau is reached. These findings are consistent with those found by Walker et al.
The introduction of a co-monomer into the polymer chain also affects the binding performance of polymers with thio groups to gold; P5, P6 and P7 can be compared (Fig. 9 and 10). Both pDEGMEMA (P5) and mannose containing (P7) homopolymers bind well to the gold surface, however, copolymer (P6), containing both repeat units is less stable on gold. From Fig. 9(D) both mannose containing polymers display non-linear kinetics. Furthermore P6, which is the copolymer, shows less interaction between its polymer chains in comparison to the others as shown by the steeper gradient. This correlates to less efficient packing in the random copolymer backbone to allow for association between polymer chains.
 | ||
| Fig. 9 QCM-D traces showing the shift in frequency and dissipation against time of: homopolymers (P5(A), P7(B)) and copolymer (P6,(C)). The corresponding f vs. D trace showing the adsorption profile is shown (D). | ||
 | ||
| Fig. 10 Comparison of mass adsorbed onto gold for homopolymers (P5, P7) and copolymer (P6). | ||
X-ray photoelectron spectroscopy (XPS) was carried out to confirm the presence of polymer on the surface after QCM-analysis and to investigate the interaction between the sulfo-groups (thiol, disulfide, trithiocarbonate or dithioester) and the gold surface. XPS reveals the presence of peaks attributed to O1s, C1s, S2p and Au4f7 at around 532.0 eV, 284 eV, 164 eV and 83 eV respectively. O1s and C1s spectra correspond to the expected spectra for PEG based polymers (Fig. 11). According to the ratio between the S and Au atomic composition, we can estimate the grafting density of the polymers for the different polymers (Table 3). Indeed, a high ratio indicates the presence of high grafting density as this analysis uses the same volume element. A comparison of the S/Au ratio reveals the following trend in grafting density with: thiol ∼ trithiocarbonate < dithioester < disulfide terminated polymer. These results are in good agreement with the data obtained with QCM-D where more disulfide terminated polymer mass remained bound. Fig. 12 shows the high resolution XPS spectrum of the S2p region for the different functional polymers grafted onto Au (formed in situ during QCM-D experiments). S2p spectra are typically composed of 2p3/2 and 2p1/2 peaks originating from the spin–orbit splitting effects, separated by 1.1–1.2 eV.64Thiol and disulfide terminated polymers have a similar spectrum exhibiting the presence of –S–Au bonds centred at 162.5 and 163.6 eV (Fig. 12).
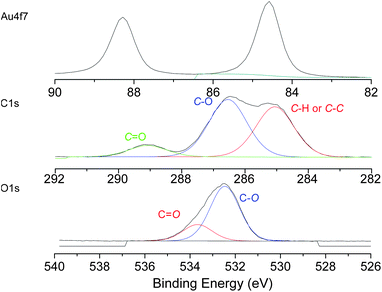 | ||
| Fig. 11 High resolution XPS spectra of Au4f7, C1s and O1s regions of PEGMEMA (Mn ∼ 5000 g mol−1) assembled onto gold surface during QCM-D experiments. | ||
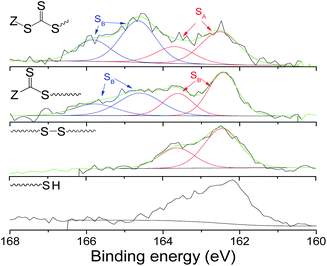 | ||
| Fig. 12 High resolution XPS spectra of S2p region of different sulfur terminated polymers (Mn ∼ 5000 g mol−1) assembled onto gold surface during QCM-D experiments. | ||
It was demonstrated in the literature that the disulfide bond can be cleaved onto the gold surface to yield a S–Au bond.65,66 In the case of dithioester and trithiocarbonate moieties, two types of sulfur compounds are detected at 162.5–163.6 eV (SA) and at 164.5–165.7 eV (SB) attributed to the S–Au bond and C–S bond, respectively.66 According to the nature of the RAFT agent (dithioester or trithiocarbonate), we observed that the ratio between SA and SB changes as expected. In conclusion, it is interesting that polymers containing a disulfide bond in the middle of the polymer chain bind more strongly than polymers where the sulfur moiety is located at the polymer chain end.
Conclusions
QCM-D experiments followed thio-functional oligo(ethylene glycol) polymers and their adsorption to goldin situ providing a deeper understanding of how sulfur groups are affected by polymer chains when adsorbing to the surface. Calculation of mass adsorbed by Sauerbrey's relation and the Voigt model obtained similar results. For DEG containing polymers it was clear that different sulfur species bound to gold in different ways; depending on the orientation which sulfur atoms take up at the molecular level to dissociate electron density. Disulfide DEG polymers result in more mass adsorbed on gold suggesting disulfides aligning on a planar axis to gold provide equal distribution of charge over the disulfide bond. In the case of thiols, only one sulfur atom is involved in the adsorption process while for the dithio and trithio species a carbon atom is between the sulfur atoms and changes the bond angle between the sulfur atoms leading to a weaker dissociation of charge. This study demonstrates that electronics, sterics of pendent side chains and chain length have a big impact on the binding of thio-functional oligo(ethylene glycol) polymers to gold surfaces. When longer oligo(ethylene glycol)s were used, the effect of each sulfur species could not be clearly described, however it was apparent that the binding of these polymers was less than that found for the corresponding DEG polymers. XPS data independently supported the binding strength trends observed by QCM-D and furthermore suggested that the disulfide group dissociates at the gold surface as the resulting spectrum is akin to that of a thiol end group with only one species observed.Acknowledgements
The authors would like to thank EPSRC, Unilever PLC, (SS), ARC-APD (CB), EU Marie Curie Fellowship (No. 235999) (CRB). Equipment used in part was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).References
- R. G. Nuzzo and D. L. Allara, J. Am. Chem. Soc., 1983, 105, 4481 CrossRef CAS.
- E. B. Troughton, C. D. Bain, G. M. Whitesides, R. G. Nuzzo, D. L. Allara and M. D. Porter, Langmuir, 1988, 4, 365 CrossRef CAS.
- E. Sabatani, J. Cohenboulakia, M. Bruening and I. Rubinstein, Langmuir, 1993, 9, 2974 CrossRef CAS.
- M. A. Bryant, S. L. Joa and J. E. Pemberton, Langmuir, 1992, 8, 753 CrossRef CAS.
- W. Hill and B. Wehling, J. Phys. Chem., 1993, 97, 9451 CrossRef CAS.
- T. T. T. Li, H. Y. Liu and M. J. Weaver, J. Am. Chem. Soc., 1984, 106, 1233 CrossRef CAS.
- K. Uvdal, P. Bodö and B. Liedberg, J. Colloid Interface Sci., 1992, 149, 162 CrossRef CAS.
- A. Ihs, K. Uvdal and B. Liedberg, Langmuir, 1993, 9, 733 CrossRef CAS.
- T. Arndt, H. Schupp and W. Schrepp, Thin Solid Films, 1989, 178, 319 CrossRef CAS.
- J. A. Mielczarski and R. H. Yoon, Langmuir, 1991, 7, 101 CrossRef CAS.
- T. R. G. Edwards, V. J. Cunnane, R. Parsons and D. Gani, J. Chem. Soc., Chem. Commun., 1989, 1041 RSC.
- A. J. Arduengo, J. R. Moran, J. Rodriguez-Parada and M. D. Ward, J. Am. Chem. Soc., 1990, 112, 6153 CrossRef CAS.
- G. Xue, X.-Y. Huang, J. Dong and J. Zhang, J. Electroanal. Chem., 1991, 310, 139 CrossRef CAS.
- S. Bharathi, V. Yegnaraman and G. P. Rao, Langmuir, 1993, 9, 1614 CrossRef CAS.
- M. G. Samant, C. A. Brown and J. G. Gordon, Langmuir, 1992, 8, 1615 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321 CrossRef CAS.
- S. Svedhem, D. Dahlborg, J. Ekeroth, J. Kelly, F. Hook and J. Gold, Langmuir, 2003, 19, 6730 CrossRef CAS.
- S. E. Moya, A. A. Brown, O. Azzaroni and W. T. S. Huck, Macromol. Rapid Commun., 2005, 26, 1117 CrossRef CAS.
- G. M. Liu, L. F. Yan, X. Chen and G. Z. Zhang, Polymer, 2006, 47, 3157 CrossRef CAS.
- A. K. Dutta and G. Belfort, Langmuir, 2007, 23, 3088 CrossRef CAS.
- E. J. Park, D. D. Draper and N. T. Flynn, Langmuir, 2007, 23, 7083 CrossRef CAS.
- M. Dobbelin, G. Arias, I. Loinaz, I. Llarena, D. Mecerreyes and S. Moya, Macromol. Rapid Commun., 2008, 29, 871 CrossRef CAS.
- N. Nordgren, J. Eklof, Q. Zhou, H. Brumer and M. W. Rutland, Biomacromolecules, 2008, 9, 942 CrossRef CAS.
- D. Patton, W. Knoll and R. C. Advincula, Macromol. Chem. Phys., 2011, 212, 485 CrossRef CAS.
- C. Bonnans-Plaisance, G. Levesque and V. Toulin, React. Funct. Polym., 2001, 47, 77 CrossRef CAS.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47, 1449 RSC.
- A. H. Soeriyadi, C. Boyer, J. Burns, C. R. Becer, M. R. Whittaker, D. M. Haddleton and T. P. Davis, Chem. Commun., 2010, 46, 6338 RSC.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- J. A. Syrett, M. W. Jones and D. M. Haddleton, Chem. Commun., 2010, 46, 7181 RSC.
- C. Boyer, A. H. Soeriyadi, P. J. Roth, M. R. Whittaker and T. P. Davis, Chem. Commun., 2011, 47, 1318 RSC.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595 CrossRef CAS.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105 CrossRef CAS.
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2005, 101, 47 CrossRef CAS.
- J. -H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. J. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–47 CrossRef CAS.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641 CrossRef CAS.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207 CrossRef CAS.
- M. Rodahl, F. Hook, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924 CrossRef CAS.
- D. M. Haddleton, A. J. Clark, M. C. Crossman, D. J. Duncalf, A. M. Heming, S. R. Morsley and A. J. Shooter, Chem. Commun., 1997, 1173 RSC.
- G. Mantovani, V. Ladmiral, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 2089 RSC.
- D. M. Haddleton, C. B. Jasieczek, M. J. Hannon and A. J. Shooter, Macromolecules, 1997, 30, 2190 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823 CrossRef CAS.
- M. I. Gibson, N. Vinson, Y. Z. Gou, C. R. Becer and D. M. Haddleton, Polym. Chem., 2011, 2, 107 RSC.
- A. J. Limer, A. K. Rullay, V. San Miguel, C. Peinado, S. Keely, E. Fitzpatrick, S. D. Carrington, D. Brayden and D. M. Haddleton, React. Funct. Polym., 2006, 66, 51 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS.
- A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815 RSC.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
- K. Kanazawa and G. Gordon, Anal. Chem., 1985, 57, 1770 CrossRef CAS.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917 CrossRef CAS.
- C.-A. Fustin, C. Colard, M. Filali, P. Guillet, A.-S. Duwez, M. A. R. Meier, U. S. Schubert and J.-F. Gohy, Langmuir, 2006, 22, 6690 CrossRef CAS.
- A.-S. Duwez, P. Guillet, C. Colard, J.-F. Gohy and C.-A. Fustin, Macromolecules, 2006, 39, 2729 CrossRef CAS.
- J. W. Hotchkiss, A. B. Lowe and S. G. Boyes, Chem. Mater., 2006, 19, 6 CrossRef.
- F. Hook, M. Rodahl, P. Brzezinski and B. Kasemo, Langmuir, 1998, 14, 729 CrossRef.
- A. Monkawa, T. Ikoma, S. Yunoki, T. Yoshioka, J. Tanaka, D. Chakarov and B. Kasemo, Biomaterials, 2006, 27, 5748 CrossRef CAS.
- I. G. Sedeva, R. Fetzer, D. Fornasiero, J. Ralston and D. A. Beattie, J. Colloid Interface Sci., 2010 Search PubMed.
- H. Grönbeck, A. Curioni and W. Andreoni, J. Am. Chem. Soc., 2000, 122, 3839 CrossRef.
- R. G. Nuzzo, B. R. Zegarski and L. H. Dubois, J. Am. Chem. Soc., 1987, 109, 733 CrossRef CAS.
- P. Fenter, A. Eberhardt and P. Eisenberger, Science, 1994, 266, 1216 CAS.
- S. Lusse and K. Arnold, Macromolecules, 1996, 29, 4251 CrossRef.
- M. L. Walker, D. J. Vanderah and K. A. Rubinson, Colloids Surf., B, 2011, 82, 450 CrossRef CAS.
- M. Zolk, F. Eisert, J. Pipper, S. Herrwerth, W. Eck, M. Buck and M. Grunze, Langmuir, 2000, 16, 5849 CrossRef CAS.
- J. Noh, S. Jang, D. Lee, S. Shin, Y. J. Ko, E. Ito and S.-W. Joo, Curr. Appl. Phys., 2007, 7, 605 CrossRef.
- J. C. Munro and C. W. Frank, Polymer, 2003, 44, 6335 CrossRef CAS.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, ACS Nano, 2008, 2, 757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sm06410j |
| This journal is © The Royal Society of Chemistry 2012 |