Selective liquid phase oxidation with supported metal nanoparticles
Nikolaos
Dimitratos
,
Jose A.
Lopez-Sanchez†
and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 30th September 2011
Abstract
Over the past twenty years there has been intense interest in the design and understanding of catalysis by gold. More recently, it has been observed that alloying gold with a second metal greatly enhances the catalytic efficacy. These supported nanoparticles offer great potential as catalysts for the synthesis of fine chemicals and they are now at the stage where they can contribute to the sustainable development of chemical processes, in particular selective oxidation. A number of factors have contributed to this increased interest; namely, environmental issues promoting the need for more atom efficient processes and the new advances in the synthesis of nanoparticles as well as the characterisation methods available for their study. New catalytic materials obtained by careful control of the morphology of the metal nanoparticles and their use under solvent-free conditions all contribute to the latest developments. Most importantly their use can obviate the need to use stoichiometric oxidants. In this perspective we demonstrate the recent advances in these materials for selective oxidation reactions.
Introduction
Catalysis is a crucial enabling technology permitting the functionalisation and utilisation of a myriad of raw materials ensuring they can be used to form products of great value and usefulness for society as a whole. Most people that are unaware of chemical science are probably ignorant of the key role played by catalysis in ensuring the goods and services they enjoy in everyday life function efficiently. In many respects this is a great pity as the central chemical beauty of catalysis remains hidden to the greater majority of society but it is essential that we try to ensure this knowledge gap is addressed in some way. A key example of catalysis that has caught the interest of chemists has been the observation of catalysis by gold nanoparticles. This has engendered a wide range of studies aimed at making and using small gold nanoparticles. The field of gold catalysis is one that perhaps could be taken to a wider audience, but this may have to wait until gold catalysis finds its place in the commercial production of chemical intermediates.One of the key features of chemical catalysis is that it regularly undergoes rapid advances. One key area experiencing such advances is the use of catalysis to access milder reaction conditions, which enable selectivity control to be more readily achieved. Historically, many approaches to the industrial production of chemicals, fuels and intermediates tend to utilise high temperatures and pressures. For example the conversion of methane to methanol,1 the synthesis of ammonia2 or modern gas to liquids technology all employ the production of synthesis gas, which uses high pressures and temperature. The use of synthesis gas as a central processing route tends to create a mind-set that this is the most viable way to proceed. At present, society is becoming increasingly aware of sustainability issues and in the longer term alternative technologies will be required. In some cases this requires the need to use bio-available and sustainable materials, mainly based on carbohydrates such as sugars.3,4 Of course the use of bio-available materials creates a new tension between fuels and food, but there are plenty of sustainable resources that cannot be used as foodstuffs, for example algae. A number of people advocate that bio-derived carbohydrates could readily be converted to synthesis gas thereby reinforcing the pre-eminence of this high temperature approach. However, such an approach neglects the chemical complexity that is available in these molecules and transforming them to the basic building blocks of CO and H2 will be inappropriate in many cases. There is therefore a need to develop catalytic processes that operate under milder conditions. This has been the focus of the new branch of green chemistry and has required the identification of new catalytic materials that can be active under mild conditions. One class of these materials are supported metal nanoparticles and in this perspective we demonstrate the recent advances in these materials for selective oxidation reactions under mild liquid phase conditions.
Selective oxidation of hydrocarbons
The selective utilisation of hydrocarbons represents one of the greatest challenges for catalysis, as the activation of C–H bonds typically requires forcing conditions. However, it is a field where supported metal nanoparticles can make a major impact. As this is such a major area for new green chemistry approaches we start our discussion of oxidation catalysis with these molecules that present the greatest challenge. In the last ten years there has been significant progress in this research area, and we describe this in sections related to the structure of the substrate. It must be noted that liquid phase selective oxidation of hydrocarbons is a broad area of research, and relevant heterogeneously catalysed industrial processes are mostly carried out with mixed oxides or ordered porous catalytic systems. General trends in industrial and technological developments in commercial liquid phase oxidation reactions can be found in the recent literature.5–7Selective oxidation of cycloalkanes
The selective oxidation cyclohexane under mild conditions is highly desired from a commercial viewpoint (Scheme 1). The synthesis of cyclohexanol and cyclohexanone from cyclohexane is currently operated industrially to produce a first step in the production of adipic acid and caprolactam, both essential intermediates in nylon manufacture. | ||
| Scheme 1 | ||
Recent reports highlighting the ability of supported gold nanoparticles to selectively oxidise alkenes and alcohols have drawn attention to using gold in the direct activation of cyclohexane. Zhao et al.8,9 have shown that gold can activate cyclohexane at 150 °C. Gold nanoparticles on MCM-41 and ZSM-5 displayed selectivities in higher than 90% which changed from cyclohexanone to cyclohexanol as the catalysts deactivated. Wu et al.10 reported more recently a catalyst comprising of gold nanoparticles of sizes in the range 2–4 nm dispersed on ordered mesoporous silica prepared via a one-pot synthesis. This method afforded catalysts with high catalytic activity and selectivity using molecular oxygen, which performance was dependent on the porous structure, the amount of Au loading and the Au particle size. The sintering of the Au nanoparticles during high temperature calcination and reaction is avoided thanks to the protective effect of the silica and this confers additional stability to the catalyst. In a similar fashion, Liu et al. prepared silver nanoparticles on MCM-41 in a one pot synthesis method and showed that the catalyst is very active in the absence of solvent, with oxygen and in the absence of initiators.11 The authors obtained 83.4% selectivity to cyclohexanol and cyclohexanone at 10.7% conversion of cyclohexane over the Ag/MCM-41 catalyst at 155 °C for 3 h.
High selectivities can be expected when the selective oxidation is performed at lower temperatures, and with this in mind, Xu et al.12 investigated the oxidation of cyclohexane with gold, platinum and palladium catalysts at temperatures well below 100 °C. The selectivity to cyclohexanone and cyclohexanol was very high at low conversion, but it decreased rapidly with conversion at longer reaction times. No substantial differences were observed between the catalytic performances of gold, platinum and palladium. The same authors later reported that titania can actually promote the activity of gold on silica and prepared a very stable catalyst with selectivity of 90% at ca. 10% conversion.13 Weckhuysen et al.14 recently aimed to clarify the effect of gold in the reaction mechanism and carried out reaction studies over Au/Al2O3. Au/TiO2, and Au/SBA-15 and compared their reactivity with the autoxidation reaction. They concluded that gold-based catalysts do not exhibit excellent catalytic performance, and that the process is not catalytic, to the contrary, that the oxidation follows a radical-chain mechanism instead. Their claims are based upon the observation that product distributions observed are typical for an autoxidation process. They used hydroquinone as a radical scavenger supporting this conclusion. Their observations can actually explain the low selectivity observed at increasing conversion and they claim that the discrepancies of their work with published literature is a consequence of the difficulty of performing a full analysis of the reaction products, which other researchers have not achieved. Indeed it is well known that supported gold nanoparticles are very effective at decomposing peroxides to form radicals in solution and as the cyclohexane hydroperoxide is a key intermediate in cyclohexane oxidation it can be expected that a radical process operates.15 Recent work from Neuenschwander et al.16 also identifies a radical chain oxidation mechanism in the selective oxidation of α-pinene.
More recent research however emphasises the effect of gold further. Liu et al.17 synthesized gold clusters, Aun (n = 10, 18, 25, 39), with controlled cluster size on hydroxyapatite (HAP) and showed that these could efficiently oxidize cyclohexane to cyclohexanol and cyclohexanone at 150 °C and 1 bar of oxygen. No reaction was observed in the absence of gold and it additionally required the presence of a radical initiator (TBHP). The turnover frequency increased with an increase in the cluster size, reaching values as high as 18 500 h−1/Au atom.
Selective oxidation of alkylaromatics
The selective oxidation of alkylaromatics with heterogeneous catalysts is highly desired in industry as currently these reactions are carried out mostly with homogeneous catalysts. Benzaldehyde is produced by the chlorination of toluene followed by saponification,18 whereas benzoic acid is produced by the liquid-phase cobalt-catalysed oxidation of toluene using oxygen at 165 °C with acetic acid as solvent, but the conversion has to be limited to <15% to retain high selectivities.19 The use of halogens and acidic solvents makes these processes environmentally unfriendly, which is a motivation for developing new catalytic systems and/or finding alternative processes and reaction conditions. Heterogeneous catalysts could be readily used in flow reactors, facilitating the efficient production of materials using continuous processes. For the oxidation of toluene, there have been many attempts to find a suitable liquid-phase oxidation catalyst, and to date these have used oxides of copper and manganese,20–22cobalt in SBA-15,23 or chromium in chromium silicalite-1 (CrS-1)24catalysts, but all of these perform very poorly even at temperatures in excess of 190 °C. Thomas and co-workers have recently reported an elegant single-site solid catalyst, comprising microporous manganese-doped aluminum phosphate (e.g., Mn0.1Al0.9PO-5) which shows significant activity for toluene oxidation with air under solvent free conditions.25Benzaldehyde and benzoic acid were the main reaction products. Against this background, there is a clear need to develop heterogeneous catalysts for toluene oxidation that can improve activity while retaining or improving selectivity.Carboxylic acids as initiators have been utilised by Beier et al. recently.26 They reported that silver supported on silica catalysed the solvent-free aerobic side-chain oxidation of alkyl aromatics even at atmospheric pressure when carboxylic acids were used as initiators.26Benzoic acid or p-toluic acid were added (3% molar) as promoters whereas the role of additional CeO2 was more complex and showed both promoting and inhibiting effects depending on substrate and reaction conditions. Overall, they highlighted that addition of a Ce precursor to a catalyst prepared by flame spray pyrolysis results in significantly smaller Ag nanoparticles that exhibit a superior catalytic performance with TONs up to 2000 together with higher stability compared to impregnated catalysts. In the selective oxidation of toluene, 3mol% benzoic acid was added as a promoter in addition to ceria and biphenyl and the reaction was carried out with 10 bar air for 1 h to give a combined yield of 2.6% at 170 °C after one hour. Benzyl alcohol, benzaldehyde and benzoic acid were the only significant products.
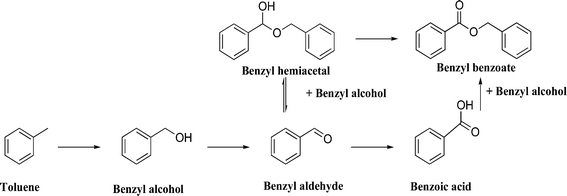
Au–Pd alloy nanoparticles are known to be very effective for the direct synthesis of hydrogen peroxide27–30 and the oxidation of primary alcohols using oxygen.31 This catalyst is considered to operate by producing a reactive hydroperoxy intermediate and, because these intermediates are known to be involved in the enzymatic oxidation of primary carbon–hydrogen bonds,32 it was reasoned that Au–Pd nanoparticles could be active for the oxidation of toluene. Rossi and Prati first showed the applicability of colloidal methods for the preparation of gold catalysts and their use in liquid phase oxidation reactions33,34 and subsequently Hutchings and co-workers have intensively studied this methodology and found that a sol immobilization method produces more active Au–Pd catalysts for a range of selective oxidation reactions with an effective control of morphology and particle composition.28–30,35–38 Consequently, Hutchings and co-workers prepared Au–Pd alloyed nanoparticles by sol immobilization and showed that they are very active for the oxidation of toluene with molecular oxygen and unexpectedly selective to benzyl benzoate under solvent-free conditions (Scheme 2).39
 | ||
| Scheme 2 | ||
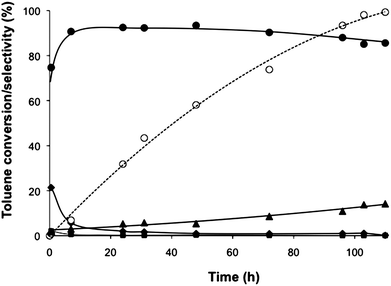
Wet impregnation is a very attractive method to support metal catalysts due to its simplicity and ease to scale-up for industrial commercialisation. However, Au–Pd nanoparticles synthesized by the impregnation method have a very heterogeneous mixture of morphologies and compositions and the overall particle size can be relatively large (typically >6 nm), particularly after the heat treatment process required to produce the nanoparticles from the metal salts. They also have substantial compositional variations, which are very difficult to affect during the synthesis and this can limit their reactivity. Hutchings and co-workers initially investigated Au–Pd/TiO2 catalysts, prepared by impregnation; however, these catalysts were not very active for toluene oxidation.39 The catalysts prepared by sol-immobilisation were then tested and the only products observed were benzyl alcohol, benzaldehyde, benzoic acid, and benzyl benzoate. The monometallic gold catalyst was not active, but the addition of Pd brought up conversion, and a clear synergistic effect was shown with an optimum catalyst composition of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Au:Pd molar ratio. For this catalyst, the turnover frequency (TOF: mole of product formed per mole of metal per hour) after 7 h of reaction was ∼50. To show general applicability, the selective oxidation of xylenes was studied and, the catalyst formed the aldehyde, acid, and esters, with the relative amounts being dependent on conversion levels. The reaction profile was also investigated using a lower substrate/metal molar ratio and conversion continued to increase steadily, fully depleting the toluene after 110 h, while the selectivity to benzyl benzoate increased (Fig. 1). The results are summarised in Table 1. TONs that are a factor of ∼30 greater than those of previous heterogeneous catalysts for this reaction were reported. The key feature of this catalytic reaction is the high selectivity to benzyl benzoate that is observed and this is formed via the formation and oxidation of a hemiacetal (Scheme 2).
:2 Au:Pd molar ratio. For this catalyst, the turnover frequency (TOF: mole of product formed per mole of metal per hour) after 7 h of reaction was ∼50. To show general applicability, the selective oxidation of xylenes was studied and, the catalyst formed the aldehyde, acid, and esters, with the relative amounts being dependent on conversion levels. The reaction profile was also investigated using a lower substrate/metal molar ratio and conversion continued to increase steadily, fully depleting the toluene after 110 h, while the selectivity to benzyl benzoate increased (Fig. 1). The results are summarised in Table 1. TONs that are a factor of ∼30 greater than those of previous heterogeneous catalysts for this reaction were reported. The key feature of this catalytic reaction is the high selectivity to benzyl benzoate that is observed and this is formed via the formation and oxidation of a hemiacetal (Scheme 2).
 | ||
| Fig. 1
Toluene conversion and selectivity to partial oxidation products. Reaction conditions: 160 °C, 0.1 MPa pO2, 20 ml toluene, 0.8 g of catalyst (1 wt% AuPd/C prepared by sol immobilization with 1:1.85 Au/Pd ratio), toluene/metal molar ratio of 3250 and reaction time: 110 h. Key: ○ conversion ■ selectivity to benzyl alcohol, ♦ selectivity to benzaldehyde, ▲ selectivity to benzoic acid, ● selectivity to benzyl benzoate. | ||
:Pd bimetallic catalysts in this table were prepared with a 1:1.85 mol ratio. (Table modified from ref. 39)
| Entry | Catalyst | T/°C | Time (h) | Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Benzyl alcohol | Benzaldehyde | Benzoic acid | Benzyl benzoate | TON | |||||
| a Reaction conditions: toluene = 20 ml, substrate/metal mol ratio = 6500, mass of catalyst variable. b Reaction of a physical mixture comprising 1 wt % Au/C and 1 wt % Pd/C, toluene 20 ml, substrate/metal mol ratio 6500. c Reaction conditions: toluene = 10 ml, substrate/metal = 6500, mass of catalyst = 0.2 g. | |||||||||
| 1 | Au–Pd/Ca | 160 | 7 | 4.8 | 0.9 | 12.7 | 10.3 | 76.1 | 310 |
| 2 | Pd/Ca | 160 | 7 | 1.6 | 3.9 | 56.4 | 3.3 | 36.4 | 105 |
| 3 | Au/C + Pd/Cb | 160 | 7 | 1.6 | 0.8 | 26.2 | 11.4 | 61.6 | 105 |
| 4 | Au–Pd/TiO2a | 160 | 7 | 2.1 | 2.9 | 6.6 | 1.0 | 89.5 | 135 |
| 5 | Au–Pd/Cc | 160 | 48 | 50.8 | 0.1 | 1.1 | 4.5 | 94.3 | 3300 |
| 6 | Au–Pd/TiO2c | 160 | 48 | 24.1 | 0.5 | 1.2 | 2.8 | 95.5 | 1570 |
| 7 | Au–Pd/Cc | 120 | 48 | 10.6 | 0.2 | 7.1 | 13.1 | 79.7 | 690 |
| 8 | Au–Pd/TiO2c | 120 | 48 | 4.0 | 1.1 | 6.0 | 4.8 | 88.1 | 260 |
| 9 | Au–Pd/Cc | 80 | 48 | 0.9 | 8.6 | 34.2 | 0.1 | 57.2 | 60 |
| 10 | Au–Pd/Cξ | 160 | 7 | 21.5 | 0.3 | 1.8 | 3.1 | 94.8 | 350 |
| 11 | Au–Pd/Cξ | 160 | 27 | 94.4 | 0.2 | 1.0 | 13.3 | 85.5 | 1534 |
Characterisation and reactivity studies confirmed that any sintering or structural modification of these highly active catalysts is minimal, and the catalysts are stable and reusable. The effect of the oxidant was studied and, at 80 °C and using tert-butyl hydroperoxide, it was found that the conversion increased appreciably up to TON values of 850 to 1200. However, at long reaction times, benzoic acid became the main product, together with appreciable amounts of benzaldehyde and benzyl alcohol rather than the benzyl benzoate obtained with O2.
The effect of the support in activity was important, and carbon-supported catalysts gave improved performance when compared with titania as a support (Table 1). To determine the origin of the differences in activity between the two catalysts, the starting sol and the two sol-immobilised materials were characterised using scanning transmission electron microscopy (STEM) and high angle annular dark field electron microscopy (HAADF). The starting sol displays nanoparticles that have a mean particle size of 2.9 nm which were found to be homogeneous Au–Pd alloys. More than 80% of the particles were icosahedral or decahedral (i.e. multiply twinned), with the remainder being cub-octahedral or just single/double twinned in character. They were also distinctly rounded in shape as they were still coated with protective PVA ligands. After deposition of the colloids onto activated amorphous carbon or TiO2, the sol-immobilised material was dried at 120 °C for 3h in an oven, which caused a very modest size increase in the Au–Pd particles leading to HAADF measured mean sizes for the TiO2 and carbon supported catalysts of 3.9 nm and 3.7 nm, respectively. Representative micrographs of the supported nanoparticles are displayed in Fig. 2.
 | ||
| Fig. 2 Representative scanning transmission electron microscopy (STEM) high angle annular dark field (HAADF) micrographs of AuPd nanoparticles in the Au + Pd/TiO2 (A–C) and Au + Pd/C (D–F) catalysts. (Modified from ref. 39). | ||
For the nanoparticles supported on titania, many of the smaller particles were found to be highly facetted, and primarily cub-octahedral (Fig. 2 A–C) or singly twinned in character. Such particles preferentially exposed distinct {111}- and {200}-type facets. The larger particles still tended to be multiply twinned and exclusively exposed {111}-type facet planes. In addition, the Au–Pd particles tended to form an extended flat interface structure with the crystalline TiO2 substrate, which might improve particle adhesion and stability, and inhibit sintering. The corresponding HAADF images (Fig. 2 D–F) from the carbon supported samples show that the Au–Pd particles are more rounded and have lower ability to wet the amorphous carbon support. The distribution of Au–Pd particle morphologies on the carbon support was much closer to that of the starting colloid, with icosahedral and decahedral (i.e. five-fold twinned) particles predominating (Fig. 2 D–F) and comparatively few single/double twinned and cub-octahedral particles present. The authors indicate that simple metal surface area considerations are not dominating the catalytic activity in this instance since the total number of exposed surface atoms are almost identical as the TON per surface exposed atoms for the most active catalysts are 1.03 × 104 for Au–Pd/C and 0.56 × 104 for AuPd/TiO2. XPS does not provide a clue as to the source of the activity difference and a very similar surface composition was observed. It is possible that the disparity in catalytic activity may be related to the fact that the nanoparticles supported on carbon have a higher number of low co-ordination facet-edge and corner sites due to being more irregular and rougher in morphology. It was also observed that the AuPd/C catalyst predominantly has multiply twinned (icosahedral and decahedral) particles, which tend to have {111} facet terminations, whereas the AuPd/TiO2 materials show an increased fraction of cub-octahedral and singly/doubly twinned particles which exhibit mixed {100}/{111} facet terminations. It is reasonable to think that the increasing proportion of {100}-type facets in the AuPd/TiO2 sample correlates with a decrease in catalytic activity and this be the reason behind the lower activity observed for the titania supported catalysts.
Selective oxidation of alkenes
The epoxidation of cycloalkenes has been found to be effectively catalysed using supported metal nanoparticles and with gold catalysts in particular. Hughes et al. have shown that gold nanoparticles supported on graphite can epoxidise cyclic alkenes using molecular oxygen when using a peroxide in catalytic amounts.40 In these studies it was demonstrated that cis-cyclooctene could be epoxidised with a selectivity of >80% under mild solvent-free conditions (Scheme 3), but also cyclohexene (Scheme 4), styrene (Scheme 5) and cis-stilbene (Scheme 6). | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
 | ||
| Scheme 6 | ||
In the absence of the catalyst, some oxidation was observed but this was minor and non-selective giving very low selectivities to the epoxide. The radical initiator, typically a peroxide, was considered to be important to observe high selectivity, although the reaction could also be carried out in the absence of an initiator.33 In a recent report, both activity and selectivity where enhanced using a colloidal preparation method instead of impregnation or deposition–precipitation and this was ascribed to the smaller particle size distribution produced with the colloidal method.41Tert-butyl hydroperoxide (TBHP) is found to be the most effective initiator but, under the reaction conditions, it does not persist, setting up the reactive radical species that propagate the catalytic oxidation.42 The catalyst was found to be fully reusable.
Cai et al.43 used promoted manganese oxide octahedral molecular sieves as supports for gold and found that catalysts denoted Au/OMS-2 and Au/La-OMS-2 were very active in the liquid phase selective oxidation of cyclohexene without using an initiator. La was exchanged in K-OMS-2 and gold nanoparticles were incorporated by deposition–precipitation with NaOH yielding gold loadings between 0.1 and 1.5 wt.%. High cyclohexene conversions of up to 48% were obtained with Au/La-OMS-2 (0.24) with over 85% selectivity towards C6oxidation products (2-cyclohexene-1-ol and 2-cyclohexene-1-one made-up >80% selectivity). The reactions were performed under solvent-free conditions at 80 °C for 24 h.
Patil et al.44 reported the selective epoxidation of styrene with tert-butyl hydroperoxide using gold nanoparticles deposited on MgO, CaO, SrO, and BaO. The catalysts were prepared by deposition–precipitation, and the authors highlighted the importance of the use of urea as precipitating agent as compared with NaOH. This is attributed to the fact that with urea, higher gold loading and small particle sizes can be obtained. Styrene conversion >53%, and selectivities towards styrene oxide between 45–60% were reported. Yb2O3, Sm2O3, Eu2O3, and Tb2O3 were used as supports in a subsequent work.45 These novel catalysts are highly active, selective and reusable and they reported styrene conversions and selectivities to styrene oxide over 70% with re-usable catalysts containing 6.6 wt% of gold with an average particle size of 11 nm. Additional research is, however, required to understand the promotional role of water as there appears to be a promoting effect when aqueous TBHP was utilised compared to its anhydrous counterpart.
The reactivity of the solvent can sometimes play as an advantage in catalytic reactions and, Lignier et al.46 discovered that gold catalysts can promote the liquid phase epoxidation of trans-stilbene in methylcyclohexane by taking part in a chain reaction which involves a radical formed from the solvent. The participation of the solvent adds complexity to the study of the reaction, but the authors have fully investigated the mechanism.15,46,47 The reaction was performed at atmospheric pressure and using catalytic amounts of TBHP with a Au/TiO2 reference gold catalysts provided by the World Gold Council.48 Another level of complexity is added and the overall activity is product of the combination of the oxidation catalytic activity of gold and titania in a bifunctional catalyst where both sites play a role.47TBHP acts as a radical initiator and oxygen from air is the oxidant whereas the solvent is also oxidised and acts as propagating radical. The authors suggest that both titania and gold play roles in the production of the active radicals as well as trapping unselective radicals and stabilising the reaction intermediate. Subsequently, they prepared their gold catalysts on other supports including carbon, alumina or iron oxide and titania47 and described how the nature of the radical initiator has a critical influence on the reaction selectivity.15 In particular, TBHP leads to high yields of epoxide; whereas, hydrogen peroxide and di-tert-butylperoxide cause the undesired degradation of trans-stilbene. The use of alkyl-substituted cyclohexanes as solvents lead to high yields of epoxide, whereas more polar solvents or cyclohexane resulted in lower activity and epoxide yields.
Interestingly, Mendez et al. found that in the same reaction the support has little influence on gold intrinsic activity but affects the apparent reaction rates, which are a combination of catalytic activity and diffusion limitations.49 The rate-determining step was the gold-catalysed homolytic decomposition of TBHP generating radicals. The authors used gold supported on gadolinium-doped titania nanocrystallites obtained by mild hydrolysis of a new Gd4TiO(OiPr)14 bimetallic oxo alkoxide. It is suggested that this choice of support leads to enhanced wetting of gold particles in the TBHP-initiated epoxidation of stilbene in methylcyclohexane. However, the reaction can also be catalysed by naked gold colloids of about 2 nm under the same reaction conditions with superior activity than the reference catalyst supported on titania.50 It is suggested that the protecting ligand forms a siloxane polymer which plays an active role supplying the radicals during reaction.
Lambert and co-workers51 deposited Au55 nanocrystals (ca. 1.4 nm) in inert supports and showed that they are very active catalysts for styrene oxidation with oxygen under conditions where catalysts prepared by impregnation or sol methods were inactive. Interestingly, they found only a relatively minor selectivity to the epoxide whereas the major product was the benzaldehyde. The authors highlight the importance of the particle morphology and in particular, they proposed that particles larger than 2 nm are completely inactive and suggest that the origin of catalytic activity lies in the altered electronic structure intrinsic to small gold nanoparticles. Unfortunately, the supported Au55 clusters are not retained after the cluster is supported and a range of particle sizes are observed and this makes it difficult to establish definitive structure–activity relationships.
Recent advances in the application of new carbon materials has been reflected in reports in the epoxidation of cyclooctene over gold nanoparticles supported on carbon nanotubes,52 and also in the epoxidation of cyclohexene and trans-stilbene with gold supported on mesoporous carbon (CMK-3). The authors propose that the carbon nanotubes offer a better combination of conversion/selectivity than active carbon or other typical oxide supports,52 whereas the mesoporous carbon was not superior to a silica analogue structure.53
Selective oxidation of oxygenate compounds
Selective oxidation of alcohols
Ruthenium supported nanoparticles have been used for the aerobic oxidation of alcohols to carbonyl compounds (see Table 2). Ru supported catalysts were synthesised by Kaneda and co-workers, using monomeric Ru cation species that were uniformly fixed on the surface of calcium hydroxyapatite. It was reported that the synthesised Ru3+–hydroxyapatite was an effective heterogeneous catalyst for the oxidation of various alcohols using molecular oxygen (Scheme 7).59 The oxidation of various alcohols such as benzylic and allylic alcohols was carried out at 80 °C using toluene as solvent, with yields in the range of 92–99%. Reusability of catalyst was demonstrated without any detectable leaching. Kaneda and co-workers proposed a mechanism that is based via initiation of the oxidation of the alcohol by a ligand exchange between alcohol and a chlorine species of the Ru–hydroxyapatite to form a Ru–alcoholate species, which undergoes a β-hydride elimination to produce the corresponding carbonyl compound and a Ru–hydride species. Further reaction of the hydride species with oxygen affords a Ru–hydroperoxide species, followed by a ligand exchange to regenerate the Ru–alcoholate species with the formation of oxygen and water.
| Substrate | Catalyst | T/°C | Solvent | Conv. % | Ref |
|---|---|---|---|---|---|
| a In brackets the selectivities to (aldehyde/ketone) are reported. b Note that selectivity to the aldehydes/ketone was ca. 98–99% in all entries. | |||||
| Benzyl alcohol | RuHAP | 80 | Toluene | 100 | 59 |
| 2-Octanol | Ru/Al2O3 | 83 | Trifluorotoluene | 91 | 60 |
| Cyclohexanol | RuO2-FAU | 80 | Toluene | 17 | 62 |
| 2-Thiophenmethanol | Ru–Co(OH)2–CeO2 | 60 | Benzotrifluoride | 100 | 63 |
| Benzyl alcohol | RuCoHAP | 90 | Toluene | 100 | 64 |
| Benzyl alcohol | Ru/C | 50 | Toluene | 100 | 65 |
| Benzyl alcohol | Ru(III)/HAP–Bacid | 60 | Toluene | 100 | 67 |
| Benzyl alcohol | Ru–CHNAP–MgO | 80 | Toluene | 100 | 68 |
| Benzyl alcohol | Ru(OH)x/TiO2 | 80 | Toluene | 100 | 69 |
| Cinnamyl alcohol | Pd561phen60(OAc)180/TiO2 | 60 | Acetic acid | 100 | 71 |
| Benzyl alcohol | Pd(II)/Hydrotalcite | 65 | Toluene | 98 | 72 |
| 1-phenylethanol | Pd/HAP-0 | 90 | Trifluorotoluene | 99 | 73 |
| Cinnamyl alcohol | Pd–OMC | 80 | Supercritical CO2 | 82.5 | 74 |
| Cinnamyl alcohol | Pd/PEG/SiO2 | 80 | Supercritical CO2 | 96.8 | 75 |
| Benzyl alchohol | Pd/MS | 60 | Supercritical CO2 | 92.5 | 76 |
| 1-Octanol | Pd/NiZn | 80 | Trifluorotoluene | 99 | 77 |
| Benzyl alcohol | Pd–G/SBA–16-G | 80 | Solvent free/K2CO3 | 99 | 78 |
| Glycerol | Pt–Bi/C | 50 | Water | 95(80) | 79 |
| Cinnamyl alcohol | Pt–Bi/Al2O3 | 40 | Water/Li2CO3/dodecylbenzenesulfonic acid sodium salt | 99(97) | 80 |
| Cyclohexanol | Pt–Bi/C | 50 | Water-dioxane | 99(83) | 81 |
| Cinnamyl alcohol | Pt–Bi/Graphite | 60 | ethanol | 65(95) | 82 |
| 1-Octanol | Pt–Bi/C | 60 | Toluene | 60(81) | 83 |
| Cinnamyl alcohol | Pt–Bi/Al2O3 | 65 | Toluene | 38 | 84 |
| 1-Phenylethanol | Pt/AC | 60 | Water | 81 | 85 |
 | ||
| Scheme 7 | ||
Mizuno et al. reported one of the first examples of solvent-free oxidation of alcohols.60 By using a Ru/Al2O3 catalyst the group demonstrated that aerobic heterogeneous oxidation of alcohols, which can possess sulphur atom, a nitrogen atom or a carbon–carbon double bond could be highly effective. Moreover, the reusability of the catalyst was confirmed as it was reused several times without loss of activity and without any leaching. Mechanistic investigation were performed and showed that the oxidation of alcohols proceed firstly via the formation of Ru–alcoholate species via the formation by ligand exchange between Ru–hydroxide and alcohol. Secondly, alcohol undergoes β elimination to form the corresponding carbonyl compound and Ru–H species, which was finally re-oxidised by molecular oxygen (Fig. 3).61 The rate of the reactions was of zero order dependence on the pressure of molecular oxygen. A zeolite-confined nanometre-sized RuO2 with mean particle size of 1.3 nm was synthesised by White and co-workers by using a one-step hydrothermal method.62 The confined RuO2 nanoclusters exhibited high conversion and selectivity in the aerobic oxidation of various activated (benzylic and allylic) and unactivated (saturated) alcohols under mild conditions (80 °C, toluene or chlorobenzene as solvent and atmospheric pressure) with TONs values of 5–15 (Scheme 8). Moreover, it was demonstrated the bifunctional role of the catalyst due to the fact that the zeolitic framework displayed substrate shape-selectivity and the encapsulated RuO2 nanoparticles showed high activity, which was attributed to the much higher density of active sites in nanoRuO2.
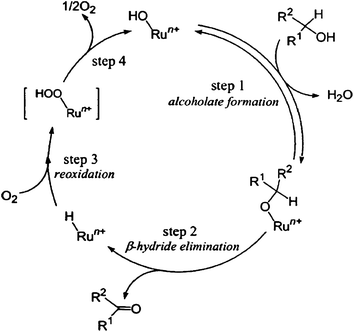 | ||
| Fig. 3 Proposed mechanism for the oxidation of alcohols using Ru/Al2O3 catalyst. Reproduced with permission from ref. 61. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. | ||
 | ||
| Scheme 8 | ||
Kaneda and co-workers developed a new bimetallic catalyst which combined Ru cation with cobalt hydroxide and cerium oxide (RuCo(OH)2–CeO2 by using a co-precipitation method.63 The obtained catalyst exhibited high catalytic activity for the oxidation of alcohols in the presence of molecular oxygen. The authors suggested that a combination of Ru with Co and Ce elements is essential for achieving high conversion levels. Significant enhancement of the formation of carboxylic acids was observed by the addition of water whereas the authors suggest that the possible role of Co was to maintain the high oxidation state of Ru. Baiker and co-workers developed a new methodology based on the hypothesis that the isolated Ru species should be close to the surface of the hydroxyapatite particles to improve the catalytic activity.64 To achieve this goal, different metal promoters were added, the amount of ruthenium used was reduced and, the pre-treatment conditions were varied. By following this approach the efficient oxidation of aromatic and aliphatic alcohols was demonstrated using toluene as solvent at 1 bar of O2 pressure and at 90 °C with TOF values in the range 15–80 h−1. The higher activity of the promoted Ru catalysts was attributed (i) the presence of Ru(OH)2 species and (ii) to the steric effect of promoter. A Ru/C commercial catalyst was used as a heterogeneous catalyst for the oxidation of primary and secondary benzylic alcohols to the corresponding carbonyl compounds without any additives such as bases and using toluene as solvent, molecular oxygen and temperature in the range 50–90 °C.65 Moreover, the authors demonstrated the general applicability of the commercial catalyst by oxidising a range of allylic and aliphatic alcohols with good yields without confirmation of re-usability studies.
Mechanistic studies confirm that the alcohol oxidation proceeds through the following steps: (i) via the formation of Ru–alcoholate species, which undergo β-hydride elimination to produce the carbonyl compound and a hydrido–ruthenium species, and finally is re-oxidised by molecular oxygen to close the catalytic cycle.66 In subsequent studies Baiker and co-workers developed Ru supported on organically modified hydroxyapatite catalysts.67 The enhanced catalytic activity observed was due to the higher intrinsic activity of the Ru species owed to their improved location and coordination in the organically modified hydroxyapatite. Ru species were located mainly on the outer surface and anchored to phosphate and hydroxyl groups, therefore these sites were more accessible to the alcohol substrate and as a consequence the catalytic activity was enhanced. The role of the organic modifiers was to act as templating agents for the controlled location and coordination of the Ru species. For the deposition and stabilisation of ruthenium species an aerogel-prepared nanocrystalline MgO (NAP–MgO) modified by the incorporation of choline hydroxide was used.68 The authors claimed that the development of Ru3+ took place during the preparation method and the Ru3+ species were distributed on the outer surface by a combination of strong electrostatic interaction and coordination between the surface functionalised MgO and Ru3+. The catalytic performance of the material showed high conversion of aromatic alcohols with yields approximately of 95% to ketones when toluene was used as solvent at 80 °C and molecular oxygen as the oxidant. No significant loss of catalytic activity was found during reusability tests. Deposition of ruthenium hydroxide (Ru(OH)x) on a variety of supports (titania and silica) by Mizuno et al.69 and showed high catalytic activity for the aerobic oxidation of alcohols including a variety of benzylic, allylic, aliphatic and heteroatom-containing ones into the corresponding carbonyl compounds. The catalytic activity was dependent on the coordination number of nearest-neighbour Ru atoms (the size of Ru(OH)x species) and the reusability of Ru(OH)x/TiO2 catalyst was successful without an appreciable loss of this catalytic activity. Subsequent theoretical studies70 confirmed that the reaction mechanism took place viaalcoholate formation and β-hydride elimination steps. Moreover, it was demonstrated that the alcoholate formation was promoted by the “concerted activation” of an alcohol by the Lewis acid and Brønsted bases on Ru(OH)x/support and the formed Ru–H species reacted with molecular oxygen to form Ru–OOH, which could further react with an alcohol or water regenerating the alcoholate or hydroxide species with simultaneous formation of hydrogen peroxide.
The catalytic performance of Pd based catalysts has been shown successfully in a variety of oxidation reactions, including hydroxylation of benzenes, oxidation of alkanes, oxidative coupling reactions, epoxidation of alkenes and oxidation of alcohols/aldehydes (see Table 2).7 A large variety of different synthetic approaches have been designed and utilised for the synthesis of active Pd based catalysts. Immobilisation of giant Pd clusters with five shells, Pd561phen60(OAc)180 on the surface of metal oxides, i.e.TiO2 and the catalytic performance was studied by Kanedaet al.71 The immobilised giant Pd clusters (2–3 nm mean particle size) efficiently catalysed the oxidation of primary allylic alcohols in the presence of molecular oxygen with conversion levels higher than 90%. The active sites are suggested to be Pd–Pd paired sites with oxidation state smaller than +2, where the oxidations took place via multiple interactions of these sites with the allylic alcohols. In another report, a heterogenised Pd catalyst was developed by Kakiuchi et al.,72 where a Pd(II)-hydrotalcite catalyst was synthesised by supporting a palladium(II) acetate–pyridine complex on commercially available hydrotalcite. The oxidation of benzyl alcohol in the presence of toluene and pyridine at mild conditions (65 °C) to benzaldehyde with 98% yield was reported and the general applicability of the Pd(II)-hydrotalcite in the oxidation of aliphatic and alkenic alcohols as well as of diols was demonstrated. Moreover, the authors attributed the efficient conversion of the alcohol due to the addition of pyridine and that the temperature affects the final oxidation state of Pd. It was shown that Pd(0) is not active due to the fact that at high temperature (80 °C) the reduction of Pd(II) to Pd(0) took place and resulted in an inactive catalyst (Scheme 9). Reusability tests showed Pd leaching and decrease of catalytic activity.
 | ||
| Scheme 9 | ||
Kaneda and co-workers73 extended their initial work with palladium nanoclusters71 (Fig. 4) and demonstrated the synthesis of palladium-grafted hydroxyapatite, where Pd supported nanoclusters can be synthesised in the presence of alcohol. The aerobic oxidation of aromatic alcohols such as 1-phenylethanol was successfully demonstrated by using nanoclustered Pd(0) species, with TOF (turnover frequency) of up to 9800 h−1 and TON (turnover number) of up to 236000. Moreover, Kanedaet al. demonstrated that the catalyst is recyclable and catalytic activity was dependent on particle size (Scheme 10). Finally, they proposed that low-coordinated Pd atoms are responsible for the activation and oxidation of alcohols (Fig. 5), therefore the alcohol oxidation is considered “structure sensitive”.
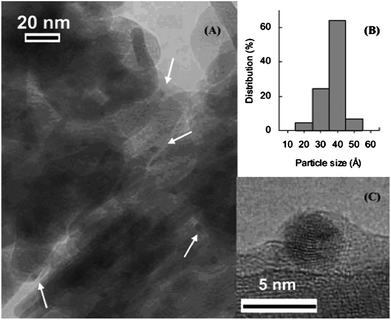 | ||
| Fig. 4 (A) TEM image, (B) size distribution diagram of the Pd nanoparticles, and (C) HR-TEM image of an eight-shell Pd nanocluster for the recovered PdHAP-0 catalyst after oxidation of 1-phenylethanol. Reproduced with permission from ref. 73. Copyright American Chemical Society. | ||
 | ||
| Scheme 10 | ||
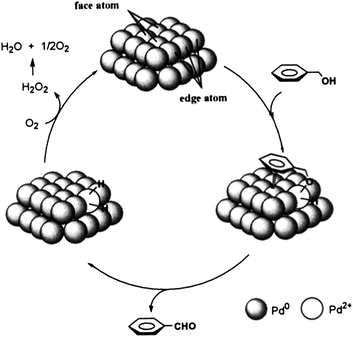 | ||
| Fig. 5 Possible reaction mechanism for the oxidation of alcohols on the surface of Pd nanocluster. Reproduced with permission from ref. 73. Copyright American Chemical Society. | ||
The synthesis of well-dispersed Pd metallic nanoparticles on ordered mesoporous carbon (Pd-OMC), was reported by Schüth and co-workers.74 Moreover, it was demonstrated that Pd clusters were highly thermally stable and fine dispersed Pd clusters below 1 nm were uniformly embedded in the carbon walls. High catalytic activity was reported at mild conditions (80–100 °C) for the oxidation of aromatic alcohols, such as benzyl alcohol, cinnamyl alcohol and 1-phenylethanol, with selectivity around 99%, using supercritical CO2 as reaction medium (Scheme 11).
 | ||
| Scheme 11 | ||
The utilisation of supercritical CO2 for the oxidation activity of palladium was also reported by Leitner and co-workers.75 By using batch as well as continuous-flow process they reported efficient and stable catalysts for the selective aerobic oxidation of benzylic and allylic alcohols to aldehydes and ketones with selectivities over 98% and TONs values in the range 22–47 using supercritical CO2 as the mobile phase. Stabilisation of the palladium nanoparticles by polyethylene glycol (PEG)-modified silica was reported and the authors claim that this procedure resulted in a lower rate of agglomeration of the metal nanoparticles. In following studies76 they reported the immobilisation of palladium nanoparticles on mesoporous silica using 2,2′-dipyridylamine as a linker. The role of the linker was to facilitate immobilisation and stabilisation of the Pd nanoparticles. They observed that particle size as well as oxidation state were affected by the method of reduction, either using benzyl alcohol under reflux conditions or molecular hydrogen. They attributed the high catalytic activity due to the formation of very small palladium particles with metallic oxidation state. The hydrogen treatment resulted in average particle size of approximately 6 nm, larger than the 2 nm particles obtained by the alcohol reduction approach.
Shimazu and co-workers77 used a new approach for the synthesis of Pd(II) catalysts supported on Ni–Zn mixed basic salt. Specifically, their strategy was based at the intercalation of an anionic [Pd(OH)4]2− species into an interlayer of NiZn. The intercalated anionic Pd hydroxide complex was rigidly fixed by the strong electrostatic interactions characteristic of the NiZn. The formed catalyst (Pd/Ni/Zn) was an efficient heterogeneous catalyst for the aerobic oxidation of benzylic, aliphatic and allylic alcohols to the corresponding aldehydes/ketones without the need of any additives with yields of around 90% and using trifluorotoluene as a solvent at 80 °C. The authors proposed a reaction mechanism based on the following steps: (i) initial binding of the alcohol to the Pd(II) hydroxide species to form a Pd(II)-alkoxide species and the carbonyl product, (ii) then the Pd(II)-hydride species could reductively eliminate water to form Pd(0), which could then re-oxidised by molecular oxygen to form Pd-peroxide species. A grafting method was used by Ji and co-workers78 for the synthesis of supported Pd catalysts. Using SBA-16 as the chosen support, a mixture of palladium–guanidine complex and guanidine was grafted on SBA-16via a one-pot silylation. In this way the Pd–quanidine complex was introduced into the interior of SBA-16 and the mesoporous material structure of SBA-16 remained intact during the preparation method. The aerobic oxidation of alcohols using toluene as a solvent and K2CO3 as a base at 80–100 °C was carried out with selectivities to aldehydes/ketones of 99%. After 10 cycles only a minimum loss of activity was observed without affecting selectivity. Characterisation of the materials by means of TEM showed the formation of Pd nanoparticles that were stabilised by the nanocages of SBA-16 and consequently preventing aggregation and agglomeration of Pd nanoparticles.
Baiker and co-workers86 carried out systematically mechanistic and structural studies in the selective oxidation of benzyl alcohol, cinnamyl alcohol and 1-phenylethanol using Pd supported catalysts. The oxidation of the aforementioned substrates using site-selective blocking by CO and isotope labelling with in situ attenuated total reflection infrared spectroscopy (ATR-IR) proved to be highly informative. A commercial Pd/Al2O3 catalyst with mean metal particle size of 3–4 nm was used for discriminating the active sites. High-resolution transmission electron microscopic images (HR-TEM) of the commercial Pd/Al2O3 showed that the palladium particles are round-shaped particles with defined (111) and (100) crystalline planes and possessing a cuboctahedron morphology (Fig. 6). The authors conclude that the active sites responsible for the dehydrogenation reaction during the liquid phase oxidation of benzyl alcohol are all planes exposed by the Pd particles, whereas those sites catalysing the undesired decarbonylation of the product are (111) planes. Moreover, the possible role of the oxidation state of Pd was investigated for exploring the desired oxidation state that will enhance aerobic alcohol oxidation.87 By means of a combination of in situ-X-ray adsorption spectroscopy with on-line catalytic measurements and FT-IR spectroscopy the authors identified that in the presence of metallic palladium a significant increase in activity was observed with respect to palladium oxide. The increased catalytic activity observed with the metallic palladium was a factor of 50 times higher than the palladium oxide. To optimise the conditions for the oxidation of alcohols it is essential that first the Pd constituent should remain in metallic state and secondly the presence of oxygen is required to remove the formed hydrogen and degradation products from the catalyst surface.
 | ||
| Fig. 6 High-resolution transmission electron microscopy (TEM, left) a commercial Pd/Al2O3 catalyst. Right is a representation of the idealized cuboctahedron shape of a Pd crystallite exposing (111) and (100). Reproduced with permission from ref. 86. Copyright American Chemical Society. | ||
The utilisation of Pt-based catalysts for the liquid phase oxidation of alcohols to highly valuable products has been extensively explored by academia in recent years.54,55 Baiker and co-workers synthesized Bi–Pt supported catalysts by selective deposition of Bi onto supported Pt particles of 3–4nm (see Table 2). The synthesized Bi–Pt catalysts were tested in the selective aerobic oxidation of cinnamyl alcohol to cinnamaldehyde as the desired product and using water as solvent. It was concluded that by increasing Bi/Pt surface ratio a higher Bi coverage was observed, which suppressed the hydrogen adsorption on Pt and therefore improved significantly not only activity but also selectivity to cinnamaldehyde. The increased activity of the Bi–Pt supported catalyst in comparison to the monometallic Pt catalyst was attributed to the presence of Bi particles. The role of the Bi particles is the effective blockage of Pt sites (geometric blocking) and overall of suppressing deactivation that are responsible for over-oxidation of cinnamaldehyde.80 The liquid phase oxidation of cyclohexanol to adipic acid using Pt/C catalysts was investigated by Gallezot and co-workers and high activity was obtained using water as solvent at moderate temperature and pressure (150 °C, 5 MPA air). During their investigation they showed that high conversion of cyclohexanol is achievable (90%) with selectivity to adipic acid of 50% and major byproducts glutaric and succinic acids. The production of adipic acid using a green process and a heterogeneous catalyst is highly desirable due to the highly industrial importance of adipic acid as an important intermediate for the manufacture of nylon but also as adipic acid is used as plasticizer and food additive. However, the main drawback of this method is the low solubility of cyclohexanol in water, nevertheless the utilisation of water as green solvent and air as oxidant makes this process environmentally cleaner and friendly.88 In subsequent studies the same group showed the efficient liquid aerobic oxidation of unsaturated alcohols (9-decen-1-ol) using Pt supported catalysts at mild conditions.81 The desired product was 9-decenoic acid which has been used in the preparation of flavour and fragrance ingredients. By performing the oxidation at 50 °C using molecular oxygen, NaOH as base and water/dioxane as solvent, high conversion (99%) was reported with selectivity of 83%. Moreover addition of Bi on the Pt catalyst minimized deactivation and improved activity and resistance of the catalyst. The oxidation of allylic alcohols was in addition carried out by Lee and co-workers using Pt–Bi/graphite catalysts.82 They performed systematic studies for understanding the influence of the double bond position within the chain in terms of activity and selectivity. After performing systematic studies they concluded that the carbon double bond is playing a role in the control of oxidation and depending the position of the double bond within the chain can facilitate the anchoring of the hydroxyl group to the catalyst surface and therefore to influence the oxidative dehydrogenation step to the aldehyde. Further investigation of the effect of air pressure, catalyst mass and stirrer speed showed that aldehyde formation was zero-order in dioxygen and the main role of oxygen was in the removal of carbonaceous species from the catalyst surface while the reaction was not mass-transport limited. A high throughput screening technique was used by Griffin and co-workers for identifying the effect of different composition of Pt/Bi supported catalysts in terms of catalyst activity and product selectivity.83 Using water as solvent and air as oxidant they identified that the most efficient catalyst for the transformation of a variety of alcohols was 5%Pt–1%Bi/C. In addition, they demonstrated the utilisation of hydrogen peroxide as green oxidant with high efficiency. Baiker and co-workers investigated the effect of promoter by using a simple methodology.84 The catalytic performance in the presence and absence of molecular oxygen was carried out using various promoted (Bi, Pb) and unpromoted Pt-group catalysts in the oxidation of aliphatic, aromatic and allylic alcohols. From their studies it was reported that the role of the promoter depends on the substrate used and, it is either to influence the reaction rate and selectivity of the alcohol dehydrogenation reaction or the adsorption and transfer of oxygen. Moreover, the role of oxygen was identified as to participate in the oxidation of the co-product hydrogen to water and the oxidative removal of surface impurities which results in the improved resistance of Pt metal against over-oxidation. Ikeda and co-worker reported a methodology that facilitates the incorporation of Pt nanoparticles of 3nm particle size inside the framework of porous carbon and in this way minimizes particle aggregation, movement and leaching. In this way active and reusable catalyst for the oxidation of a range of alcohols was reported.85 The synthesized Pt on carbon catalyst selectively transformed at high yield aromatic alcohols to aldehydes/ketones such as benzyl and 1-phenylethanol at mild conditions, using atmospheric oxygen, at 60 °C and water as the solvent. The high activity observed is due to the incorporation of Pt nanoparticles inside the pores of the carbon, which consist of three-dimensional hydrophobic channels where efficient mass transfer and preferential adsorption of the reaction substrate is possible.
Au based catalysts have been explored extensively the last 15 years in the selective oxidation of alcohols, polyols,89,90 and especially in the last decade (see Table 3).
| Substrate | Catalyst | T/°C | Solvent | Conv. (%) | Sel.% (Aldehyde/ketone) | Ref |
|---|---|---|---|---|---|---|
| Propane-1,2-diol | Au/C | 90 | Water/Sodium Hydroxide | 78 | 100(Acid) | 33 |
| Ethane-1,2-diol | Au/C | 70 | Water/Sodium Hydroxide | 100 | 96(Acid) | 34 |
| D-Glucose | Au/C | 50 | Water/Sodium Hydroxide | 99 | 99(Acid) | 91 |
| Glycerol | Au/G | 60 | Water/Sodium Hydroxide | 56 | 100(Acid) | 92 |
| Glycerol | Au/C | 60 | Water/Sodium Hydroxide | 90 | 83(Acid) | 93 |
| Glycerol | Au/CeO2 | 60 | Water/Sodium Hydroxide | 33 | 44 (Acid) | 94 |
| Salicylic alcohol | Au/Fe2O3 | 60 | Water | 90 | 90 | 95 |
| 3-Octanol | Au/CeO2 | 80 | Solvent-free | 97 | 99 | 96 |
| Ethanol | Au/MgAl2O4 | 150 | Water | 97 | 86 | 97 |
| Benzyl alcohol | Au/GMS | 130 | Toluene | 99 | 79 | 98 |
| 4-Methylbenzyl alcohol | Au/Cu5Mg1Al2Ox | 90 | Mesitylene | 98 | 99 | 99 |
| 1-Phenyethanol | Au-Microgel | 60 | Water | 75 | 100 | 100 |
| Benzyl alcohol | Au/MnO2 nanorods | 120 | Solvent-free | 41 | 99 | 101 |
| Benzyl alcohol | Au/γ-Ga2O3 | 130 | Solvent-free | 40 | 98 | 102 |
| Benzyl alcohol | Au/Ga3Al3O9 | 80 | Toluene | 98 | 99 | 103 |
| Benzyl alcohol | Au/PoPD | 25 | Water | 99 | 99(Acid) | 104 |
| 1-Phenylethanol | Au/Zn | 90 | Solvent-free | 6.5 | 99 | 105 |
| Benzyl alcohol | Au/SBA-15 | 80 | Water | 100 | 91(Acid) | 106 |
| 1-Phenylethanol | Au/PNIPAM | 80 | Water | 99 | 99 | 107 |
| Cyclohexanol | Au/Fe3O4@SiO2 | 100 | Toluene | 42 | 100 | 108 |
| Benzyl alcohol | Au/ZrO2 | 94 | Solvent free | 59.5 | 81.5 | 109 |
| 1-Phenylethanol | Au/TiO2 | 90 | Water | 99 | 100 | 110 |
| Benzyl alcohol | Au/SBA-15 | 60 | Water/K2CO3 | 99 | 87 (Acid) | 111 |
Galvagno and co-workers, by synthesizing Au/Fe2O3 catalyst using a co-precipitation method, performed catalytic tests in the liquid-phase oxidation of o-hydroxybenzylalcohol under mild conditions (50 °C, PO2 = 1 Atm). By increasing the gold loading activity followed an increase, whereas the selectivity to aldehyde at similar levels of conversion was higher with lower metal loading. The reaction is first order with respect to the organic substrate and zero order with respect to oxygen partial pressure.95 The combination of small gold particles (2–5 nm) and nanocrystalline ceria (5 nm) produced a highly active, selective and recyclable catalyst for the oxidation of alcohols into aldehydes and ketones with high TON numbers at solvent free conditions (Scheme 12) as it was demonstrated by Corma and co-workers. Moreover, the same group investigated the possible mechanism and they attributed the high activity due to the deposition of gold nanoparticles onto the support that transform nanocrystalline cerium oxide from a stoichiometric oxidant into a catalytic material (Fig. 7).96
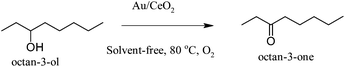 | ||
| Scheme 12 | ||
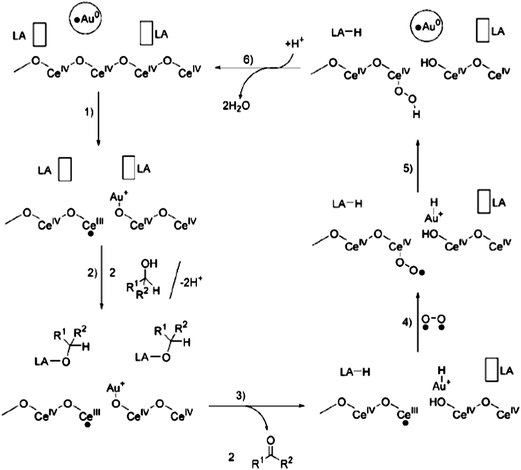 | ||
| Fig. 7 Proposed mechanism for the oxidation of alcohols using Au/CeO2 catalyst (LA = Lewis acid). Reproduced with permission from ref. 96. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The feasible formation of acetic acid was demonstrated by Christensen and co-workers using aqueous-phase oxidation of ethanol with air in the presence of gold supported nanoparticles at mild conditions (Scheme 13). MgAl2O4 was a very effective support for the deposition of gold nanoparticles using deposition–precipitation methodology for the deposition of gold nanoparticles. High conversion of ethanol was achieved in the range of 90–200 °C with yield above 80%.97
 | ||
| Scheme 13 | ||
By confining gold nanoparticles in the walls of mesoporous silica, Richards and co-workers reported the effective aerobic oxidation of alcohols with high activity and selectivity to aldehyde (79–94%) using toluene as solvent and K2CO3. Moreover, reusability tests showed that the catalyst was reusable and prevention of sintering of the gold nanoparticles was demonstrated due to the fact that gold nanoparticles were inside the pore channels of the mesoporous silica (GMS).98 Baiker and co-workers deposited gold nanoparticles (6– 9 nm range) on Cu-Mg-Al mixed oxides using a deposition–precipitation method99 and it was reported that the activity strongly depended on the composition of the support, (Cu/Mg molar ratio) with Au/Cu5Mg1Al2Ox showing the highest catalytic activity. XANES analysis revealed the presence of gold reduced and oxidized species on the ternary mixed oxide support before and after reaction. The oxidation of several alcohols was demonstrated at full conversion and at high selectivity (98%). In subsequent studies, Baiker and co-workers focused in studies for determining the oxidation state of gold during aerobic alcohol oxidation. By using XANES it was observed that the catalytic activity increased with the increase in the reduction of the gold component112 therefore, the main active species in catalytic aerobic oxidation is metallic gold. The use of microgels for stabilizing gold nanoclusters and their use as “guasi-homogeneous” catalysts for the aerobic oxidation of primary and secondary alcohols in water was reported by Prati and co-workers.100 Microgels containing gold can showed high and comparable activity with known and well used gold supported catalysts. Deposition of gold nanoparticles (4– 5 nm) onto MnO2 nanorods was reported by Cao and co-workers and they demonstrated the high catalytic efficiency of these catalysts in the liquid phase oxidation of benzyl alcohol and 1-phenylethanol.101 Moreover, comparison with commercial Au/MnO2 showed that the Au/MnO2 nanorods showed much higher catalytic performance and it was attributed to the collaborative effects resulting from the interaction of the gold nanoparticles and the well-defined reactive surface of MnO2 nanorods. Au nanoparticles supported on different polymorphs of gallia (α-β- and γ-Ga2O3) were active in the solvent-free liquid phase aerobic oxidation of benzyl alcohol.102 Different activity was obtained among the gold supported on different polymorphs of Gallia, suggesting that the support could influence catalytic activity. The most active gold supported catalyst was obtained when γ-Ga2O3 was the chosen support. General applicability of the Au/γ-Ga2O3 was demonstrated for oxidizing a range of aromatic and aliphatic alcohols with selectivities up to 98% to their corresponding ketones and aldehydes as well as reusability. The high activity of Gallia-supported gold nanoparticles is considered to be due to the high hydrogenation capability of γ-Ga2O3 in comparison to other supports. In subsequent studies, a series of binary mesostructured Ga-Al mixed-oxide supports (GaxAl6−xO9, x = 2, 3, 4) were synthesized and gold was deposited.103 The Au/GaxAl6−xO9 material showed high catalytic performance in the aerobic oxidation of alcohols at mild conditions (80 °C) and high selectivity to the aldehyde/ketone formation (Scheme 14). Comparison with other conventional oxide-supported systems showed that Au/GaxAl6−xO9 was superior and the authors concluded that for achieving optimum activity the collaborative interaction between gold and the mixed-oxide support is essential. Finally, reusability tests confirmed that the catalyst is reusable and the spinel structure of the oxide was retained during the oxidation process.
 | ||
| Scheme 14 | ||
Guo and co-workers developed a novel method for the synthesis of gold nanoparticles with different particle size (3 to 15 nm) and shape supported on both inner and outer surfaces of poly(o-phenylenediamine hollow microspheres (PoPD).104 High catalytic activity was achieved using the supported gold nanoparticles in the liquid phase aerobic oxidation of alcohols using water as a solvent and K2CO3, at very mild conditions (room temperature) with yields in the range 91–95%. Hensen and co-workers reported the synthesis of supporting gold nanoparticles (4–6 nm range) onto basic hydrozincite or bismuth carbonate and the efficient oxidation of alcohols in the liquid phase.105 The catalytic performance of a series of metal (Zn, Bi, Ce, La, Zr) carbonate supported gold catalysts was highly dependent on the basicity (presence of strong basic sites) of the supported materials, where the initial O–H bond cleavage is likely on the support basic sites. Using this approach it is evident the possibility to develop heterogeneous catalysts possessing strong base sites for alcohol oxidation and therefore it is not necessary to utilise soluble bases. Immobilisation of around 1 nm Au clusters within mesoporous silicas (SBA-15, MCF, HMS) using triphenylphosphione-protected Au clusters (Au11:TPP) as precursors was reported by Tsukuda and co-workers. Gold (Au11:TPP) clusters were homogeneously dispersed and by controlled calcinations the removal of the protecting ligands was achieved without aggregation of the resulting Au clusters. The catalytic properties of the gold supported clusters were studied in water with addition of K2CO3 and using benzyl alcohol as model reaction high yield of 91% to benzoic acid was achieved (Scheme 15).106
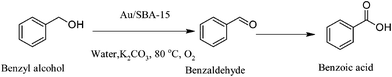 | ||
| Scheme 15 | ||
Size-controlled Au nanoparticles (2.6–6.3 nm) within a porous chelating hydrogel of poly(N-isopropylacrylamide)-co-poly[2-methacrylic acid 3-(bis-carboxymethylamino)-2-hydroxypropyl ester] referred as [PNIPAM-co-PMACHE] were reported by Zhang and co-workers.107 The encapsulated Au nanoparticles were tested for aerobic alcohol oxidation (oxidation of 1-phenyethanol) in the presence of water and KOH and it was demonstrated that the catalytic activity depends on the particle size of the Au particles. Moreover, the Au encapsulated nanoparticles in the hydrogel were highly efficient and reusable catalysts. Au nanoparticles immobilized on a magnetically recoverable support (core-shell Fe3O4@SiO2) were synthesized by Rossi and co-workers. High catalytic activity (conversions of 100%) of the synthesized Au supported catalysts was reported in the liquid phase aerobic oxidation of benzyl alcohol, 1-phenylethanol and cyclohexanol using toluene as solvent, K2CO3 at 100 °C.108 Reusability tests showed a decrease in activity due to growth of particle size and change in the morphology of the support. A magnetically recoverable support could potential offer advantages for the recovery of the catalyst.
Synthesis of Au–Pd supported catalysts and their utilisation in the aerobic liquid phase oxidation of alcohols has attracted significant levels of research. The main reasons for the academic and industrial interest relies on the fact that (i) gold and palladium can form solid solutions in the whole range of gold/palladium atomic ratio and (ii) the addition of second metal can alter the electronic and geometrical properties of the synthesised particle, therefore will affect in a positive manner catalytic activity, catalyst stability and distribution of products.
Prati and co-workers reported the catalytic performance of Au–Pt and Au–Pd single phase catalysts (Fig. 8) in the selective oxidation of various primary alcohols (benzyl alcohol, cinnamyl alcohol and 1-octanol) with Au–Pd catalysts were by far more active than Au–Pt catalysts (Scheme 16). A significant improvement in catalytic activity was found when water instead of toluene was used as a solvent, with TOFs values to increase by a factor of 1.5–6.115 Moreover, using the same preparation method they demonstrated the general applicability of the single-phase alloy Au–Pd/C catalysts and studied systematically the effect of AuxPdy molar ratio in a range of alcohols. They concluded that the most efficient Au/Pd compositions was Au80/Pd20 in terms of catalytic activity (see Table 4).116
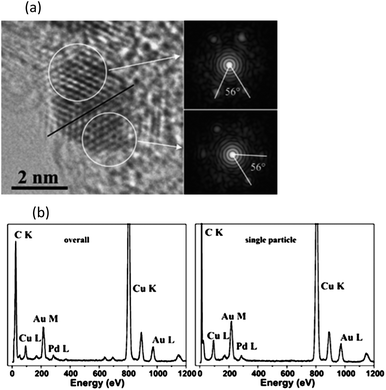 | ||
| Fig. 8 (a) Representative HRTEM image of small particles from Au–Pd/AC and (b) Overall EDX spectrum and the representative one for individual single particles for Au–Pd/AC. Reproduced with permission from ref. 115. Copyright Elsevier Inc. | ||
 | ||
| Scheme 16 | ||
| Substrate | Catalyst | T/°C | Solvent | Conv.% | Sel.% (Aldehyde/ketone) | Ref |
|---|---|---|---|---|---|---|
| Benzyl alcohol | Au–Pd/TiO2 | 100 | Solvent-free | 90 | 95 | 31 |
| Benzyl alcohol | Au–Pd/C | 120 | Solvent-free | 80 | 65 | 37 |
| Cinnamyl alcohol | Au–Pd/C | 60 | Water | 95 | 83 | 115 |
| Benzyl alcohol | Au80–Pd20/C | 60 | Water/Sodium Hydroxide | 90 | 99 | 116 |
| Benzyl alcohol | Au–Pd/SBA-15 | 80 | Water/Na2CO3 | 40 | 99 | 121 |
| Benzyl alcohol | Au–Pd/polystyrene | 100 | Toluene | 99 | 98 | 123 |
| 1-Phenyethanol | Au–Pt/polystyrene | 25 | Water/Benzotrifluoride | 99 | 99 | 124 |
| 1,2-propanediol | Au–Pd/TiO2 | 60 | Water/NaOH | 94 | 96 (Acid) | 36 |
| D-Sorbitol | Au–Pd/C | 50 | Water/Sodium Hydroxide | 90 | 90 (Acid) | 138,139 |
| Glycerol | Au–Pd/C | 50 | Water/Sodium Hydroxide | 90 | 90 (Acid) | 140–143 |
| Glycerol | Au–Pt/C | 50 | Water/NaOH | 70 | 60 (Glyceric acid) | 144 |
| Glycerol | Au–Pt/Mordenite | 100 | Water | 70 | 83 (Glyceric acid) | 145 |
Bimetallic Au–Pd supported catalysts were reported by Hutchings and co-workers, where the solvent-free liquid phase oxidation of a variety of alcohols was possible at 160 °C, using molecular oxygen without the use of initiators or solvents with a Au–Pd/TiO2 catalyst synthesised by the impregnation method (Scheme 17).31 The TOFs values reached up to 269000 h−1 and the high activity of the Au–Pd supported catalyst was due to the gold core–palladium shell structure on the support surface and the electronic promotion of Au for Pd (Fig. 9). Moreover, the Au/Pd weight ratio was studied and it was found that the most active catalyst was the one with Au/Pd weight ratio of 1/1, whereas the highest selectivity to benzaldehyde was observed with Au-rich catalysts.117
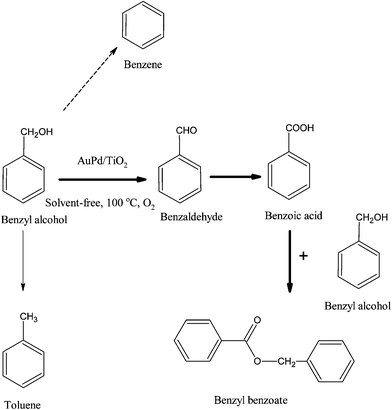 | ||
| Scheme 17 Solvent-free oxidation of benzyl alcohol using Au–Pd supported nanoparticles. | ||
 | ||
| Fig. 9 (a) Montage showing the ADF-STEM image of a bimetallic particle and the corresponding reconstructed MSA-filtered Au–Pd–Ti composition map (Ti, red; Au, blue; and Pd, green). Reproduced with permission from ref. 31. Copyright AAAS. | ||
In following studies Hutchings and co-workers investigated the synthesis of Au–Pd supported catalysts using a colloidal method and they demonstrated the efficient aerobic oxidation of benzyl alcohol with very high TOFs at lower reaction temperatures.28 STEM-XDS and XPS analysis showed the presence of random homogeneous alloys with metallic oxidation state for Au and Pd. The higher activity of the Au–Pd supported catalysts synthesised by the colloidal method instead of the impregnation method was attributed to the smaller particle size, narrow particle size distribution and metallic oxidation state.
A preparation strategy for the synthesis of bimetallic hydrosols with the formation core-shell structures, involving the sequential addition and reduction of the metal and deposition of the bimetallic sols on carbon and titania was used for the synthesis of bimetallic supported catalysts.37 It was found that the catalytic activity of the aerobic oxidation can be achieved at mild conditions (120 °C, PO2 = 10 bar) and the order of metal addition has a marked effect on activity. The choice of support (carbonversustitania) was shown to affect significantly the activity and distribution of products with carbon supported materials to give an increase of activity by a factor of 2 and a lower selectivity to benzaldehyde at iso-conversion compared to the titania-supported catalysts. It was found that the reaction is zero order in oxygen and the oxidation of benzaldehyde is dependent on the concentration of oxygen at the surface. Sankar et al. studied in detail the mechanism of benzyl alcohol oxidation particularly the formation of toluene.118 Initial rate measurements, deuterium labeling, kinetic isotope effects, and the study of substituent effects were the basis for proposing a multiple reaction pathway in this heterogeneous system. Subsequently119 the two pathways were identified as the sources of the principal product, benzaldehyde: the direct catalytic oxidation of benzyl alcohol to benzaldehyde by O2, and the disproportionation of two molecules of benzyl alcohol to give benzaldehyde and toluene. Furthermore, it was found possible to switch off the disproportionation reaction and thereby completely stop the toluene formation by optimising the support. In a recent report, Miedziak et al. showed that for the same catalytic system, sol immobilization is superior to deposition–precipitation techniques.120 They additionally report that the utilisation of glass reactors at mild reaction conditions (low oxygen pressure and low temperature) can suppress significantly the financial cost of equipment and provide an accurate and easy way of transforming alcohols to useful chemical intermediates. Qiao and co-workers reported the synthesis of Au–Pd/SBA-15 material by impregnation and grafting methods.121 It was found that by using the grafting method metal nanoparticles (5 nm mean particle size) could be highly dispersed in mesoporous channels of SBA-15, showing high catalytic performance in the selective oxidation of benzyl alcohol to benzaldehyde at mild reaction conditions (80 °C, water as solvent and Na2CO3). It was demonstrated that agglomeration and leaching of metal nanoparticles was avoided by restricting the nanoparticles inside the mesopores of SBA-15, therefore leading to enhanced stability and reusability of Au–Pd/SBA-15. The effective confinement of Au–Pd nanoparticles was also reported by Y. Yang and co-workers using SBA-16 as support. Au–Pd nanoparticles were supported on SBA-16 using an adsorption method. The authors demonstrated the synergistic effect by of the bimetallic Au–Pd supported nanoparticles by showing enhanced catalytic performance of the Au–Pd catalyst compared to the Au and Pd monometallic catalysts. STEM and EDX analysis revealed the alloyed structure of the bimetallic nanoparticles with Pd rich shell and Au core structure.122
Mechanistic studies were performed in the oxidation of benzyl alcohol using a Au–Pd alloy catalyst synthesised by sol-immobilisation method and it was found that in the absence of oxygen benzyl alcohol was transformed into benzaldehyde and toluene at initial equal rates.118 Introduction of oxygen significantly increased the rate of benzyl alcohol disappearance and appearance of benzaldehyde at the expense of toluene formation. It was found that at low partial pressures (below 3 bar of oxygen) rates are dependent on oxygen, suggesting that oxygen can participate in the reaction pathway as an adsorbed species (Fig. 10). The beneficial interaction of gold and palladium in bimetallic catalysts was also demonstrated by Baiker and Marx by synthesizing bimetallic catalysts using a colloidal route, where the admixing of Pd to Au resulted in the synthesis of bimetallic nanoparticles in a narrow range (2.4–3.7 nm) and a Au rich core and a Pd rich shell.123 The synthesised Au–Pd supported nanoparticles on polyaniline showed high activity in the liquid phase oxidation of benzyl alcohol and high selectivity to benzaldehyde (98%) using toluene as solvent and aqueous solution of NaOH.
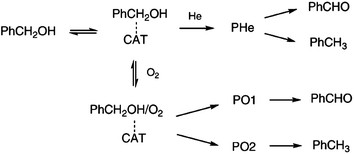 | ||
| Fig. 10 Proposed reaction scheme for oxidation of alcohols using AuPd supported nanoparticles synthesised by colloidal method. Reprinted with permission from ref. 118. Copyright RSC. | ||
The utilisation of Au–Pt supported nanoparticles for the liquid phase oxidation of a variety of alcohols has been presented with high yields (90–99%) of the corresponding aldehydes or ketones at room temperature using atmospheric pressure and benzotrifluoride/water as solvent (Scheme 18).124 A colloidal method was used for the synthesis of the metal nanoparticles. X-Ray spectroscopy confirmed the presence of both metals in each nanoparticle and the particle size distribution was in the range 1.5–5 nm. Catalyst reusability showed that the catalysts are reusable several times without loss of activity.
 | ||
| Scheme 18 | ||
Selective oxidation of oxygenated compounds derived from renewable feedstocks
Oxidation of polyols
Hutchings and co-workers demonstrated for the first time the utilization of gold based catalysts in the selective oxidation of glycerol to glycerate using alkaline conditions (Scheme 19). By synthesizing gold supported nanoparticles of around 25 nm mean particle size and varying parameters, such as pressure, sodium hydroxide and catalyst amount, they optimized conversion of glycerol and yield to glycerate.92 It was proposed that the role of sodium hydroxide was essential for the initial dehydrogenation pathway, since in the presence of base the hydrogen is readily abstracted from one of the primary hydroxyl groups of glycerol and becomes the rate determining step. In subsequent studies they demonstrated the superior performance of the gold catalyst with respect to Pd and Pt based catalysts in terms of high selectivity to glycerate and higher catalytic activity.131
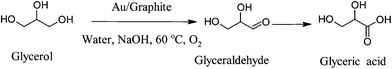 | ||
| Scheme 19 | ||
Prati and Porta investigated the effect of different preparation methods where a variation of particle size could be achieved and from their studies they concluded that small gold nanoparticles of 6 nm mean diameter and well-dispersed were responsible for high activity to glycerate,93 however the initial selectivity could not be maintained due to the consecutive oxidation of glycerate to tartronate. Large nanoparticles of around 20 nm were responsible for high selectivity to glycerate without over-oxidation of glyceric acid.93 From mechanistic studies they concluded that the overall selectivity of the reaction is affected from a combination of factors such as initial selectivity of the catalyst, base-catalyzed inter-conversion and stability of the products.
Claus and co-workers investigated the effect of support as well as of the gold particle size and it was concluded that under the same reaction conditions and with similar particle size, the carbon supported gold catalysts are more active than the oxide supported gold catalysts.132 From the investigation of the gold particle size they concluded that the reaction is structure-sensitive in agreement with the previous observations by Hutchings and Prati. In subsequent studies they investigated the effect of ceria as support, although the ceria supported catalysts were active, there was a considerable decrease in catalytic activity during recycling tests due to gold leaching.94 The effect of gold particle was also studied by Davis and co-workers and it was confirmed that small gold nanoparticles are more active than large gold nanoparticles and the selectivity to glycerate dropped with gold particle size decreased. Most importantly, Davis et al. observed the formation of hydrogen peroxide over all gold catalysts and that the concentration of hydrogen peroxide had a direct relationship with the selectivity to glycerate; lower amounts of hydrogen peroxide were associated with higher selectivity to glycerate and less formation of glycolate. They concluded that the formation of glycolic acid, which is due to C–C cleavage may be unavoidable over monometallic gold catalysts.133,134
The effect of the preparation method was studied by Prati and co-workers and it was found that a low temperature chemical reduction of the gold supported catalyst enhances activity due to the formation of gold particles in the range of 2–5 nm in a narrower particle size distribution. The use of a higher pre-treatment temperature (400 °C) resulted in lower activity due to the increase of gold particle size and also to higher selectivity to glycerate due to the suppression of the over-oxidation.135 Inaya and co-workers reported the selective aerobic oxidation of glycerol to dihydroxyacetone using a Pt supported on charcoal at mild reaction conditions (50 °C).79 By incorporating bismuth in platinum the selectivity to dihydroxyacetone increased remarkably from 10% to 80%. It was proposed that the formation of a bismuth submonolayer on Pt is responsible for the high selectivity towards dihydroxyacetone. Bismuth adatoms block the sites, which are responsible for unselective oxidation and control the glycerol orientation towards dihydroxyacetone formation. Comparison with the conventional fermentation process showed that the catalytic method they proposed showed higher productivity. Gallezot and co-workers in similar studies confirmed that the deposition of bismuth on platinum particles using a low Bi/Pt molar ratio, orientates the selectivity towards the oxidation of the secondary hydroxyl group to form dihydroxyacetone with selectivities of 50% to 70% conversion. No leaching of bismuth into the solution was reported.136 The synthesis of hydroxypyruvic acid, a starting material for the synthesis of L-tyrosine and its use as flavour component has been reported from Bekkum and co-workers, using a Pt/C catalyst modified by Bismuth.137 In addition, they reported selectivity of 93% with conversion of sodium glycerate of 95%. Proper control of pH was necessary for controlling the selectivity towards the desired product.
:4. From their results the authors conclude that for enhancing the catalytic activity of a bimetallic Au–Pd system a small amount of the one metal in the presence of the other seems to be enough for the creation of an active bimetallic system (see Table 4).
In the case of glycerol, the choice of bimetallic system (Au–Pd, or Au–Pt), preparation method and support (carbonversusgraphite) not only influence catalytic activity but also played a role in the distribution of products and particle size distribution.140,141,146 The utilisation of Au–Pd supported nanoparticles showed to improve the selectivity towards the products of the oxidation of the terminal hydroxyl groups (glyceric acid and tartronic acid) with selectivities over 90% at conversion levels of 90% (Scheme 20). In the case of Au–Pt supported catalysts similar TOFs values were obtained with respect to the Au–Pd supported catalysts and enhancement in the formation of glycolic acid indicate the facilitation in the oxidation of the secondary hydroxyl group of glycerol. The effect of Au/Pd molar ratio was studied by the same authors and they observed that the catalytic activity was improving by increasing the Au/Pd molar ratio with the highest activity corresponded to a rich Au system (Au/Pd = 9/1).142,143 From their studies they conclude that surface Pd monomer in contact with Au has a prominent promoting effect on activity and stability.
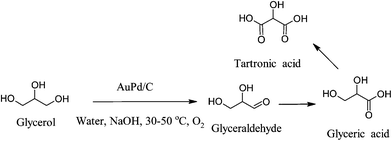 | ||
| Scheme 20 | ||
Synthesis of single-phase Au–Pd catalysts showed to exhibit higher activity and reusability than random Au–Pd catalysts in the selective oxidation of glycerol.147,148
Comparison of two preparation methods (impregnation and sol-immobilisation methods) for the synthesis of Au–Pd supported catalysts in the liquid phase oxidation of glycerol led to the conclusion that high activity coupled with high selectivity to the desired product (glycerate) can be achieved by gold-rich surface bimetallic nanoparticles with mean particle size of 3–5 nm and metallic oxidation state (Fig. 11), whereas larger particles (over 6 nm) led to a significant lower catalytic activity as it was observed with the impregnation method.149 In subsequent studies Hutchings and co-workers demonstrate the excellent catalytic performance of Au–Pd supported nanoparticles and the strong synergistic effect of the addition of gold into the palladium metal in the liquid phase oxidation of 1,2-propanediol to the sodium salt of lactic acid, which is an important monomer for the synthesis of biodegradable polymers (Scheme 21).36 It was shown that high selectivity to lactate was possible (96%) at 94% conversion and the usage of oxidants such as molecular oxygen as well as of hydrogen peroxide was achievable at mild conditions (60 °C and PO2 = 10 bar or atmospheric pressure).
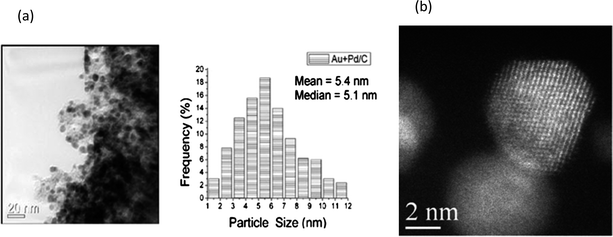 | ||
| Fig. 11 (a) TEM micrograph (b) Particle size distribution data and (C) STEM-HAADF image for Au–Pd/C, synthesised by colloidal method. Reproduced with permission from ref. 149. Copyright RSC. | ||
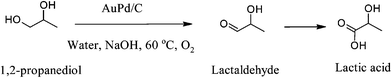 | ||
| Scheme 21 | ||
The utilisation of Au–Pt catalysts has been reported for efficient aerobic liquid phase oxidation of alcohols and polyols.144 Au–Pt supported nanoparticles were immobilised on carbon using colloidal methodology with mean particle size in the range 3–6 nm and the catalytic performance was investigated for the aerobic liquid phase oxidation of glycerol. It was found that the effect of reducing agent and the nature of Pt precursor were affecting activity and selectivity. Au–Pt supported nanoparticles showed a higher activity and resistance to poisoning in comparison to the monometallic Pt catalysts and a substantially increase of activity with respect to the monometallic Au or Pt supported nanoparticles. It was also demonstrated the oxidation of polyols (glycerol, 1,2-propanediol) is possible without the use of base at mild conditions with high selectivity to the carboxylic acid using Au–Pt supported nanoparticles on mordenite, whereas monometallic Au could not be activated without the use of base and Pt was showing high conversion but with low selectivity to glyceric acid.145 A colloidal method was used for the synthesis of the Au–Pt nanoparticles and from STEM analysis the mean particle size was in the range 3.7–4 nm.
Prüße and co-workers used a deposition–precipitation methodology for the synthesis of gold catalysts for glucose oxidation.153 Specifically using NaOH or Urea as precipitation agents and alumina as support, they reported highly active and selective catalysts which showed an excellent long-term stability. DP-Urea was found to be a better method due to the fact that no losses of gold occurred during the preparation.
Haruta and co-workers154 reported on the effect of support and size of the gold particles on glucose oxidation using a new preparation method, which is based on the solid grinding, by using a volatile organogold complex [Me2Au(acac)] (acac = acetylacetonate). Following this methodology it is possible to deposit gold clusters smaller than 2 nm in diameter onto porous coordination polymers, several kinds of metal oxides and carbon supports. From their studies they concluded that the most active catalyst was gold on ZrO2 and the control of particle size is critical in the liquid phase oxidation.
| Substrate | Catalyst | T/°C | Solvent | Conv. % | Sel. % | Ref |
|---|---|---|---|---|---|---|
| 5-Hydroxymethyl-furfural | Au/TiO2 | 130 | Methanol/NaOH | 100 | 98(DMF) | 164 |
| 5-Hydroxymethyl-furfural | Au/TiO2 | 30 | Water/NaOH | 100 | 71(FDCA) | 165 |
| 5-Hydroxymethyl-furfural | Au/CeO2 | 130 | Methanol/NaOH | 100 | 100(DMF) | 166 |
| 5-Hydroxymethyl-furfural | Au/TiO2 | 130 | Water/NaOH | 100 | 95(FDCA) | 167 |
| Benzyl alcohol | Au/K2Ti6O13 | 20 | Methanol/KOH | 99 | 93(Methyl Benzoate) | 190 |
| Propane-1,2-diol | Au/Fe2O3 | 100 | Methanol/NaOH | 99 | 72(Methyl lactate) | 192 |
 | ||
| Scheme 22 | ||
Utilisation of tert-butylhydroperoxide or hydrogen peroxide as oxidant
The utilisation of TBHP or H2O2 as alternative oxidants have been investigated using gold supported nanoparticles. Choudhary and co-workers have shown the successful liquid phase oxidation of benzyl alcohol to benzaldehyde using TBHP at mild conditions 95 °C with gold supported nanoparticles synthesized by using a homogeneous precipitation method.109,169 They study the effect of the support and preparation method and concluded that the most active catalysts consist of gold supported nanoparticles with high surface ratio of Au3+/Au0, whereas gold particle size seems not to be a crucial parameter in the range 38 nm. In addition, the most effective supports were TiO2, ZrO2 and MgO.The utilisation of H2O2 as oxidant for the efficient oxidation of alcohols by gold supported nanoparticles using solvent free conditions was reported by Cao and co-workers. 1-Phenyethanol was oxidised at 90 °C using gold supported catalysts supplied by World Gold Council and the most effective support was ceria. The general applicability of the methodology was demonstrated by oxidizing a range of non-activated alcohols and the catalysts were reusable several times.110 Tsukuda and co-workers reported the oxidation of benzyl alcohol using hydrogen peroxide and microwave irradiation and studied the effect of particle size by synthesizing gold supported nanoparticles in the range of 0.8–1.9 nm. They concluded that there is a size dependence of the activity of gold clusters on SBA-15, with smaller gold particles showing higher catalytic activity.111
The one-pot synthesis approach: Combining catalytic selective oxidation with another reaction
A promising and challenging aspect in liquid phase oxidation reaction arises from the possibility of combining two functionalities or reaction steps in one pot synthesis. Several recent advances have been achieved in this area and here we specifically highlighted progress in the direct oxidative carboxylation of olefins to produce cyclic carbonates, benzene hydroxylation with in situ produced H2O2 and in the oxidative esterification of alcohols.Oxidative alkene carboxylation
There is a strong interest in developing new green synthetic methods to prepare organic carbonates, and in particular to find industrial alternatives to the use of phosgenes in synthesis.170 Supported metal nanoparticles have been already used to produce organic carbonates, and recently Hammond et al. reported that gold nanoparticles supported on oxides promoted the synthesis of glycerol carbonate from glycerol and urea without a solvent and under mild conditions.171–173 However, it would be very desirable to form carbonates directly from olefins. This can be achieved by reacting the olefin in the presence of CO2 under conditions where the epoxide is rapidly carboxylated and so avoiding the need to isolate the epoxide.174 Another advantage of this process is that the CO2 utilise does not need to be free of oxygen which would otherwise require a expensive separation process. Few homogeneous catalysts have been reported,174 but supported metal catalysts have recently been studied and shown to improve yields and avoid the use of toxic organic solvents.175–177Sun et al. reported a one pot synthesis for the oxidative carboxylation of styrene to styrene carbonate catalysed with supported metals (Scheme 23).175Styrene carbonate was obtained from the reaction of styrene with CO2 using TBHP with a multifunctional catalytic system consisting of Au/SiO2, zinc bromide, and tetrabutylammonium bromide (Bu4NBr). Au/SiO2 acts as an oxidation catalyst and is responsible for the epoxidation of styrene, while zinc bromide and Bu4NBr promote the cycloaddition of CO2 to the epoxide. They also describe an improved gold catalyst supported on a resin (R201) that affords 51% of carbonate yield in follow-up work.176 They use CO2 as solvent and reagent and the Au/R201 catalyst was fully reusable. More recently, they report that gold supported in ferric hydroxide is more efficient due to a synergistic effect between gold and the ironin.177 After 10 h reaction, styrene carbonate yields of 53% were obtained at 80 °C at 4 MPa of CO2.
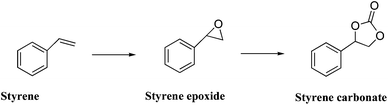 | ||
| Scheme 23 | ||
Benzene hydroxylation with in situ generated H2O2
Advances in this area are highly desired from economic and industrial views as the development of an in situ industrial process would represent a great improvement in cost and safety, as the transportation and the handling of H2O2 would be eliminated. The first step of the reaction consists on the direct catalytic formation of hydrogen peroxide on Pd and Pt from hydrogen and oxygen followed with its subsequent reaction with benzene to produce phenol. Although the applicability of this technology is matter of much debate, we have briefly summarised some of the recent progress in this area.Early studies by Miyake et al.178 indicated that Pt/V2O5 supported on silica was able to yield phenol with acetic acid as solvent. Later reports enhanced catalytic activity by combining Pd as active site for intermediate hydrogen peroxide formation with another metal as oxidation function like Ti in TS-1179 or molecular vanadium sites.180 Acidic zeolites and resins have also been used as supports for noble metals and this eliminates the need of liquid acids.181 Centi and Perathoner182 have recently highlighted some perspectives for this reaction on a context for sustainable chemical production.
Much progress in the last ten years has been achieved by using Pd membrane reactors.183–187 Mizukami and co-workers reported that methyl benzoate can be directly hydroxylated to methyl salicylate183 with a yield of 4.7% at 150 °C; whereas the same reactor achieved benzene conversion of 15% and a phenol selectivity of 95%. Although the authors concede that an increase in reaction temperature caused undesired simultaneous hydrogenation.184 In a more recent work various active metals were loaded on the alpha-Al2O3 porous tube which was the substrate of the thin Pd membrane.186 This resulted in a number of side reactions, such as complete oxidation and hydrogenation, and a decrease in the hydroxylation activity. However, the loading of Cu suppressed complete oxidation and enhanced the hydroxylation activity. Vulpescu et al. performed a technical and economic feasibility study on the application of Pd-based catalytic membranes for the formation of phenol and discussed the limitations of the available technology for commercial application.187 They concluded that the total oxidation of hydrogen to water and the total oxidation of benzene to carbon dioxide were the major limitations for industrial application.
Oxidative esterification of alcohols
Methyl esters are important products in the chemical industry, e.g. in the synthesis of flavoring agents, solvent extractants and intermediates as well as in fragrance industry. Therefore, the synthesis of esters by avoiding the traditional route which involves the reaction of carboxylic acid and methanol and using strong acid catalysts, such as sulfuric, sulfonic and p-toluenesulfonic acid or base alkoxides will be desirable due to environmental concerns.188,189 The selective oxidation of alcohols in methanol for the synthesis of the corresponding methyl esters has been reported by several groups (see Table 5). Christensen and co-workers have synthesized potassium titanate nanowires and deposited gold nanoparticles and have shown that at ambient temperature the aerobic oxidation of benzyl alcohol in methanol to give methyl benzoate using catalytic amount of base can proceed with high efficiency, (over 99% conversion and yield of 93% to methyl benzoate). They demonstrated the significance of adding base in the solution, since in the presence of base deactivation of the catalyst was prevented.190 In a subsequent work, they demonstrated the production of methyl esters by selectively oxidizing aldehydes in a primary alcohol environment and using gold supported catalysts.191 They reported the formation of methyl benzoate at temperatures even below 0 °C. The general applicability was shown by synthesizing acrylate esters at room temperature using air as oxidant, with selectivities of more than 85% at 97% conversion. The proposed mechanism indicates that the alcohol is first oxidized to an aldehyde, then the aldehyde forms an hemiacetal with methanol, which is further oxidized to the corresponding ester and the rate determining step is the aerobic oxidation of alcohols to the corresponding aldehyde. In subsequent studies Christensen et al. reported the synthesis of methyl esters using polyols such as, glycerol, 1,2-propanediol and 1,3-propanediol. Using gold supported nanoparticles in the range of 3–7 nm it was shown that the synthesis of important chemicals such as methyl lactate from 1,2-propanediol, methyl acrylate from 1,3-propanediol and dimethyl mesoxalate from glycerol can be obtained with yields in the range of 40–90%.192The oxidative esterification of alcohols was also demonstrated by the group of Rossi et al. who synthesized gold supported nanoparticles using SiO2 as support with mean gold particle size of 5.7 nm and for the synthesis of the supported metal nanoparticles the Brust method was used.193 They demonstrated the general applicability of the oxidation of alcohols to the corresponding methyl esters and the reusability of the synthesized catalyst obtaining high yields to the corresponding methyl esters in the range 70–95%.
Other selective oxidation reactions
Selective oxidation of amines
It is only very recent that heterogeneous catalysts are being developed for the selective oxidation of amines using oxygen. Also for these reactions, gold appears to be superior to palladium and platinum nanoparticles.Della Pina et al. first reported the oxidation of tertiary amines using molecular oxygen with Au, Pt and Rh catalysts in aqueous solution producing mainly the corresponding N-oxides.194 The activity and selectivity of gold, made it the catalyst of choice and 100% yields were obtained with triethylamine, pyridine and 3-dimethylaminopropan-1-ol using Au/C. Zhu et al. demonstrated that gold does not need to be finely derived to be an effective catalyst for the aerobic oxidative dehydrogenation of amines (CH–NH) to imines (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N).195 The reactions were carried at 1 atm O2 and 100 °C (Scheme 24).
N).195 The reactions were carried at 1 atm O2 and 100 °C (Scheme 24).
 | ||
| Scheme 24 | ||
In particular, a 5% Au/Al2O3 catalyst, comprising of large gold particles (50–150 nm) and prepared by impregnation was very efficient. The large particle size was no deterrent for activity for a reaction in which the authors report that gold can catalyse even in powder form. The higher dispersion of gold in the supported catalyst does however increase activity and the catalytic activity of 5 mg of gold in the Au/Al2O3 catalyst is greater than that of 1 g of gold powder.
More recently, Aschwanden et al.196 utilised Au(OAc)3 for the selective oxidation of dibenzylamine to dibenzylimine using molecular oxygen in toluene at mild temperatures. The TOF observed were lower than unity for the homogeneous catalyst. Immobilising Au(OAc)3 onto CeO2, did however result in more active catalysts, yielding 100% conversion and 91% selectivity at 108 °C (TOF = 7.2 h−1; related to the total amount of gold). The authors then describe that Au(OAc)3 is first reduced by the amine and this produces metallic gold on the support, which is the active phase of the reaction.
So et al. also report catalytic studies in the selective oxidation of cyclic and acyclic benzylic amines to imines with graphite-supported gold catalysts and find that this activity can be translated to substituted quinolines.197 Conversions between 43–100% and product yields between 66–99% were obtained with a reusable catalyst. A hydrogen-transfer mechanism from the amine to the metal followed by the oxidation of the M–H was suggested. More recently, the same authors reported a gold catalyst able to react anilines with aldehydes to form quinolines in a one-pot reaction with yields up to 95%.198 The oxidation of benzylamines to N-benzylidene benzylamines with gold catalysts was also investigated by Corma et al.,199 who additionally reported that para-substituted benzylamines as well as heterocyclic methanamines undergo oxidative condensation. Pd and Pt catalysts with much higher metal loading (5wt%) appear to be less active, and gold is again the preferred metal. The authors highlight that the efficiency of gold increases exponentially as the average particle size is reduced and the TOF increases accordingly for a gold catalyst supported on titania. They also report that oxidative condensation also occurs selectively with sulphur-containing heterocyclic amines and that the selective formation of secondary benzylamines is catalysed by a gold catalyst through a one-pot, two-step reaction which involves oxidation and hydrogenation steps. More recently, the same authors show that gold supported on titania is also able to selectively oxidize aromatic anilines to azo compounds with yields above 98%.200
Christensen and co-workers have shown that gold nanoparticles supported on titania operate as an excellent bifunctional catalyst for the aerobic oxidation of amines.201 They could oxidise n-hexyl amine and 1,6-hexanediamine with high selectivity into the corresponding amides, N-hexyl hexanoic amide and caprolactam, respectively. The authors claim these new methodologies open up two new and environmentally benign routes to caprolactam and cyclohexanone oxime, both of which are precursors for nylon-6.
Haruta et al. have carried out the one-pot N-formylation of amines with methanol and molecular oxygen, to produce formamides obtaining selectivities of 90%.202 The authors show that the reaction of amine is tunable by the proper selection of metal oxide as support and the solvent. Among a number of oxide supports, gold on NiO exhibited the highest catalytic activity as well as the highest selectivity to formamide due to the stronger affinity for alcoholic groups compared to amine groups. The authors propose a mechanism whereby methyl formate is generated in situ and subsequently reacted with the amines.
Selective oxidation of oximes
Supported nanoparticles of gold, palladium and platinum can catalyse the oxidative transformation of carvone oxime into carvone (Scheme 25) as recently reported by Grirrane et al.203 | ||
| Scheme 25 | ||
The reaction is of general applicability, and represents just one example of the use of metal nanoparticles for the aerobic oxidation of oximes to the corresponding carbonylic compounds. It is however a reaction of industrial interest as Carvone is an essential oil utilized in the fragrance industry. Current synthetic methods are difficult due to the reluctance of the oxime to undergo hydrolysis. The discovery by Grirrane et al.203 affords an alternative route via the oxidation of the CN bond. The preferred solvents were aqueous solutions of ethanol but the reaction can be carried out in several solvents. Oxygen is the oxidising agent, and the reaction is carried out without the need of corrosive Brønsted acids and without significant wastes inherent in the current synthetic methods. The effect of the support was investigated and ceria was superior to titania and carbon. Gold and platinum both showed very similar activity, but superior to platinum.
Selective oxidation of silanes to silanols
Silanols are utilised mainly in the preparation of silicon-based polymeric materials. However, traditional methods for the synthesis of silanols utilize toxic reagents and are non environmentally-friendly, whereas other recently reported synthetic methods suffer the main drawback of the production of disiloxanes. Kanedaet al.204 overcome these problems by using water as solvent and a silver catalyst and report very little condensation to the disiloxanes. The reaction can also be catalysed by homogeneous silver, although the supported nanoparticles on hydroxyapatite were superior in activity with the additional advantage of being fully reusable.Conclusions and final remarks
The utilisation of metal supported catalysts as an integral part of the methodology for the synthesis of fine chemicals has shown significant development in recent years. Indeed this has been motivation behind this perspective article. We have tried to demonstrate the wide applicability of heterogeneous catalysts based on gold and gold bimetallic alloys as efficient selective oxidation catalysts. However, it is important to note that there has been significant recent progress in the selective oxidation using gold complexes as homogeneous catalysts. As the scope of this article has been devoted to highlighting the advances made in the use of supported metal nanoparticles, the advances in homogeneous catalysis have fallen outside of our focus. However, it should be noted that the successful heterogenization of gold homogenous catalysts could be very beneficial and could provide a valuable new research area for exploitation. For example, Hashmi et al. have recently provided the first experimental evidence that mononuclear gold complexes are active in the selective oxidative esterification of aldehydes and alcohols205 whereas the epoxidation of alkenes has been reported by Cinellu et al.,206 These are notable observations, which further emphasise that the immobilisation of homogeneous complexes on solid supports should be an area of future interest.The field of heterogeneous catalysis based on gold nanoparticles is now getting to the stage where it can now contribute to the development of sustainable chemical processes. The use of supported metal nanoparticles together with molecular oxygen instead of stoichiometric oxidising reagents will clearly represent a major advance in this field. Generally, a greater effort is needed for developing new technologies, which can compete with the traditional selective oxidation industrial processes, and at the same time remain economically feasible. However, as we note much progress has been made in this field recently. The incorporation of two metals as an alloy on the surface of the support seems to be a key parameter for enhancing catalytic activity and resistance to poisoning, thereby lengthening the lifetime of the catalyst. In most selective oxidations, the synthesis of nanoparticles with relatively small particle sizes (2–6 nm) seems to be the key factor ensuring high catalytic activity as well as selectivity. The utilisation of gold based catalysts can improve existing chemical processes as high conversion, improved selectivity and longer life time of the catalytic systems has been demonstrated. However, there are still challenges to be solved, such as the improvement in the stability of the supported nanoparticles and one promising option could be confinement within mesoporous materials. Also, the high cost and volatility in the price of gold and other precious metals could still raises concerns when the commercialisation of these catalysts, however it is considered that using very small metal loadings will overcome this problem. There are still challenges in the synthesis of supported metal nanoparticles with precise particle size and morphology and, in particular, in the case of alloyed nanoparticles. Another important challenge is the understanding of the mechanism by which bimetallic supported nanoparticles generate enhanced catalytic performance when compared to their monometallic counterparts. Therefore, improved understanding of the active sites responsible for catalysis will be beneficial in designing and controlling the properties of metal nanoparticles. This aspect is key is further progress is to be made rapidly. It is important that we gain an understanding of how key products are formed. For example in the oxidation of alcohols in some cases hemiacetal formation occurs which drives the reaction towards esterification, wheras in other reported instances over-oxidation to the acid occurs. There are many key parameters that need to be studied and controlled; for example, reactions can be solvent-free or be carried out in aqueous base and this will certainly open up different reaction channels. This is therefore an area presenting a key opportunity for further study and advances. Finally, there is still room for the discovery of new supported metal nanoparticles in a range of chemical processes especially considering the great advances in recent years regarding the control in catalyst preparation and the discovery of the reactivity of gold and its alloys.
Acknowledgements
We would like to thank Ramchandra Tiruvalam, Qian He, Dr. Andrew A. Herzing and Prof. Christopher J. Kiely for useful discussions on aspects of this work and particularly the interpretation of the high quality TEM images.References
- D. A. Hickman and L. D. Schmidt, Science, 1993, 259, 343–346 CrossRef CAS.
- R. Schlogl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Top. Catal., 2009, 52, 258–268 CrossRef CAS.
- F. Rosowski, S. Storck and J. Zühlke, Oxyfunctionalization of Alkyl Aromatics, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- J. M. Bregeault, Dalton Trans., 2003, 3289–3302 RSC.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- R. Zhao, D. Ji, G. M. Lv, G. Qian, L. Yan, X. L. Wang and J. S. Suo, Chem. Commun., 2004, 904–905 RSC.
- G. M. Lu, R. Zhao, G. Qian, Y. X. Qi, X. L. Wang and J. S. Suo, Catal. Lett., 2004, 97, 115–118 CrossRef.
- P. P. Wu, P. Bai, Z. B. Lei, K. P. Loh and X. S. Zhao, Microporous Mesoporous Mater., 2011, 141, 222–230 CrossRef CAS.
- H. Zhao, J. C. Zhou, H. Luo, C. Y. Zeng, D. H. Li and Y. J. Liu, Catal. Lett., 2006, 108, 49–54 CrossRef CAS.
- Y. J. Xu, P. Landon, D. Enache, A. Carley, M. Roberts and G. Hutchings, Catal. Lett., 2005, 101, 175–179 CrossRef CAS.
- L. X. Xu, C. H. He, M. Q. Zhu, K. J. Wu and Y. L. Lai, Catal. Lett., 2007, 118, 248–253 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef CAS.
- P. Lignier, S. Mangematin, F. Morfin, J. L. Rousset and V. Caps, Catal. Today, 2008, 138, 50–54 CrossRef CAS.
- U. Neuenschwander, F. Guignard and I. Hermans, ChemSusChem, 2010, 3, 75–84 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 Search PubMed.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef CAS.
- Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS.
- X. Li, J. Xu, L. Zhou, F. Wang, J. Gao, C. Chen, J. B. Ning and H. Ma, Catal. Lett., 2006, 110, 149–154 CrossRef CAS.
- F. Wang, J. Xu, X. Q. Li, J. Gao, L. P. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- J. Gao, X. L. Tong, X. Q. Li, H. Miao and J. Xu, J. Chem. Technol. Biotechnol., 2007, 82, 620–625 CrossRef CAS.
- R. L. Brutchey, I. J. Drake, A. T. Bell and T. D. Tilley, Chem. Commun., 2005, 3736–3738 RSC.
- A. P. Singh and T. Selvam, J. Mol. Catal. A: Chem., 1996, 113, 489–497 CrossRef CAS.
- R. Raja, J. M. Thomas and V. Dreyer, Catal. Lett., 2006, 110, 179–183 CrossRef CAS.
- M. J. Beier, B. Schimmoeller, T. W. Hansen, J. E. T. Andersen, S. E. Pratsinis and J. D. Grunwaldt, J. Mol. Catal. A: Chem., 2010, 331, 40–49 CrossRef CAS.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192–9198 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. A. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, N. Glanville, L. Kesavan, C. Hammond, J. K. Edwards, A. F. Carley, C. J. Kiely and G. J. Hutchings, Appl. Catal., A, 2011, 391, 400–406 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- J. Colby, D. I. Stirling and H. Dalton, Biochem. J., 1977, 165, 395–402 CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- L. Prati and G. Martra, Gold Bull., 1999, 32, 96–101 CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. Carley, L. Prati and G. J. Hutchings, Catal. Today, 2006, 122, 317–324.
- N. Dimitratos, J. A. Lopez-Sanchez, S. Meenakshisundaram, J. M. Anthonykutty, G. Brett, A. F. Carley, S. H. Taylor, D. W. Knight and G. J. Hutchings, Green Chem., 2009, 11, 1209–1216 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142–5153 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- S. Bawaked, N. F. Dummer, N. Dimitratos, D. Bethell, Q. He, C. J. Kiely and G. J. Hutchings, Green Chem., 2009, 11, 1037–1044 RSC.
- S. Bawaked, N. F. Dummer, D. Bethell, D. W. Knight and G. J. Hutchings, Green Chem., 2011, 13, 127–134 RSC.
- Z. Y. Cai, M. Q. Zhu, J. Chen, Y. Y. Shen, J. Zhao, Y. Tang and X. Z. Chen, Catal. Commun., 2010, 12, 197–201 CrossRef CAS.
- N. S. Patil, B. S. Uphade, P. Jana, S. K. Bharagava and V. R. Choudhary, J. Catal., 2004, 223, 236–239 CrossRef CAS.
- N. S. Patil, B. S. Uphade, P. Jana, S. K. Bhargava and V. R. Choudhary, Chem. Lett., 2004, 33, 400–401 CrossRef CAS.
- P. Lignier, F. Morfin, S. Mangematin, L. Massin, J. L. Rousset and V. Caps, Chem. Commun., 2007, 186–188 RSC.
- P. Lignier, F. Morfin, L. Piccolo, J. L. Rousset and V. Caps, Catal. Today, 2007, 122, 284–291 CrossRef CAS.
- Gold Bull., 2003, 36, p. 24 Search PubMed.
- V. Mendez, K. Guillois, S. Daniele, A. Tuel and V. Caps, Dalton Trans., 2010, 39, 8457–8463 RSC.
- M. Boualleg, K. Guillois, B. Istria, L. Burel, L. Veyre, J. M. Basset, C. Thieuleux and V. Caps, Chem. Commun., 2010, 46, 5361–5363 RSC.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–U931 CrossRef CAS.
- B. Li, P. He, G. Yi, H. Lin and Y. Yuan, Catal. Lett., 2009, 133, 33–40 CrossRef CAS.
- F. Kerdi, V. Caps and A. Tuel, Microporous Mesoporous Mater., 2011, 140, 89–96 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196–214 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- C. P. Vinod, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2011, 86, 161–171 CrossRef CAS.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353–4361 CrossRef CAS.
- B. Z. Zhan, M. A. White, T. K. Sham, J. A. Pincock, R. J. Doucet, K. V. R. Rao, K. N. Robertson and T. S. Cameron, J. Am. Chem. Soc., 2003, 125, 2195–2199 CrossRef CAS.
- H. B. Ji, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2002, 43, 7179–7183 CrossRef CAS.
- Z. Opre, J. D. Grunwaldt, M. Maciejewski, D. Ferri, T. Mallat and A. Baiker, J. Catal., 2005, 230, 406–419 CrossRef CAS.
- S. Mori, M. Takubo, K. Makida, T. Yanase, S. Aoyagi, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. Commun., 2009, 5159–5161 RSC.
- Z. Opre, J. D. Grunwaldt, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 2005, 242, 224–232 CrossRef CAS.
- Z. Opre, D. Ferri, F. Krumeich, T. Mallat and A. Baiker, J. Catal., 2006, 241, 287–295 CrossRef CAS.
- M. L. Kantam, U. Pal, B. Sreedhar, S. Bhargava, Y. Iwasawa, M. Tada and B. M. Choudary, Adv. Synth. Catal., 2008, 350, 1225–1229 CrossRef CAS.
- K. Yamaguchi, J. W. Kim, J. L. He and N. Mizuno, J. Catal., 2009, 268, 343–349 CrossRef CAS.
- F. Nikaidou, H. Ushiyama, K. Yamaguchi, K. Yamashita and N. Mizuno, J. Phys. Chem. C, 2010, 114, 10873–10880 CrossRef CAS.
- K. Ebitani, Y. Fujie and K. Kaneda, Langmuir, 1999, 15, 3557–3562 CrossRef CAS.
- N. Kakiuchi, Y. Maeda, T. Nishimura and S. Uemura, J. Org. Chem., 2001, 66, 6620–6625 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS.
- A. H. Lu, W. C. Li, Z. S. Hou and F. Schuth, Chem. Commun., 2007, 1038–1040 RSC.
- Z. S. Hou, N. Theyssen and W. Leitner, Green Chem., 2007, 9, 127–132 RSC.
- Z. Hou, N. Theyssen, A. Brinkmann, K. V. Klementiev, W. Grunert, M. Buhl, W. Schmidt, B. Spliethoff, B. Tesche, C. Weidenthaler and W. Leitner, J. Catal., 2008, 258, 315–323 CrossRef CAS.
- T. Hara, M. Ishikawa, J. Sawada, N. Ichikuni and S. Shimazu, Green Chem., 2009, 11, 2034–2040 RSC.
- H. Q. Yang, X. J. Han, Z. C. Ma, R. Q. Wang, J. Liu and X. F. Ji, Green Chem., 2010, 12, 441–451 RSC.
- H. Kimura, K. Tsuto, T. Wakisaka, Y. Kazumi and Y. Inaya, Appl. Catal., A, 1993, 96, 217–228 CrossRef CAS.
- T. Mallat, Z. Bodnar, P. Hug and A. Baiker, J. Catal., 1995, 153, 131–143 CrossRef CAS.
- A. B. Crozon, M. Besson and P. Gallezot, New J. Chem., 1998, 22, 269–273 RSC.
- A. F. Lee, J. J. Gee and H. J. Theyers, Green Chem., 2000, 2, 279–282 RSC.
- R. Anderson, K. Griffin, P. Johnston and P. L. Alsters, Adv. Synth. Catal., 2003, 345, 517–523 CrossRef CAS.
- C. Keresszegi, T. Mallat, J. D. Grunwaldt and A. Baiker, J. Catal., 2004, 225, 138–146 CrossRef CAS.
- Y. H. Ng, S. Ikeda, T. Harada, Y. Morita and M. Matsumura, Chem. Commun., 2008, 3181–3183 RSC.
- D. Ferri, C. Mondelli, F. Krumeich and A. Baiker, J. Phys. Chem. B, 2006, 110, 22982–22986 CrossRef CAS.
- J. D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586–25589 CrossRef CAS.
- J. C. Beziat, M. Besson and P. Gallezot, Appl. Catal., A, 1996, 135, L7–L11 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696–697 RSC.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397–403 CrossRef.
- S. Demirel, P. Kern, M. Lucas and P. Claus, Catal. Today, 2007, 122, 292–300 CrossRef CAS.
- C. Milone, R. Ingoglia, G. Neri, A. Pistone and S. Galvagno, Appl. Catal., A, 2001, 211, 251–257 CrossRef CAS.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS.
- C. H. Christensen, B. Jorgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651 CrossRef CAS.
- J. C. Hu, L. F. Chen, K. K. Zhu, A. Suchopar and R. Richards, Catal. Today, 2007, 122, 277–283 CrossRef CAS.
- P. Haider and A. Baiker, J. Catal., 2007, 248, 175–187 CrossRef CAS.
- A. Biffis, S. Cunial, P. Spontoni and L. Prati, J. Catal., 2007, 251, 1–6 CrossRef CAS.
- L. C. Wang, Y. M. Liu, M. Chen, Y. Cao, H. Y. He and K. N. Fan, J. Phys. Chem. C, 2008, 112, 6981–6987 CrossRef CAS.
- F. Z. Su, M. Chen, L. C. Wang, X. S. Huang, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Catal. Commun., 2008, 9, 1027–1032 CrossRef CAS.
- F. Z. Su, Y. M. Liu, L. C. Wang, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2008, 47, 334–337 CrossRef CAS.
- J. Han, Y. Liu and R. Guo, Adv. Funct. Mater., 2009, 19, 1112–1117 CrossRef CAS.
- J. Yang, Y. J. Guan, T. Verhoeven, R. van Santen, C. Li and E. J. M. Hensen, Green Chem., 2009, 11, 322–325 RSC.
- Y. M. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 13457–13461 CrossRef CAS.
- Y. Wang, R. Yan, J. Z. Zhang and W. Q. Zhang, J. Mol. Catal. A: Chem., 2010, 317, 81–88 CrossRef CAS.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144–149 RSC.
- V. R. Choudhary, D. K. Dumbre and S. K. Bhargava, Ind. Eng. Chem. Res., 2009, 48, 9471–9478 CrossRef CAS.
- J. Ni, W. J. Yu, L. He, H. Sun, Y. Cao, H. Y. He and K. N. Fan, Green Chem., 2009, 11, 756–759 RSC.
- Y. M. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Lett., 2010, 39, 159–161 CrossRef CAS.
- P. Haider, J. D. Grunwaldt, R. Seidel and A. Baiker, J. Catal., 2007, 250, 313–323 CrossRef CAS.
- L. F. Liotta, A. M. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay and L. Guczi, Catal. Today, 2001, 66, 271–276 CrossRef CAS.
- M. J. Beier, T. W. Hansen and J. D. Grunwaldt, J. Catal., 2009, 266, 320–330 CrossRef CAS.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. S. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS.
- D. I. Enache, D. Barker, J. K. Edwards, S. H. Taylor, D. W. Knight, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 407–411 CrossRef CAS.
- S. Meenakshisundaram, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. W. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341–356 RSC.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem.–Eur. J., 2011, 17, 6524–6532 CrossRef CAS.
- P. Miedziak, M. Sankar, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Catal. Today, 2011, 164, 315–319 CrossRef CAS.
- C. Y. Ma, B. J. Dou, J. J. Li, J. Cheng, Q. Hu, Z. P. Hao and S. Z. Qiao, Appl. Catal., B, 2009, 92, 202–208 CrossRef CAS.
- Y. T. Chen, H. M. Lim, Q. H. Tang, Y. T. Gao, T. Sun, Q. Y. Yan and Y. H. Yang, Appl. Catal., A, 2010, 380, 55–65 CrossRef CAS.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191–6201 CrossRef CAS.
- H. Miyamura, R. Matsubara and S. Kobayashi, Chem. Commun., 2008, 2031–2033 RSC.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231–236 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi, S. Coluccia and G. Martra, Catal. Today, 2000, 61, 165–172 CrossRef CAS.
- P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- C. H. C. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329–1336 RSC.
- S. Demirel-Gulen, M. Lucas and P. Claus, Catal. Today, 2005, 102, 166–172 CrossRef.
- W. C. Ketchie, Y. L. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94–101 CrossRef CAS.
- W. C. Ketchie, M. Murayama and R. J. Davis, Top. Catal., 2007, 44, 307–317 CrossRef CAS.
- N. Dimitratos, A. Villa, C. L. Bianchi, L. Prati and M. Makkee, Appl. Catal., A, 2006, 311, 185–192 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- A. Abbadi and H. vanBekkum, Appl. Catal., A, 1996, 148, 113–122 CrossRef CAS.
- N. Dimitratos and L. Prati, Gold Bull., 2005, 38, 73–77.
- N. Dimitratos, F. Porta, L. Prati and A. Villa, Catal. Lett., 2005, 99, 181–185 CrossRef CAS.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102, 203–212 CrossRef.
- N. Dimitratos, F. Porta and L. Prati, Appl. Catal., A, 2005, 291, 210–214 CrossRef CAS.
- A. Villa, C. Campione and L. Prati, Catal. Lett., 2007, 115, 133–136 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, L. Prati and D. S. Su, J. Phys. Chem. C, 2008, 112, 8617–8622 CrossRef CAS.
- N. Dimitratos, C. Messi, F. Porta, L. Prati and A. Villa, J. Mol. Catal. A: Chem., 2006, 256, 21–28 CrossRef CAS.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499–4502 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147–153 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, D. S. Su and L. Prati, Chem. Commun., 2006, 1956–1958 RSC.
- L. Prati, A. Villa, F. Porta, D. Wang and D. S. Su, Catal. Today, 2007, 122, 386–390 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, J. M. Anthonykutty, G. Brett, A. F. Carley, R. C. Tiruvalam, A. A. Herzing, C. J. Kiely, D. W. Knight and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 4952–4961 RSC.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2006, 244, 122–125 CrossRef CAS.
- Y. Onal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef CAS.
- C. Baatz, N. Thielecke and U. Prusse, Appl. Catal., B, 2007, 70, 653–660 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- A. S. K. Hashmi, M. Wölfle, F. Ata, M. Hamzic, R. Salathé and W. Frey, Adv. Synth. Catal., 2006, 348, 2501–2508 CrossRef CAS.
- A. S. K. Hashmi, F. Ata, E. Kurpejovic, J. Huck and M. Rudolph, Top. Catal., 2007, 44, 245–251 CrossRef CAS.
- A. S. K. Hashmi, M. Wölfle, J. H. Teles and W. Frey, Synlett, 2007, 1747–1752 CrossRef CAS.
- A. S. K. Hashmi, M. Rudolph, J. W. Bats, W. Frey, F. Rominger and T. Oeser, Chem.–Eur. J., 2008, 14, 6672–6678 CrossRef CAS.
- A. S. K. Hashmi, S. Pankajakshan, M. Rudolph, E. Enns, T. Bander, F. Rominger and W. Frey, Adv. Synth. Catal., 2009, 351, 2855–2875 CrossRef CAS.
- A. S. K. Hashmi, Nachr. Chem., 2009, 57, 379–382 Search PubMed.
- A. S. K. Hashmi, M. Rudolph, J. Huck, W. Frey, J. W. Bats and M. Hamzic, Angew. Chem., Int. Ed., 2009, 48, 5848–5852 CrossRef CAS.
- A. S. K. Hashmi, M. Wölfle, F. Ata, W. Frey and F. Rominger, Synthesis, 2010, 2297–2307 CrossRef CAS.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109–116 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS.
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchings and F. Cavani, Green Chem., 2011, 13, 2091 RSC.
- V. R. Choudhary and D. K. Dumbre, Catal. Commun., 2009, 10, 1738–1742 CrossRef CAS.
- A. A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS.
- J. A. Lopez-Sanchez and G. J. Hutchings, 2010, WO/2010/097585.
- C. Hammond, J. A. Lopez-Sanchez, M. H. Ab Rahim, N. Dimitratos, R. L. Jenkins, A. F. Carley, Q. A. He, C. J. Kiely, D. W. Knight and G. J. Hutchings, Dalton Trans., 2011, 40, 3927–3937 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, M. H. Ab, J. M. RahimAnthonykutty, A. F. Carley, R. C. Tiruvalam, C. J. Kiely, D. W. Knight and G. J. Hutchings, Prepr. Symp.-Am. Chem. Soc., Div. Fuel Chem., 2010, 55, 415–416 Search PubMed.
- J. Sun, L. Liang, J. Sun, Y. Jiang, K. Lin, X. Xu and R. Wang, Catal. Surv. Asia, 2011, 15, 49–54 CrossRef CAS.
- J. M. Sun, S. I. Fujita, F. Y. Zhao, M. Hasegawa and M. Arai, J. Catal., 2005, 230, 398–405 CrossRef CAS.
- D. Xiang, X. F. Liu, J. S. Sun, F. S. Xiao and J. M. Sun, Catal. Today, 2009, 148, 383–388 CrossRef CAS.
- Y. L. Wang, J. H. Sun, D. Xiang, L. Wang, J. M. Sun and F. S. Xiao, Catal. Lett., 2009, 129, 437–443 CrossRef CAS.
- T. Miyake, M. Hamada, Y. Sasaki and M. Oguri, Appl. Catal., A, 1995, 131, 33–42 CrossRef CAS.
- T. Tatsumi, K. Yuasa and H. Tominaga, J. Chem. Soc., Chem. Commun., 1992, 1446–1447 RSC.
- J. E. Remias, T. A. Pavlosky and A. Sen, J. Mol. Catal. A: Chem., 2003, 203, 179–192 CrossRef CAS.
- W. Laufer, J. P. M. Niederer and W. F. Hoelderich, Adv. Synth. Catal., 2002, 344, 1084–1089 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 143, 145–150 CrossRef CAS.
- K. Sato, S. Niwa, T. Hanaoka, K. Komura, T. Namba and F. Mizukami, Catal. Lett., 2004, 96, 107–112 CrossRef CAS.
- K. Sato, T. A. Hanaoka, S. Niwa, C. Stefan, T. Namba and F. Mizukami, Catal. Today, 2005, 104, 260–266 CrossRef CAS.
- S. Y. Ye, S. Hamakawa, S. Tanaka, K. Sato, M. Esashi and F. Mizukami, Chem. Eng. J., 2009, 155, 829–837 CrossRef CAS.
- K. Sato, S. Hamakawa, M. Natsui, M. Nishioka, T. Inoue and F. Mizukami, Catal. Today, 2010, 156, 276–281 CrossRef CAS.
- G. D. Vulpescu, M. Ruitenbeek, L. L. van Lieshout, L. A. Correia, D. Meyer and P. Pex, Catal. Commun., 2004, 5, 347–351 CrossRef CAS.
- M. Hudlicky, Oxidations in Organic Chem., American Chem. Soc., Washington, DC, 1990 Search PubMed.
- R. W. Taft, M. S. Newman and F. H. Verhoek, J. Am. Chem. Soc., 1950, 72, 4511–4519 CrossRef CAS.
- S. K. Klitgaard, A. T. DeLa Riva, S. Helveg, R. M. Werchmeister and C. H. Christensen, Catal. Lett., 2008, 126, 213–217 CrossRef CAS.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168–170 RSC.
- E. Taarning, A. T. Madsen, J. M. Marchetti, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 408–414 RSC.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366–1370 RSC.
- C. Della Pina, E. Falletta and M. Rossi, Top. Catal., 2007, 44, 325–329 CrossRef CAS.
- B. L. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1–6 CrossRef CAS.
- L. Aschwanden, T. Mallat, J. D. Grunwaldt, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 300, 111–115 CrossRef CAS.
- M. H. So, Y. G. Liu, C. M. Ho and C. M. Che, Chem.–Asian J., 2009, 4, 1551–1561 CrossRef CAS.
- M. H. So, Y. G. Liu, C. M. Ho, K. Y. Lam and C. M. Che, ChemCatChem, 2011, 3, 386–393 Search PubMed.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138–144 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, Nat. Protoc., 2010, 5, 429–438 Search PubMed.
- S. K. Klitgaard, K. Egeblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419–423 RSC.
- T. Ishida and M. Haruta, ChemSusChem, 2009, 2, 538–541 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 268, 350–355 CrossRef CAS.
- T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938–7940 CrossRef CAS.
- A. S. K. Hashmi, C. Lothschuetz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem.–Eur. J., 2010, 16, 8012–8019 CAS.
- M. A. Cinellu, G. Minghetti, F. Cocco, S. Stoccoro and A. Zucca, Angew. Chem., Int. Ed., 2005, 44, 6892–6895 CrossRef CAS.
Footnote |
| † Present address: Stephenson Institute for Renewable Energy, The Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. |
| This journal is © The Royal Society of Chemistry 2012 |