Water-hydroxyl phases on an open metal surface: breaking the ice rules†
Matthew
Forster
a,
Rasmita
Raval
a,
Javier
Carrasco
b,
Angelos
Michaelides
c and
Andrew
Hodgson
*a
aSurface Science Research Centre and Department of Chemistry, University of Liverpool, Oxford Street, Liverpool, UK L69 3BX. E-mail: ahodgson@liverpool.ac.uk
bInstituto de Catálisis y Petroleoquímica, CSIC, Marie Curie 2, E-28049, Madrid, Spain
cLondon Centre for Nanotechnology and Department of Chemistry, University College London, London, UK WC1E 6BT
First published on 25th August 2011
Abstract
Hydroxyl is a key reaction intermediate in many surface catalyzed redox reactions, yet establishing the phase diagram for water/hydroxyl adsorption on metal surfaces remains a considerable challenge for interfacial chemistry. While the structures formed on close packed metal surfaces have been discussed widely, the phase diagram on more reactive, open metal surfaces, is complex and the H-bonding structures are largely unknown. Based on scanning tunnelling microscopy and density functional theory calculations, we report the phase diagram for water/hydroxyl on Cu(110), providing a complete molecular description of the complex hydrogen bonding structures formed. Three distinct phases are observed as the temperature is decreased and the water/hydroxyl ratio increased: pure OH dimers, extended 1H2O:1OH chains, aligned along the close-packed Cu rows, and finally a distorted 2D hexagonal c(2 × 2) 2H2O:1OH network. None of these phases obey the conventional ‘ice rules’, instead their structures can be understood based on weak H donation by hydroxyl, which favours H-bonding structures dominated by water donation to hydroxyl, and competition between hydroxyl adsorption sites. Hydroxyl binds in the Cu bridge site in the 1D chain structures, but is displaced to the atop site in the 2D network in order to accommodate water in its preferred atop binding geometry. The adsorption site and stability of hydroxyl can therefore be tuned simply by changing the surface temperature and water content, giving a new insight as to how the open metal template influences the water/hydroxyl structures formed and the activity of hydroxyl.
1. Introduction
Developing a qualitative understanding of water–metal and hydroxyl–metal interactions underpins our ability to devise molecular models for the structure of wet metal interfaces and, therefore, to model reactions that occur in these environments, including important catalytic1,2 and electrochemical reactions.3,4 The recent surge in discussion of water and water/hydroxyl adsorption at metal surfaces has been driven by the combination of new experimental insights and the development of theoretical techniques that allow us to examine the inter-relationship between hydrogen bond formation and the adsorbate–metal interaction.5–7 These experiments have demonstrated that the structure and behavior of the interface layer depends sensitively on the hydrogen bonding networks formed, which may show either local or extended order, just as in aqueous phases.8–10 Complexity arises because some of the water molecules may dissociate,11,12 particularly at steps or by electron exposure, to yield hydrogen bonded water/hydroxyl structures. Hydroxyl coadsorption can also be induced by reaction with O, creating water/hydroxyl phases that are stabilized compared to pure water.13 The majority of recent studies have examined close packed surfaces, where the symmetry of the metal template matches that of a hexagonal bulk ice film, and have shown that the structures formed are determined by a delicate balance between intermolecular hydrogen bonding and adsorbate–metal bonding. Instead of simply adopting a bulk ice arrangement, water forms hydrogen bonding structures that depend sensitively on the metal involved.13 The arrangement of intact14,15 and partially dissociated structures16–18 can often be rationalized based on optimizing the water–metal interaction by forming flat water clusters, while maintaining a strong, complete 2D hydrogen bonding network, but it is not yet clear how transferable these ideas are to other systems.In contrast to the close packed faces, the f.c.c. (110) metal surfaces offer the opportunity to explore how a different surface symmetry influences the wetting behavior. Moreover, these surfaces have an enhanced reactivity compared to close packed surfaces and are active in a number of important catalytic reactions involving water, for example in the low temperature water gas shift reaction.1,2,19 Since hydroxyl is a key intermediate in many of these reactions, understanding its thermodynamic and kinetic behaviour is of direct practical relevance, yet we have only a very limited picture of the water/hydroxyl phases formed on open f.c.c. (110) faces.13 The Cu(110) surface has received the most study, but the structure and composition of the water/hydroxyl phases remains the source of debate.6,20–26Water adsorbs in its molecular form at low temperature, desorbing intact above 160 K.6,21–25Low energy electron diffraction (LEED) studies reported a c(2 × 2) structure, which was initially attributed to a distorted water bilayer,24–26 but recent work shows this structure is formed by electron induced dissociation of water.6,22,23Scanning tunnelling microscopy (STM) images reveal a dramatic change in structure with coverage,22water initially forming 1D chains along the [001] direction, then 2D islands and finally completion of the (7 × 8) layer. A comparison of STM images and vibrational spectra with density functional theory (DFT) structure calculations identifies the 1D chain structure as a face sharing arrangement of water pentagons.6 This structure is favored because the Cu(110) template is too compact to accommodate a water hexagon, whereas the pentamer chain structure allows two thirds of the water to bond in its favored site, in a flat geometry atop Cu, optimizing the water–metal bonding whilst still maintaining a strong hydrogen bonding interaction. It is now believed that the wetting structure formed on other surfaces, such as Pt(111),14,27 may contain H bonded water rings of different size, revealing a surprising flexibility in the structure adopted by water at different surfaces.
The behavior of the mixed water/hydroxyl phases on Cu(110) is less well established, with debate over both their composition and structure. At high water coverage an extended c(2 × 2) mixed water/hydroxyl phase is formed below 180 K. Recent STM results find this phase contains at least two water molecules per hydroxyl within a distorted hexagonal network,17 consistent with the composition found in high pressure X-ray photoelectron spectroscopy (XPS) measurements.28 DFT calculations indicate that the excess water is stabilized by the formation of D type Bjerrum defects within the hydrogen bonding network. These defects have two H atoms between an adjacent pair of oxygen atoms, in this case two hydroxyl molecules being H bonded such that their H atoms lie just off the hydroxyl–hydroxyl axis. This structure breaks the well known Bernal–Fowler–Pauling (BFP) ice rules, sacrificing a complete H bonding network in order to maximize the number of strong bonds formed by water donation to hydroxyl.17 In addition to the extended c(2 × 2) structure, low dimensional structures have been reported following water dissociation or reaction with adsorbed O atoms. Kumagai et al. investigated the stability of small water clusters29 and the tunnelling barrier in water,30water/hydroxyl31 and hydroxyl dimers32 at low temperature by STM, while Lee et al.33 recently reported chains of water/hydroxyl form parallel to [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] and tentatively attributed these to a 2H2O:1OH structure.
0] and tentatively attributed these to a 2H2O:1OH structure.
In this study we combine high resolution STM images and state of the art DFT structure calculations to investigate the stability and structure of water/hydroxyl phases with different composition on Cu(110). Three different water/hydroxyl phases form as the surface temperature is decreased and the water content increased: a pure hydroxyl phase consisting of OH dimers, aligned along the [001] direction, two related 1H2O:1OH chain structures and finally 2D islands of the c(2 × 2) 2H2O:1OH phase.17 Excellent agreement is found between simulations of the minimum energy structures and experimental STM images, allowing us to obtain a detailed picture of the structures formed. Instead of obeying the Bernal–Fowler–Pauling ice rules and forming three H bonds, two as an acceptor and one as a donor, hydroxyl coadsorbed with water acts as an acceptor but not as a donor, maximizing the number of strong H bonds formed by water molecules donating to hydroxyl, rather than the overall number of H bonds. Hydroxyl in the 1H2O:1OH chain structures accepts a single H bond from a water chain, whereas increasing the water content to form the c(2 × 2) 2H2O:1OH phase creates D type Bjerrum defects,17 with each hydroxyl accepting H bonds from two water molecules. Associated with the change in structure from a 1D chain to 2D network, hydroxyl moves from its optimum short bridge site into an atop geometry, allowing water to bond in the favored atop adsorption geometry and form the defective 2D c(2 × 2) network. The rich range of adsorption behaviour found on Cu(110) is qualitatively different to that found on the close packed Pt(111)18,34,35 and Pd(111)36,37 surfaces, where hydroxyl adsorbs flat atop the metal atom, forming a stoichiometric 2D 1H2O:1OH phase. Our results indicate that simple structural models, based on the BFP ice rules and optimizing the H bonding coordination,5,38 are not adequate to describe the structure of water/hydroxyl phases on open, reactive metal surfaces; instead the water/hydroxyl structures formed optimize both the water and hydroxyl binding sites and the number of strong H bonds formed. This behavior has analogies to the over-coordination of hydroxide by H-donation in bulk water,39 where it may be energetically favorable to leave the hydroxyl proton uncoordinated at some sites.40 Other metal surfaces, such as Ru(0001)41,42 and Ag(110),43,44 also form non-stoichiometric or low dimensional structures, with a reduced H bonding coordination, and further studies will be necessary to explore how generally the ideas developed here for hydroxyl on Cu(110) can be transferred to other metal surfaces.
2. Methods
STM images were recorded at 100 K in an ultra high vacuum STM (Specs 150 Aarhus) operated in constant current mode with an electrochemically etched tungsten tip. The copper surface was prepared by argon ion sputtering at 500 eV, followed by annealing to 800 K, yielding an average terrace size of approx. 800 Å. Ultra-pure (107 Ω cm) water was dosed from background gas at a constant pressure of 2 × 10−9 mbar. Water purity during dosing was monitored by mass spectrometry. The relative water coverage was determined by calibrating the water dose against that required to form a complete c(2 × 2) layer, while the O coverage was calibrated against completion of the (2 × 1)O added row structure. Temperature programmed desorption (TPD) measurements were made in a separate chamber, described elsewhere,45 using a molecular beam to control the oxygen and water dose.In order to explore possible structures associated with the different phases, DFT calculations were performed for a wide variety of pure water and water–hydroxyl overlayers. The calculations were made using the VASP code46,47 and employed the standard PBE functional48 and an accurate version of the non-local van der Waals density functional of Dionet al.,49 referred to as “optB88-vdW”.50 Atomic geometries and simulated STM images shown correspond to the PBE functional (in this respect the optB88-vdW results do not differ to any great extent). In addition, the relative stability of the different structures considered was not altered by inclusion of dispersion forces. The metal slabs were 4 layers thick, separated by 14 Å, with the two bottom layers fixed in their bulk PBE optimal position, aCu = 3.627 Å. Core electrons were treated with the projector augmented-wave method,51 whilst valence electrons were expanded in a plane wave basis with 600 eV cut-off energy. Monkhorst–Pack52 k-point meshes of at least 12 × 12 × 1 per (1 × 1) surface unit cell were employed, as was a dipole correction along the direction perpendicular to the surface. All adsorption energies (including those for pure OH adsorption systems) are referenced to H2O as a gas phase water molecule, with dissociated H atoms adsorbed in a separate slab. STM images have been simulated using the Tersoff–Hamann approach53 at 3.7 Å from the metal surface and V = −200 meV
3. Results
3.1 Composition of the different water/hydroxyl structures
Fig. 1 shows the characteristic four peak temperature programmed desorption (TPD) profile that is obtained when water is adsorbed onto a Cu(110) surface that has been pre-treated with a low coverage of oxygen (0.1 ML or less). In the absence of coadsorbed O, the water layer desorbs at 165 K with minimal dissociation.23 In addition to the peak near 160 K, due to intact water clusters,21,23TPD shows three new water desorption peaks at 185, 220 and 265 K, characteristic of mixed hydroxyl/water coadsorption. STM results, described in the following sections, confirm that the 185, 220 and 265 K TPD peaks are associated with the decomposition of three distinct phases, whose structure we describe in detail below based on the STM and DFT results. Unlike the other dissociated phases on Cu(110), there is general agreement in the literature that the 260 K peak originates from decomposition of pure hydroxyl, assigned from STM and XPS studies.32,54 Based on this assignment, and the relative amount of water desorbing from the different peaks shown in Fig. 1, it is possible to estimate the composition of the different water/hydroxyl structures formed by reaction of water and O as a function of surface temperature. A series of water TPD measurements were recorded following reaction of excess water with adsorbed O, at coverages of up to 0.1 ML, and the amount of water desorbing during decomposition of the three high temperature phases was determined by integrating the TPD peaks. On this basis, the water/hydroxyl phase giving rise to the desorption peak at 220 K has a composition (0.9 ± 0.2)H2O:OH, while the 185 K peak, associated with the well known c(2 × 2) phase, contains (2.0 ± 0.6)H2O:1OH. The error limits represent the full range of compositions obtained from different runs and are limited by the uncertainty in separating the different peaks, particularly that for the c(2 × 2) phase at 185 K from the 150 K peak. Identification of the c(2 × 2) phase to a 2H2O:1OH structure is also supported by STM measurements directly comparing the number of adsorbed O atoms originally on the surface with the number of water/hydroxyl groups present after reaction.17 The final low temperature peak at 165 K in Fig. 1 has previously been assigned to intact water which does not interact directly with oxygen or hydroxyl.21STM images show excess water is stabilized by adsorption on the c(2 × 2) phase,17 desorbing as a broad peak between 165 and 180 K when excess water is present, Fig. 1.![TPD traces for water from an O (0.1 ML) pre-covered surface following adsorption of 0.05–0.4 ML water at 140 K. The 185 K peak is assigned to decomposition of the c(2 × 2) 2H2O:1OH structure, with peaks at 220 K and 260 K from decomposition of the H2O:OH [11̄0] chain and OH dimer structures respectively, as marked. Heating rate 1 K s−1.](/image/article/2012/SC/c1sc00355k/c1sc00355k-f1.gif) | ||
| Fig. 1
TPD traces for water from an O (0.1 ML) pre-covered surface following adsorption of 0.05–0.4 ML water at 140 K. The 185 K peak is assigned to decomposition of the c(2 × 2) 2H2O:1OH structure, with peaks at 220 K and 260 K from decomposition of the H2O:OH [10] chain and OH dimer structures respectively, as marked. Heating rate 1 K s−1. | ||
3.2 The pure hydroxyl phase
Fig. 2a shows an STM image of the surface following partial reaction of a (2 × 1)O island with water at 240 K. In addition to the regular added row CuO chains of the (2 × 1)O structure, we observe formation of bright features that are elongated along the [001] direction and surrounded by a decrease in intensity. High resolution images show these features contain pairs of bright protrusions, either as isolated features or sometimes adjacent to each other along [001]; one such example is shown in detail in Fig. 2b. The protrusions have a spacing of ca. 3.2 Å, slightly less than the 3.6 Å lateral spacing of the close packed Cu rows. These features are assigned to OH dimers, in line with recent STM results showing OH dimers are formed by reaction between water and adsorbed O at 6 K.32 Although the dimers are often found as pairs or short chains, they are never observed to grow into extended chains. STM images show the dimers do not approach closer than 2aCu along the [10] direction, consistent with the literature findings of a weak (2 × 1) LEED pattern for this phase.24,25
![STM images showing (a) partial reaction of an O(2 × 1) island (continuous bright chains in centre of image) with water at 240 K to form hydroxyl dimers, lying perpendicular to the close packed [11̄0] rows (135 × 140 Å2, V(t) = −190 mV, I(t) = −0.26 nA), (b) high resolution image showing 3 adjacent hydroxyl dimers formed by complete reaction at 230 K (22 × 20 Å2, V(t) = −190 mV, I(t) = −0.36 nA).](/image/article/2012/SC/c1sc00355k/c1sc00355k-f2.gif) | ||
| Fig. 2
STM images showing (a) partial reaction of an O(2 × 1) island (continuous bright chains in centre of image) with water at 240 K to form hydroxyl dimers, lying perpendicular to the close packed [10] rows (135 × 140 Å2, V(t) = −190 mV, I(t) = −0.26 nA), (b) high resolution image showing 3 adjacent hydroxyl dimers formed by complete reaction at 230 K (22 × 20 Å2, V(t) = −190 mV, I(t) = −0.36 nA). | ||
Density functional calculations for OH adsorption on Cu(110) find that single hydroxyl groups adsorb preferentially at bridge sites along Cu close packed rows,55 the next most stable adsorption site being the bridge between two close packed rows, about 360 meV less stable. The isolated hydroxyl points along the [001] azimuth with a tilt angle of about 60° with respect to the Cu(110) surface normal (Fig. 3). Flipping the H orientation has a barrier of 0.17 eV, consistent with the recent DFT results reported by Kumagai et al.32 who measured the rate at low temperature by STM. Coalescence of hydroxyls to form an (OH)2 dimer is favoured by ca. 214 meV, one hydroxyl group rotating towards the surface to form the hydrogen bond. As a result of this stabilization, hydroxyls are invariably found as dimers at the temperatures studied here. Simulated STM images show two bright features, slightly closer than the Cu spacing, surrounded by a dark region, similar to the images observed experimentally (Fig. 2). Arranging hydroxyls as a continuous chain along the [001] direction results in only a small energy gain, less than 10 meV (Table 1). The separation of adjacent Cu rows (3.6 Å) is too large to allow OH to form a continuous hydrogen bonded chain, instead hydroxyls remain as discrete dimers, with an O–O separation of 2.8 Å. The small cohesive energy between (OH)2 dimers is consistent with the experimental observation of a few short dimer chains along with isolated dimers, as shown in Fig. 2.
![Calculated binding geometry and simulated STM images for (a) an isolated OH group, (b) an OH dimer and (c) an array of OH forming a dimer chain along [001].](/image/article/2012/SC/c1sc00355k/c1sc00355k-f3.gif) | ||
| Fig. 3 Calculated binding geometry and simulated STM images for (a) an isolated OH group, (b) an OH dimer and (c) an array of OH forming a dimer chain along [001]. | ||
| PBE | optB88-vdW | |
|---|---|---|
| Isolated OH | 428 | 645 |
| OH dimer | 535 | 766 |
| OH dimer chain | 542 | 770 |
3.3 [1![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) 0] H2O:OH chain structures
0] H2O:OH chain structures
Decreasing the temperature at which water is dosed to below 200 K stabilizes the 1H2O:1OH phase. STM images reveal the disappearance of the OH dimers and the formation of 1D branched chain structures, aligned along the [10] direction, as shown in Fig. 4. Two co-existing chain structures are found, designated Z (zigzag) and P (pinch) in Fig. 4b and 4c respectively. The Z type chain has a regular branched structure, with a spacing of 5.2 Å, or two lattice constants in the [10] direction. It is noticeable that the side branches do not lie exactly perpendicular to the [10] direction, instead they invariably point slightly along the chain, in one direction or the other (see ESI†). This regular branched chain structure can change smoothly into an irregular structure (P), as can be seen at the bottom of Fig. 4b. The arrangement of the irregular chain (P) is shown in more detail in Fig. 4c. This structure has spacings that alternate between 5.1 and 5.5 Å in the [10] direction, i.e. a repeat of four lattice constants, with the branches pointing alternately up and down the chains. Despite their different internal arrangement, both regular and irregular chain structures have a similar width, ca. 7.7 ± 0.3 Å, just over twice the spacing of the Cu close packed rows (7.2 Å). STM images in which the chain structures coexist with the O(2 × 1) added row structure (see Fig. S4, ESI†) show that both P and Z types of chains are aligned symmetrically above a Cu close packed row. Since TPD shows just one decomposition peak in this temperature range, we conclude that the two structures have a similar stability and, in all likelihood, the same 1H2O:1OH composition. Both types of chain were observed near step edges during water adsorption on a clean Cu(110) surface, indicating that water dissociation occurs at step sites, even under conditions where the majority of the water adsorbs and desorbs intact from the terraces.23
![STM images of co-existing [11̄0] chain structures formed at 200 K. (a) (255 × 88 Å2), (b) “zig-zag” (Z) type chain with 2aCu repeat and regular branched spacing (41 × 21 Å2), (c) “pinched” (P) type chain with irregular branches and a 4aCu repeat.](/image/article/2012/SC/c1sc00355k/c1sc00355k-f4.gif) | ||
| Fig. 4
STM images of co-existing [10] chain structures formed at 200 K. (a) (255 × 88 Å2), (b) “zig-zag” (Z) type chain with 2aCu repeat and regular branched spacing (41 × 21 Å2), (c) “pinched” (P) type chain with irregular branches and a 4aCu repeat. | ||
Based on the periodicity of these chains, and their approximate composition, we investigated the stability of different possible [10] chain structures using DFT. From our previous analysis of the c(2 × 2) 2H2O:1OH phase,17 we expect hydroxyl, which is a poor H donor but good acceptor,18,28,56,57 to accept an H from water but not to act as a donor. Based on this idea, we examined chains containing a zig-zag water backbone, with each water bonded to hydroxyl and either a 2aCu or 4aCu repeat along [10]. Geometry optimization of the resulting chains gave the structures shown in Fig. 5a and 5b, with binding energies of 690 and 682 meV for chains with a two unit (Z) and four unit (P) period respectively. Despite the different arrangement of water along the chain, both structures have very similar adsorption sites and bond lengths for water and hydroxyl. Hydroxyl is adsorbed in the short bridge site, close to the surface (dCu–O = 2.02 Å), with the water slightly further from the surface (dCu–O = 2.14 Å), adsorbed roughly flat, displaced ca. 0.7 Å from the atop Cu site. This structure allows both hydroxyl and water to adopt adsorption geometries very similar to those found for the isolated, non-hydrogen bonded species. In both types of chain, the H bond formed by water donation to hydroxyl is short (dO–O = 2.63 Å) and that between the water molecules slightly longer, 2.77 to 2.86 Å. The offset between the bridge adsorption site of the hydroxyl and the atop site of the water causes the branches to point slightly along the [10] direction, rather than perpendicular to the chain. STM simulations for the Z structure, Fig. 5a, reproduce the alignment of the branches up or down the chain that is observed experimentally in Fig. 4b (see also Fig. S2 and S3†). For P type chains (Fig. 5b) the offset in water and hydroxyl binding site along [10] results in the characteristic alternation between open and partially closed rings shown in Fig. 4c. The offset between the hydroxyl and water species along [10] is quite distinct from the situation for the isolated OH/H2O pairs formed by manipulation at low temperature by Kumagai et al.,31 where the dimer forms a symmetric unit with both species adsorbed in the bridge site, aligned directly along the [001] direction. In that case the proton is believed to be symmetrically distributed between the hydroxyl groups, reducing the zero point vibrational energy sufficiently to stabilise this symmetric structure over the asymmetric form. In the thermodynamically stable OH/H2O chains reported here, creation of the water–water hydrogen bonding chain increases the average coordination number sufficiently to make water adsorb flat in its atop site and forgo the proton delocalisation seen in an isolated OH/H2O pair.
![Calculated structures (left) and simulated STM images (right) for the two most stable chain structures containing 1H2O:1OH. Each chain contains a central water backbone with H bonds donated to hydroxyl groups arranged with either (a) a 2aCu or (b) a 4aCu repeat along [11̄0]. These structures are referred to as “Z” (zig-zag) and “P” (pinch) respectively in the text. The solid line shows the (4 × 5) unit cell employed and binding energies are given in Table 2.](/image/article/2012/SC/c1sc00355k/c1sc00355k-f5.gif) | ||
| Fig. 5 Calculated structures (left) and simulated STM images (right) for the two most stable chain structures containing 1H2O:1OH. Each chain contains a central water backbone with H bonds donated to hydroxyl groups arranged with either (a) a 2aCu or (b) a 4aCu repeat along [10]. These structures are referred to as “Z” (zig-zag) and “P” (pinch) respectively in the text. The solid line shows the (4 × 5) unit cell employed and binding energies are given in Table 2. | ||
| Structure | H2O/OH | PBE | optB88-vdW |
|---|---|---|---|
| 5(a) | 1 | 690 | 895 |
| 5(b) | 1 | 682 | 890 |
| 6(a) | 3 | 659 | 835 |
| 6(b) | 2 | 624 | 808 |
| 6(c) | 3 | 609 | 791 |
| 6(d) | 1 | 576 | 763 |
| 6(e) | 2 | 575 | 752 |
| 6(f) | 2 | 505 | 686 |
| 6(g) | 1 | 470 | 669 |
| 6(h) | 1 | 434 | 622 |
To confirm if the structures shown in Fig. 5 are indeed the most stable [10] chain structures obtained from calculations, we calculated minimum energy structures, binding energies and STM simulations for a number of chain structures with different compositions and H bonding arrangements (see Fig. 6 and Table 2). None of the other structures we tried had a stability comparable to the 1H2O:1OH chains shown in Fig. 5a and 5b, nor was able to satisfactorily explain the STM images. Alternative possible chain structures having 1H2O:1OH composition found experimentally are shown in Fig. 6g and 6h. These structures have an adsorption energy at least 200 meV/OH lower than the structures shown in Fig. 5a and 5b and do not resemble the observed STM images. Displacing the hydroxyl groups to into the atop site (Fig. 6d) decreases the adsorption energy by 114 meV/OH. Amongst the other structures with different compositions investigated, the closest in terms of energy is a chain of hexagonal rings with a 3H2O:1OH composition, Fig. 6a, but this structure remains 20–30 meV/OH less stable than the 1H2O:1OH chains depicted in Fig. 5a and 5b and does not match the observed STM images. Our identification of the [10] chains as 1H2O:1OH structures is at odds with Lee et al.33 who also observed zig-zag chains in STM and tentatively suggested a 2H2O:1OH structure (Fig. 6b), similar to the Z type chain (Fig. 5a) but with half the hydroxyls missing. This assignment is not consistent with the composition obtained from TPD, see section 3.1, while the adsorption energy is some 80 meV smaller than the Z type chain. The STM images reported earlier show diffuse features along one side of the chain33 and we believe the structures most likely correspond to partially resolved images of the zigzag chains (Fig. 5a). Both the Z and P type chains have hydroxyl groups pointing at 42–43° to the surface normal along [001], consistent with the sharp H+ ESDIAD emission detected at 43° in this direction by Polak25 and by Lee et al.33
![Calculated structures (left) and simulated STM images (right) for other possible [11̄0] chain structures arranged with either a 2aCu, 4aCu or 6aCu repeat along [11̄0]. The unit cells are indicted by the solid lines and the binding energies and proportion of water to hydroxyl are given in Table 2.](/image/article/2012/SC/c1sc00355k/c1sc00355k-f6.gif) | ||
| Fig. 6 Calculated structures (left) and simulated STM images (right) for other possible [10] chain structures arranged with either a 2aCu, 4aCu or 6aCu repeat along [10]. The unit cells are indicted by the solid lines and the binding energies and proportion of water to hydroxyl are given in Table 2. | ||
The H2O/OH chains are sometimes terminated by a complete ring, as shown on the right hand side of Fig. 4a. These rings are ca. 11 Å wide, roughly three times the spacing of the Cu close packed rows, with each side having a similar branched arrangement to the larger ring of the irregular P type chain. These ring type structures are also observed as defects formed during reaction of the chain structures to form c(2 × 2) islands, discussed in the next section. Based on these observations we believe that these features are small water rings with the P type water arrangement, stabilized by donation to hydroxyl in the same way as for the 1D chains. As the initial O coverage is increased the surface becomes covered in 1H2O:1OH chains that have no regular alignment between chains (see ESI†). This result is consistent with decoration of the chains by OH preventing any direct H bonding between adjacent chains.
3.4 The c(2 × 2) water–hydroxyl phase
Decreasing the adsorption temperature below 180 K and depositing further water causes the [10] 1H2O:1OH chains to disappear and be replaced by distorted 2D hexagonal islands of the c(2 × 2) water–hydroxyl structure, Fig. 7. Both a comparison of the original O coverage and final water/hydroxyl coverage by STM,17 and the TPD data shown in Fig. 1, indicate that the c(2 × 2) phase forms with a composition ca.2H2O:1OH (Section 3.1). The H bonding structure within this hexagonal network has been discussed in detail by Forster et al.17 who explained the excess of water over hydroxyl by formation of stable (H2O)2–OH trimers within the 2D network. DFT calculations find that the hydroxyl groups are not involved in H donation, instead forming D type Bjerrum defects within a disordered hexagonal network, Fig. 7. This structure maximizes the number of strong H bonds formed by donation to hydroxyl, at the expense of forming OH Bjerrum defects. Although low temperature reaction forms metastable clusters, containing structures intermediate between the [10] chains and the c(2 × 2) structure, no other stable phases were observed (see ESI†).
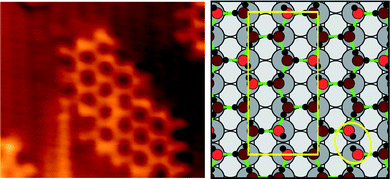 | ||
| Fig. 7 (a) STM image of the c(2 × 2) phase showing the termination of a small island (44 × 38 Å2, It = −0.3 nA, Vt = −190 mV. (b) Calculated structure of the c(2 × 2) phase showing a Bjerrum defect circled. The rectangle marks the unit cell employed in the calculation.17 | ||
Whereas intact 2D water clusters formed on close packed surfaces maximize the water H bonding coordination, forming a network of closed rings,16,58 the c(2 × 2) islands show an open edge structure. Where the structure is well resolved, e.g. in the lower right side of Fig. 7, the [10] edges have a 2aCu repeat, with the edge features displaced slightly relative to the neighbouring hexagon. This structure mimics exactly the Z type termination of 1H2O:1OH chains, shown in Fig. 4b and 5a, and allows us to assign the bright low coordinate edge features as hydroxyl groups. The offset between the edge hydroxyl features and the hexagonal network supports the assignment from DFT that the c(2 × 2) structure has O adsorbed atop Cu.17 This is an important distinction between the chain and 2D structures; in the 2D network the hydroxyl groups have been forced to adopt a less favorable atop binding site, allowing water to adsorb in its favored atop site at the expense of a reduced hydroxyl–metal interaction. In contrast the 1D chain structures have greater flexibility in the choice of water and hydroxyl adsorption site, allowing them to form a hydrogen bonding network with both species adsorbed at their favored sites.
4. Discussion
The water/Cu(110) system has been the focus of considerable study, but the complexity of this system can now be understood more clearly. The structures found on Cu(110) differ in several respects from the water and water/hydroxyl phases previously reported on close packed surfaces and offer a new insight into the influence of the surface site and symmetry on the wetting of metal surfaces. The intact and partially dissociated water structures found on close packed faces essentially follow the modified BFP ice rules, having one H between each pair of O atoms and maximizing the H bonding coordination such that each water/hydroxyl group has three H bonds. These constraints still leave considerable flexibility in the overall H bonding arrangement, allowing the adsorbate to form very different 2D networks in order to optimize the bonding on different metal surfaces.14,45,58,59 In contrast, none of the water or mixed water/hydroxyl phases analyzed so far on Cu(110) conform to an extended 2D hexagonal network of the type that have been anticipated previously,5,38 all having an average H bond coordination of less than 3 and, in the case of the c(2 × 2) phase, breaking the BFP ice rules. In this respect the absence of H donation by hydroxyl at surfaces mirrors the behavior of hydroxide in the bulk phases,39,40 where over-coordination of the O by neighboring H may leave the hydroxide H un-coordinated. These over-coordinated structures pin the hydroxide and are thought to be responsible for the reduced proton mobility of alkaline as opposed to acid doped water structures, again, something that has analogies in the pinning of mixed water/hydroxyl structures by the strong chemisorption of hydroxyl at metal surfaces.13 Here we discuss how the short metal spacing of Cu, and different adsorption sites available on the (110) surface, modify the phases formed compared to those reported on close packed surfaces. We describe how the general principles developed to explain adsorption on close packed surfaces must be adapted to describe the adsorption behavior of water and hydroxyl on Cu(110), and suggest how these may be applied more generally to the description of wetting and adsorption at other surfaces.The most obvious difference between Cu(110) and the close packed surfaces is, of course, the spacing and symmetry of the metal template, which plays a key role in determining both the local H bonding motif and the long range organization of the water and water/hydroxyl H bonding structures. DFT calculations find that a single water molecule binds in a flat geometry near the atop site on Cu(110),5,38,60,61 adopting a very similar adsorption geometry to that found on close packed metal surfaces such as Pt(111)62 and Ru(0001).12 Although the close packed faces offer the possibility of pseudomorphic ice growth, with water adopting the atop metal site to form a simple commensurate hexagonal water layer, experiments now suggest this is uncommon. A commensurate structure is found on Ru(0001),59 where the metal spacing is just 3% larger than the lateral spacing of water in bulk ice Ih(0001), but more complex unit cells are found on surfaces such as Pt(111)63 and Ni(111),64 where the metal spacing deviates further (+6% and −4.5% respectively) from the water ice repeat. The stability of large unit cell structures on Pt(111) appears to originate from the formation of small flat clusters of water, tightly bound atop Pt, embedded within a complex H-down water network.14 Templating the surface by forming a (√3 × √3)R30° surface alloy removes adjacent adsorption sites and destabilizes the chain type structures, forcing water into an H-down bilayer arrangement.65 The rectangular Cu(110) surface contains close packed rows of Cu atoms with aCu = 2.55 Å along [10], just 2% less than in bulk ice, but the match in the perpendicular direction is poor, with the Cu rows ca. 1 Å closer together along [001] than required to create an unstrained hexagonal ring of water bridging two Cu rows. Instead of forming such a strained structure, water forms 1D chains of face sharing pentamers, with 2/3 of the water bonded flat atop the Cu rows,6 maximising the O–Cu interaction while minimising the strain associated with the commensurate adsorption site. Although formation of a 2D network would increase the average H bond coordination from 8/3 to 3, the loss of the commensurate adsorption site reduces the average water–Cu interaction sufficiently that the 1D chains are favored. Increasing the coverage so that the chains approach within ca. 10–15 Å destabilizes the chains and a 2D structure forms,6 whose complex (7 × 8) H bonding network is not yet understood.23
The second key difference between Cu(110) and the close packed surfaces is the adsorption site adopted by hydroxyl. Whereas an isolated water molecule preferentially binds near the atop site on all metal faces examined, hydroxyl adopts a different site on the open Cu(110) and close packed metal faces. On Pt(111), hydroxyl slightly prefers the atop over the bridge adsorption site, by ca. 0.02 eV, with the OH axis pointing just 20° out of the surface plane.11 This common adsorption geometry for water and hydroxyl leads naturally to a planar 1H2O:1OH structure being formed on Pt(111) and Pd(111),36,66 in which each species bonds atop the metal with 3 H bonds in a commensurate (√3 × √3)R30° arrangement.35 The amount of water incorporated within this structure can be varied from ca.1H2O:2OH upwards, with no substantial change in the H bonding network. In contrast, on Cu(110) the hydroxyl potential is highly corrugated, with the short bridge site favored by 0.36 eV over the long bridge site and hydroxyl adsorbed upright, pointing at 60° from the surface along the [001] direction. The preference for a different adsorption site and orientation from that of water directly influence the structure of the water/hydroxyl phases seen on Cu(110), stabilising three very different phases depending on the water/hydroxyl ratio.
Both the 1D phases formed on Cu(110) contain hydroxyl adsorbed in the short bridge site, with a similar adsorption geometry to the isolated species. The pure hydroxyl phase, formed at high temperatures, contains dimers with one hydroxyl rotated towards the surface to form an H bond with dO–O = 2.8 Å. The spacing of the close packed Cu rows (3.6 Å) is too large to allow formation of H bonds between adjacent hydroxyl dimers, but hydroxyl prefers to retain the tilted bridge adsorption site, rather than adapting its adsorption site to increase the number of H bonds. A pure hydroxyl phase also forms on Pt(110)-(2 × 1),67Ni(110)68 and Ag(110),44 but little is known about the H bonding in these systems. Hydroxyl adsorbs in a tilted geometry on Ni(110) and Ag(110), similar to the geometry found on Cu(110). Since STM images of Ag(110) show hydroxyl forms chains parallel to the close packed rows,43 the orientation of hydroxyl along [001]69 appears to rule out H bonding stabilizing this structure. In contrast to these open f.c.c. (110) surfaces, close packed transition metal faces such as Pt(111)66 and Pd(111)36 do not form a pure hydroxyl phase, both surfaces requiring a water/O ratio of at least 2 in order for water to react to form a mixed water/hydroxyl structure. This difference can be directly correlated to the much greater exothermicity of water dissociation on Cu(110) compared to Pt(111) and Ru(0001), (0.77 eV38 compared to 0.05 eV11 and 0.38 eV70 respectively for monomer dissociation), driven by the stability of hydroxyl adsorbed in the bridge site.
Formation of stoichiometric 1H2O:1OH water/hydroxyl chains on Cu(110) allows both water and hydroxyl to adsorb in their optimum atop and bridge sites, forming strong H bonds by water donation to water and hydroxyl but giving an average coordination number of just 2. The calculated adsorption energy (PBE) of the 1D chain structures (690 and 682 meV) exceeds that of the best 2D 1H2O:1OH network by 20 to 30 meV, correctly predicting that the stoichiometric layer on Cu(110) forms 1D chains, rather than a c(2 × 2) network as proposed earlier.5,38,60,61 Formation of the 2D c(2 × 2) network occurs only once the water/hydroxyl ratio is increased and is associated with hydroxyl being displaced into the atop site to form Bjerrum defects. This structure reduces the average H bond coordination but allows water to retain the atop adsorption site while maximizing the number of strong H bonds formed by water donation to hydroxyl. This 2H2O:1OH structure is stabilized by around 45 meV compared to the best stoichiometric 1H2O:1OH c(2 × 2)network17 and is quite different to the ordered, 2D 1H2O:1OH networks formed on Pt(111)35,57 and Pd(111)36 which have all H atoms involved in hydrogen bonding. A direct consequence of the absence of uncoordinated OH groups is that the water/hydroxyl networks formed on Pt(111) and Pd(111) do not wet,36,71 whereas on Cu(110) Bjerrum defects in the c(2 × 2) network17 and uncoordinated hydroxyl in the chain structures are available to stabilize multilayer water nucleation (see for example the bright features above some of the chains in Fig. 4a).
Unlike other close packed faces studied so far, Ru(0001) also forms a water/hydroxyl structure containing an excess of water over hydroxyl,13 suggesting a parallel to Cu(110). STM images show narrow elongated stripes of a hexagonal network,41 whose edges are decorated by bright features, in an open arrangement that is superficially similar to the network found in the 1D chains and at the boundaries of the c(2 × 2) 2H2O:1OH network on Cu(110). Calculations indicate that Bjerrum D defects should also form in the Ru(0001) water/hydroxyl layer,72 analogous to the stabilization of the non-stoichiometric c(2 × 2) network on Cu(110),17 but experimental evidence for this is so far lacking. The location of hydroxyl on Ru(0001) was discussed by Tatarkhanov et al.41 who compared STM images with model DFT structures. Since water donor–hydroxyl acceptor bonding is more stable than water–water bonding by about 100 meV, it had been anticipated that hydroxyl would sit at the edge of the islands, as on Cu(110), but instead the most stable model structures had non-donor, single acceptor water molecules located at the periphery of islands and OH groups in the interior of the stripes. Reasons for this difference in behavior are not yet clear and further study is needed to understand the location of hydroxyl and the role of Bjerrum defects in the water/hydroxyl structure formed on Ru(0001).
5. Conclusions
The results described here allow us to complete the description of the different water/hydroxyl phases formed on Cu(110), making this perhaps the best understood water adsorption system of any reactive metal surface. Adsorption shows a rich behaviour, with two intact and three partially dissociated water/hydroxyl phases, stabilized by the short Cu spacing and rectangular metal template. The intact phases have been considered earlier and consist of an unusual 1D pentagonal chain structure and a 2D (7 × 8) network.6,23 A comparison of DFT simulations and STM images for water/hydroxyl structures of defined composition reveals excellent agreement between the calculated minimum energy structures and experiment, confirming the assignment of these phases and providing insight into their structure. Both 1D chain structures have hydroxyl adsorbed in the bridge site, pointing out from the surface along the [001] direction. The pure hydroxyl phase consists of dimers, with a single H bond and a weak tendency to associate into chains. The 1H2O:1OH phase is characterized by two closely related, coexisting 1D chain structures. Both structures consist of a hydrogen bonded chain of water molecules, lying flat on the surface along a close packed row, with each water molecule involved in one hydrogen bond to a terminal hydroxyl. The stability of the 1H2O:1OH chain motif can be attributed to two effects. First, it allows water and hydroxyl to bond at their preferred adsorption sites, bridge and atop respectively. Second, it maximizes the number of strong hydrogen bonds formed by water donation to hydroxyl, reflecting the greater H accepting ability of hydroxyl. The final phase is the well known c(2 × 2) structure, containing 2H2O:1OH.17 This structure is characterized by a distorted 2D hexagonal hydrogen-bonding arrangement, in which hydroxyl is displaced to the less stable atop site, forming Bjerrum D defects. DFT calculations correctly predict the transition from 1D to 2D structures upon varying composition, providing confidence in their ability to capture the influence of different surface sites and H bond arrangement on the adsorption energy. None of the water or water/hydroxyl structures formed on Cu(110) obeys the usual ice rules, instead optimizing the water and hydroxyl binding sites and number of strong H bonds, formed by water donation to water/hydroxyl, rather than simply the overall number of H bonds. This work highlights the great structural flexibility that may arise as the system seeks to maximize the water–metal interaction while maintaining strong hydrogen bonds, creating structures with differing hydroxyl binding energies and wetting properties. Future studies of adsorption on other surfaces must consider structures containing different adsorption sites, as well as both 1D and 2D H bond networks, not just the maximally coordinated networks traditionally assumed.Acknowledgements
JC is supported by the Spanish Ministerio de Ciencia e Innovación through a Ramón y Cajal Fellowship. AM's work is supported by the EURYI scheme, EPSRC, and ERC, AH by EPSRC and RR by EPSRC, BBSRC and EU. Computational resources from the London Centre for Nanotechnology and UCL Research Computing are gratefully acknowledged. Also partly via our membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/F067496), this work made use of the facilities of HECToR.References
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef CAS.
- N. Schumacher, A. Boisen, S. Dahl, A. A. Gokhale, S. Kandoi, L. C. Grabow, J. A. Dumesic, M. Mavrikakis and I. Chorkendorff, J. Catal., 2005, 229, 265 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS.
- J. Ren and S. Meng, J. Am. Chem. Soc., 2006, 128, 9282 CrossRef CAS.
- J. Carrasco, A. Michaelides, M. Forster, S. Haq, R. Raval and A. Hodgson, Nat. Mater., 2009, 8, 427 CrossRef CAS.
- K. Andersson, G. Ketteler, H. Bluhm, S. Yamamoto, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Am. Chem. Soc., 2008, 130, 2793 CrossRef CAS.
- P. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. A. Naslund, T. K. Hirsch, L. Ojamae, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995 CrossRef CAS.
- R. Ludwig, Angew. Chem., Int. Ed., 2001, 40, 1808 CrossRef CAS.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192 RSC.
- A. Michaelides and P. Hu, J. Am. Chem. Soc., 2001, 123, 4235 CrossRef CAS.
- P. J. Feibelman, Science, 2002, 295, 99 CrossRef CAS.
- A. Hodgson and S. Haq, Surf. Sci. Rep., 2009, 64(9), 381 CrossRef CAS.
- S. Nie, P. J. Feibelman, N. C. Bartelt and K. Thurmer, Phys. Rev. Lett., 2010, 105, 026102 CrossRef CAS.
- J. Carrasco, B. Santra, J. Klimes and A. Michaelides, Phys. Rev. Lett., 2011, 106, 026101 CrossRef.
- M. Tatarkhanov, D. F. Ogletree, F. Rose, T. Mitsui, E. Fomin, S. Maier, M. Rose, J. I. Cerda and M. Salmeron, J. Am. Chem. Soc., 2009, 131, 18425 CrossRef CAS.
- M. Forster, R. Raval, A. Hodgson, J. Carrasco and A. Michaelides, Phys. Rev. Lett., 2011, 106, 046103 CrossRef.
- T. Schiros, L. A. Naslund, K. Andersson, J. Gyllenpalm, G. S. Karlberg, M. Odelius, H. Ogasawara, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. C, 2007, 111, 15003 CAS.
- L. Jiang, G. C. Wang, Z. S. Cai, Y. M. Pan and X. Z. Zhao, THEOCHEM, 2004, 710, 97 CrossRef CAS.
- C. Ammon, A. Bayer, H. P. Steinruck and G. Held, Chem. Phys. Lett., 2003, 377, 163 CrossRef CAS.
- K. Andersson, A. Gomez, C. Glover, D. Nordlund, H. Ostrom, T. Schiros, O. Takahashi, H. Ogasawara, L. G. Pettersson and A. Nilsson, Surf. Sci., 2005, 585, L183 CrossRef CAS.
- T. Yamada, S. Tamamori, H. Okuyama and T. Aruga, Phys. Rev. Lett., 2006, 96, 036105 CrossRef CAS.
- T. Schiros, S. Haq, H. Ogasawara, O. Takahashi, H. Öström, K. Andersson, L. G. M. Pettersson, A. Hodgson and A. Nilsson, Chem. Phys. Lett., 2006, 429, 415 CrossRef CAS.
- K. Bange, D. E. Grider, T. E. Madey and J. K. Sass, Surf. Sci., 1984, 136, 38 CrossRef.
- M. Polak, Surf. Sci., 1994, 321, 249 CrossRef CAS.
- A. Spitzer and H. Luth, Surf. Sci., 1982, 120, 376 CrossRef CAS.
- P. J. Feibelman, G. A. Kimmel, R. S. Smith, N. G. Petrik, T. Zubkov and B. D. Kay, J. Chem. Phys., 2011, 134, 204702 CrossRef.
- K. Andersson, G. Ketteler, H. Bluhm, S. Yamamoto, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Phys. Chem. C, 2007, 111, 14493 CAS.
- T. Kumagai, H. Okuyama, S. Hatta, T. Aruga and I. Hamada, J. Chem. Phys., 2011, 134, 024703 CrossRef CAS.
- T. Kumagai, M. Kaizu, S. Hatta, H. Okuyama, T. Aruga, I. Hamada and Y. Morikawa, Phys. Rev. Lett., 2008, 100, 166101 CrossRef CAS.
- T. Kumagai, M. Kaizu, H. Okuyama, S. Hatta, T. Aruga, I. Hamada and Y. Morikawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 045402 CrossRef.
- T. Kumagai, M. Kaizu, H. Okuyama, S. Hatta, T. Aruga, I. Hamada and Y. Morikawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 035423 CrossRef.
- J. Lee, D. C. Sorescu, K. D. Jordan and J. T. Yates, J. Phys. Chem. C, 2008, 112, 17672 CAS.
- G. S. Karlberg, F. E. Olsson, M. Persson and G. Wahnstrom, J. Chem. Phys., 2003, 119, 4865 CrossRef CAS.
- G. Held, C. Clay, S. Barrett, S. Haq and A. Hodgson, J. Chem. Phys., 2005, 123, 064711 CrossRef CAS.
- C. Clay, L. Cummings and A. Hodgson, Surf. Sci., 2007, 601, 562 CrossRef CAS.
- M. J. Gladys, A. A. El Zein, A. Mikkelsen, J. N. Andersen and G. Held, Surf. Sci., 2008, 602, 3540 CrossRef CAS.
- J. Ren and S. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 054110 CrossRef.
- M. E. Tuckerman, A. Chandra and D. Marx, Acc. Chem. Res., 2006, 39, 151 CrossRef CAS.
- L. Cwiklik and V. Buch, Phys. Chem. Chem. Phys., 2009, 11, 1294 RSC.
- M. Tatarkhanov, E. Fomin, M. Salmeron, K. Andersson, H. Ogasawara, L. G. M. Pettersson, A. Nilsson and J. I. Cerda, J. Chem. Phys., 2008, 129, 154109 CrossRef CAS.
- P. Cabrera-Sanfelix, A. Arnau, A. Mugarza, T. K. Shimizu, M. Salmeron and D. Sanchez-Portal, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 155438 CrossRef.
- L. Savio, M. Smerieri, L. Vattuone, A. Gussoni, C. Tassistro and M. Rocca, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 235412 CrossRef.
- M. Canepa, P. Cantini, E. Narducci, M. Salvietti, S. Terreni and L. Mattera, Surf. Sci., 1995, 343, 176 CrossRef CAS.
- S. Haq, C. Clay, G. R. Darling, G. Zimbitas and A. Hodgson, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 115414 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schroder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS.
- J. Klimes, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. Tersoff and D. R. Hamann, Phys. Rev. Lett., 1983, 50, 1998 CrossRef CAS.
- A. Spitzer and H. Luth, Surf. Sci., 1985, 160, 353 CrossRef CAS.
- E. R. M. Davidson, A. Alavi and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153410 CrossRef.
- S. Yamamoto, K. Andersson, H. Bluhm, G. Ketteler, D. E. Starr, T. Schiros, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Phys. Chem. C, 2007, 111, 7848 CAS.
- T. Schiros, H. Ogasawara, L. A. Naslund, K. J. Andersson, J. Ren, S. Meng, G. S. Karlberg, M. Odelius, A. Nilsson and L. G. M. Pettersson, J. Phys. Chem. C, 2010, 114, 10240 CAS.
- J. Cerda, A. Michaelides, M. L. Bocquet, P. J. Feibelman, T. Mitsui, M. Rose, E. Fomin and M. Salmeron, Phys. Rev. Lett., 2004, 93, 116101 CrossRef CAS.
- M. Gallagher, A. Omer, G. R. Darling and A. Hodgson, Faraday Discuss., 2009, 141, 231 RSC.
- Q. L. Tang and Z. X. Chen, J. Chem. Phys., 2007, 127, 104707 CrossRef.
- Q. L. Tang and Z. X. Chen, Surf. Sci., 2007, 601, 954 CrossRef CAS.
- A. Michaelides, V. A. Ranea, P. L. de Andres and D. A. King, Phys. Rev. Lett., 2003, 90, 216102 CrossRef CAS.
- A. Glebov, A. P. Graham, A. Menzel and J. P. Toennies, J. Chem. Phys., 1997, 106, 9382 CrossRef CAS.
- M. Gallagher, A. Omer, S. Haq and A. Hodgson, Surf. Sci., 2007, 601, 268 CrossRef CAS.
- F. McBride, G. R. Darling, K. Pussi and A. Hodgson, Phys. Rev. Lett., 2011, 106, 226101 CrossRef.
- C. Clay, S. Haq and A. Hodgson, Phys. Rev. Lett., 2004, 92, 046102 CrossRef CAS.
- A. Shavorskiy, T. Eralp, M. J. Gladys and G. Held, J. Phys. Chem. C, 2009, 113, 21755 CAS.
- C. Benndorf, C. Nobl and T. E. Madey, Surf. Sci., 1984, 138, 292 CrossRef CAS.
- K. Bange, T. E. Madey and J. K. Sass, Surf. Sci., 1985, 152, 550 CrossRef.
- A. Michaelides, A. Alavi and D. A. King, J. Am. Chem. Soc., 2003, 125, 2746 CrossRef CAS.
- G. Zimbitas, M. E. Gallagher, G. R. Darling and A. Hodgson, J. Chem. Phys., 2008, 128, 074701 CrossRef.
- P. J. Feibelman, Chem. Phys. Lett., 2005, 410, 120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: STM images showing high coverage chains, growth of the c(2 × 2) structure from 1D 1H2O:1OH and registry of these structures to the metal surface. See DOI: 10.1039/c1sc00355k |
| This journal is © The Royal Society of Chemistry 2012 |