Phosphorus complexes of a triply-fused [24]pentaphyrin†
Tomohiro
Higashino
and
Atsuhiro
Osuka
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto, 606-8502, Japan. E-mail: osuka@kuchem.kyoto-u.ac.jp
First published on 27th September 2011
Abstract
Treatment of an N-fused [24]pentaphyrin with POCl3 in the presence of triethylamine gave a triply-fused pentaphyrin P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complex through an N-fusion reaction and oxidative C–C bond formation between the pyrrolic β-positions. This complex displays strong antiaromatic character due to its 24π-electronic network as evinced by its 1H NMR chemical shifts, absorption spectrum, and NICS calculation. The PO complex was reduced with BH3·SMe2 to provide P–BH3 complex with concurrent hydrogenation at the pyrrolic β-positions as a rare case of porphyrinoid P(III) complex.
O complex through an N-fusion reaction and oxidative C–C bond formation between the pyrrolic β-positions. This complex displays strong antiaromatic character due to its 24π-electronic network as evinced by its 1H NMR chemical shifts, absorption spectrum, and NICS calculation. The PO complex was reduced with BH3·SMe2 to provide P–BH3 complex with concurrent hydrogenation at the pyrrolic β-positions as a rare case of porphyrinoid P(III) complex.
In recent years, there are increasing interests in the chemistry of expanded porphyrins because of their unique optical, electrochemical, and coordination properties, which are mostly derived from their large conjugated circuits and conformational flexibilities that are not shared by porphyrins.1 Among these, N-fused reactions of expanded porphyrins are attractive because the resultant tricyclic ring system imposes a planar and rigid substructure, hence causing large structural and electronic perturbations.2–4N-Fused structures were formed either by nucleophilic attack of a pyrrolic nitrogen atom to the neighboring β-carbon atom2,3,4a,b or by nucleophilic substitution of ortho-halogen at a meso-aryl substituent with an adjacent pyrrolic nitrogen atom.4c–g In particular, N-fusion strategy has been extensively used to create novel structures from N-confused porphyrinoids by Furuta et al.2,3
meso-Aryl substituted pentaphyrins exist only as N-fused forms that leave a smaller cavity than those of porphyrins.4a,b As a continuation of our research toward the exploration of novel porphyrinoids,1h we examined phosphorus insertion into an N-fused pentaphyrin, since the phosphorus atom is smaller than the usual metal ions and is potentially a good fit for the small cavity of an N-fused [24]pentaphyrin. Additionally the phosphorus insertion has been recently shown to be effective in the creation of novel porphyrinoids such as triazacorroles,5a,bN-fused porphyrins,5cN-fused telluraporphyrins,5d an expanded isophlorin,5e and a Möbius antiaromatic molecule.5f In this paper, we report that phosphorus insertion into N-fused [24]pentaphyrin 1 triggered further double fusion reactions to provide a phosphorus complex of a triply-fused structure, 2. This phosphoramide PO complex 2 was transformed into a PS complex 3 and P–BH3 complex 4.
Results and discussion
Synthesis of phosphorus complex
First, we examined the reaction of meso-pentakis(pentafluorophenyl)-substituted N-fused [24]pentaphyrin 1 with POCl3. After extensive optimizing experiments, treatment of 1 with POCl3 (100 equiv) in the presence of triethylamine at 120 °C for 12 h gave the best result to yield phosphoramide PO complex 2 in 66% yield after chromatographic separation over a silica gel column (Scheme 1).
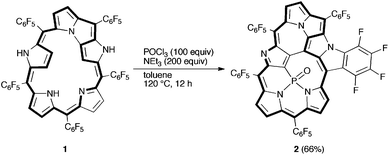 | ||
| Scheme 1 Synthesis of phosphorus complex 2. | ||
The high-resolution electrospray ionization time-of-flight (HR-ESI-TOF) mass spectrum of 2 showed the parent negative ion peak at m/z = 1273.9612 (calcd for C55H6F24N5OPCl, [M + Cl]−: 1273.9621), thus indicating the insertion of a PO moiety and the elimination of HF, and H2. Fortunately, we obtained single crystals of 2 suitable for X-ray diffraction analysis by vapor diffusion of hexane into its chlorobenzene solution.‡ The unit cell contained two independent molecules in the unsymmetric unit and the crystal structure revealed the phosphorus insertion and a triply-fused structure (Fig. 1). The inserted phosphorus atom was bound to N(1), N(2) and one β-carbon atom, C(12), with distances of 1.689(5) and 1.697(6) Å for P–N(1), 1.670(5) and 1.670(6) Å for P–N(2), and 1.732(7) and 1.735(8) Å for P–C(12), respectively. Curiously, another N-fusion reaction occurred between N(5) and the pentafluorophenyl substituent at C(55), and C–C bond formation occurred between C(13) and C(23) to form an unprecedented triply-fused skeleton. The C(13)–C(23) bond length is 1.409(9) and 1.401(10) Å, being slightly longer than the typical C–C bond length in a benzene ring (1.39 Å). The 31P NMR spectrum of 2 displays a signal at δ = 7.12 ppm consistent with the phosphoramide moiety.5 The 1H NMR spectrum of 2 exhibits five peaks due to the β-protons at δ = 6.35, 6.05, 4.59, 4.43, and 4.34 ppm (Fig. 2a). These large upfield shifts of the outer β-protons are similar to those of antiaromatic porphyrinoid, such as the doubly N-fused pentaphyrin and [28]hexaphyrin mono-AuIII complex (δ = 6.38–4.07 ppm), indicating a paratropic ring current and thus antiaromatic character.3a,6a Consistent with this conjecture, the UV/Vis absorption spectrum shows a blue-shifted Soret-like band and almost no Q-band-like absorption band (Fig. 4). These features are typical of antiaromatic expanded porphyrins.6b,c The antiaromaticity of 2 is derived from its rigid planar conformation forced by the PO-incorporated and triply-fused structure. The formation of 2 may be accounted for in terms of initial electrophilic attack of the phosphoryl group by C(12) followed by nucleophilic attack of C(23) at C(13) and subsequent oxidation, and final N-fusion reaction between N(5) of pyrrole E and the meso-pentafluorophenyl substituent at C(55) (See Supporting Information†).
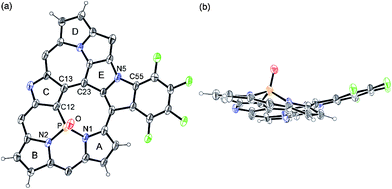 | ||
| Fig. 1 X-Ray crystal structure of PO complex 2: (a) top view and (b) side view. One of the two independent molecules in the unsymmetric unit cell is shown. Thermal ellipsoids represent 30% probability and meso-pentafluorophenyl substituents and solvent molecules are omitted for clarity. | ||
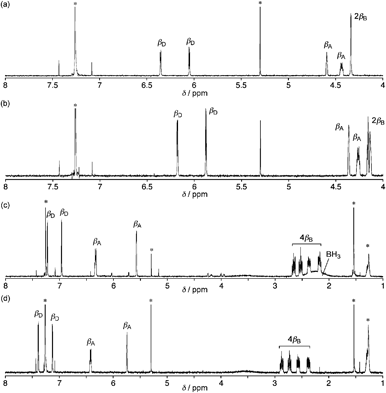 | ||
| Fig. 2 1H NMR spectra of (a) 2, (b) 3, (c) 4 and (d) 5 in CDCl3 at 25 °C. Peaks marked with * are due to residual solvents and impurities. | ||
Phosphorus(III) complex
Then, we attempted to obtain the corresponding P(III) complex, since phosphorus-incorporated porphyrinoids have been confined to P(V) species and no P(III) porphyrinoid has been reported in the literature. Trivalent phosphines are usually prepared from pentavalent phosphine oxides by reduction with silane reducing reagents.7 However, treatment of the PO complex 2 with trichlorosilane or phenylsilane resulted in complete recovery of 2 without formation of the corresponding phosphorus(III) complex. Therefore, we planned out two strategies for the synthesis of the phosphorus(III) complex, (a) conversion of the complex 2 to the corresponding phosphine sulfide (PS) complex followed by its desulfurization,8 or (b) reduction to the phosphine-borane (P–BH3) complex followed by liberation of the desired P(III) complex with a strong base.9 First, the PS complex 3 was synthesized by the reaction of 2 with Lawesson's reagent in 84% yield (Scheme 2).10 The HR-ESI-TOF mass spectrum of 3 displays the parent negative ion peak at m/z = 1289.9379 (calcd for C55H6F24N5PSCl, [M + Cl]−: 1289.9392). The 31P NMR spectrum of 3 shows a peak in a downfield region at δ = 58.55 ppm in accord with the formation of a PS moiety.10b The structure of 3 was also unambiguously revealed by X-ray diffraction analysis to be a triply-fused structure, which is essentially the same as that of 2 (Fig. 3a).‡ The 1H NMR spectrum of 3 exhibits six signals due to β-protons at δ = 6.18, 5.88, 4.36, 4.26, 4.16, and 4.13 ppm, which suggests a paratropic ring current and hence antiaromaticity for 3 (Fig. 2b). The UV/Vis absorption spectrum of 3 is similar to that of 2. Desulfurization of 3 was attempted with P(nBu)3, P(NMe2)3, and RANEY® nickel but these attempts failed, giving merely complicated mixtures.
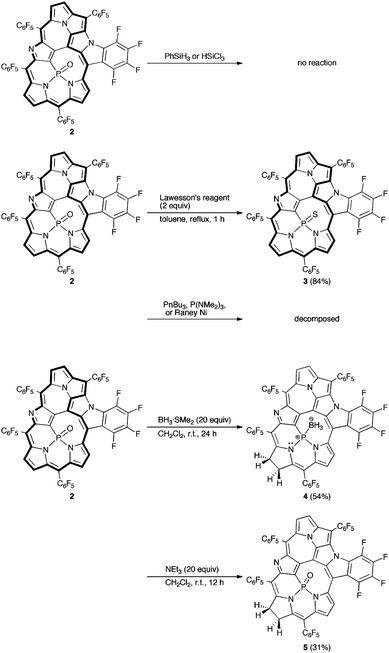 | ||
| Scheme 2 Attempts to synthesize the phosphorus(III) complex. | ||
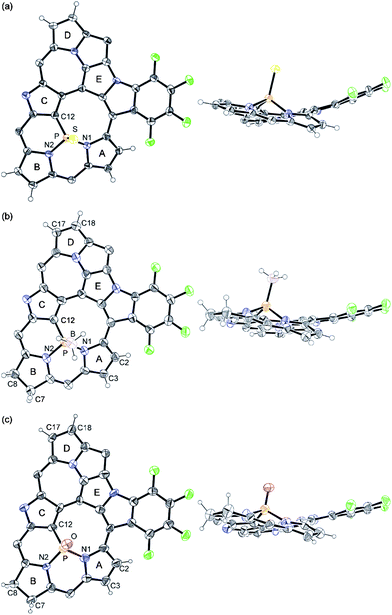 | ||
| Fig. 3 X-Ray crystal structures of (a) PS complex 3, (b) P–BH3 complex 4 and (c) chlorin-type PO complex 5: top view (left) and side view (right). Thermal ellipsoids represent (a, b) 50% and (c) 30% probability and meso-pentafluorophenyl substituents and solvent molecules are omitted for clarity. | ||
In the next step, the PO complex 2 was reduced with BH3·SMe29b to provide the P–BH3 complex 4 in 54% yield (Scheme 2). The HR-ESI-TOF mass spectrum of 4 shows the parent negative ion peak at m/z = 1238.0370 (calcd for C55H10F24N5PB, [M–H]−: 1238.0398), in line with the assigned P–BH3 structure that bears a hydrogenated pyrrole ring (pyrrole B) in Fig. 3b. X-Ray diffraction analysis of 4 reveals a C(7)–C(8) single bond with bond length of 1.544(5) Å, which is distinctly longer than those of C(2)–C(3) and C(17)–C(18) double bonds; 1.390(5) and 1.375(5) Å, respectively. In fact, two signals due to sp3-hybridized carbon atoms C(7) and C(8) are observed at δ = 27.0 and 24.5 ppm in the 13C NMR spectrum. The 1H NMR spectrum of 4 exhibits four peaks due to the β-protons of the reduced pyrrole B at δ = 2.65, 2.53, 2.37, and 2.18 ppm, and four peaks due to the β-protons of the pyrroles A and D at δ = 7.22, 6.96, 6.33, and 5.57 ppm (Fig. 2c). These data indicate that the complex 4 is a nonaromatic macrocycle. In the 31P and 11B NMR spectra, broad peaks at δ = 119.22 and −32 ppm were observed, respectively. The UV/Vis absorption spectrum of 4 shows weak and broad absorption at around 800 nm, which is similar to those of linear oligopyrroles (Fig. 4). Thus, it may be concluded that the macrocyclic conjugation becomes weak and almost vanished in 4 as a consequence of the hydrogenation at the pyrrole B.
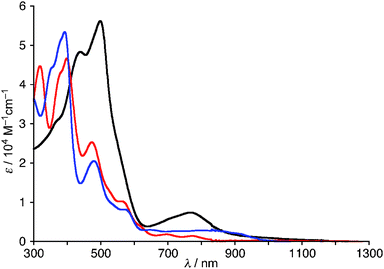 | ||
| Fig. 4 UV/Vis absorption spectra of 1 (black), 2 (red), and 4 (blue) in CH2Cl2. | ||
Many phosphorus complexes of porphyrins and expanded porphyrins were prepared so far but the phosphorus atoms within porphyrinic cavities were confined to exist as a phosphorus(V) state.11,12 Thus, we attempted to liberate the BH3 moiety from 4 to generate a free phosphorus(III) species. Treatment of 4 with an excess amount of triethylamine gave 2 in 50% yield under ambient conditions. This result suggested that the detachment of BH3 from a fused pentaphyrin P(III) complex was possible but the resultant phosphorus(III) was readily oxidized under aerobic conditions, along with concurrent dehydrogenation at the β-position. A different complex was obtained in 31% yield along with 2 when the reaction was run with freshly purified triethylamine and solvent under nitrogen atmosphere using Schlenk techniques (Scheme 2). This complex was characterized as a chlorin-type PO complex, 5, on the basis of the X-ray diffraction analysis (Fig. 3c), which revealed a PO moiety and a single bond between C(7) and C(8). The HR-ESI-TOF mass spectrum, 1H-NMR spectrum, and 31P-NMR spectrum are all consistent with the structure of 5. Therefore, it is conceivable that a free P(III) species is also quite susceptible towards oxygenation. In this regard, it is worth noting that complex 4 can be regarded as a phosphorus(III) complex, which is rare in the chemistry of phosphorus porphyrinoids.
Electrochemistry
The electrochemical properties were studied by cyclic voltammetry in CH2Cl2versusferrocene/ferrocenium ion (Fc/Fc+) with tetrabutylammonium hexafluorophosphate as an electrolyte (See Supporting Information†). The PO complex 2 underwent two reversible oxidations at 0.49 and 0.81 V, and two reversible reductions at −0.80 and −1.17 V, which allowed an estimation of the electrochemical HOMO–LUMO gap to be small, 1.29 eV. The electrochemical properties of 3 are similar to those of 2, and the HOMO–LUMO gap was estimated to be 1.27 eV. These data are in line with the assignments of 2 and 3 as antiaromatic species.1h On the other hand, since the complexes 4 and 5 are less stable than the complexes 2 and 3 under electrochemical measurement, the voltammograms were obtained with a faster scan rate (0.1 V s−1). The two reductions were observed at −1.02 and −1.54 V as reversible processes, but the two oxidations were observed as irreversible processes for 4 at 0.57 and 0.90 V. The complex 5 displayed two reversible reductions at −1.01 and −1.55 V, and one reversible oxidation at 0.60 V and one irreversible oxidation at 0.91 V. The instability of 4 and 5 under oxidative conditions suggests that the chlorin-like structures are prone to be oxidized under the electrochemical measurement conditions. The electrochemical HOMO–LUMO gaps have been estimated to be 1.59 and 1.61 eV for 4 and 5, respectively.
Theoretical calculations
We performed DFT calculations (B3LYP/6-31G(d)) for 2, 3, 4, and 5 (See Supporting Information†).13 The molecular orbitals of 2 and 3 revealed the non-degenerated HOMO/HOMO − 1 and LUMO/LUMO + 1, and the small HOMO–LUMO gaps (1.47 and 1.45 eV). The large positive nuclear independent chemical shift (NICS)14 values at the centroid of macrocycles 2 and 3 were calculated (+23.09 ppm for 2 and +22.80 ppm for 3). These computational results are consistent with the antiaromatic character of 2 and 3. In contrast, the NICS values of 4 and 5 were calculated to be +0.17 and +1.36 ppm, respectively, indicating their nonaromatic characters.Conclusions
In summary, the treatment of 1 with POCl3 gave the PO complex 2 having a triply-fused macrocycle through the N-fusion reaction and C–C bond formation between the pyrrolic β-positions. The PO complex 2 was converted to the PS complex 3 by the action of Lawesson's reagent, while 2 was reduced to the P–BH3 complex 4 with BH3·SMe2 with the concomitant hydrogenation at the pyrrolic β-position. The P–BH3 complex 4 is, to the best of our knowledge, the first phosphorus(III) complex of porphyrinoids. The complexes 2 and 3 exhibit strong 24π antiaromatic characters, while the complexes 4 and 5 are nonaromatic macrocycles due to the weak macrocyclic π-conjugation. Exploration of other phosphorus complexes of expanded porphyrins are worthy of further study.
Acknowledgements
This work was supported by Grants-in-Aid (No. 22245006 (A), and 20108001 “pi-Space”) from MEXT, Japan. T.H. acknowledges a JSPS Fellowship for Young Scientists.Notes and references
- (a) A. Jasat and D. Dolphin, Chem. Rev., 1997, 97, 2267–2340 CrossRef CAS; (b) T. D. Lash, Angew. Chem., Int. Ed., 2000, 39, 1763–1767 CrossRef CAS; (c) H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795–1804 RSC; (d) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef CAS; (e) T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676–691 CrossRef CAS; (f) A. Ghosh, Angew. Chem., Int. Ed., 2004, 43, 1918–1931 CrossRef CAS; (g) M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef CAS; (h) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS; (i) A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330–4339 RSC.
- (a) H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 1999, 121, 2945–2946 CrossRef CAS; (b) H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 2000, 122, 5748–5757 CrossRef CAS.
- (a) A. Srinivasan, T. Ishizuka and H. Furuta, Angew. Chem., Int. Ed., 2004, 43, 876–879 CrossRef CAS; (b) A. Srinivasan and H. Furuta, Acc. Chem. Res., 2005, 38, 10–20 CrossRef CAS; (c) I. Gupta, A. Srinivasan, T. Morimoto, M. Toganoh and H. Furuta, Angew. Chem., Int. Ed., 2008, 47, 4563–4567 CrossRef CAS; (d) M. Toganoh, T. Kimura, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2008, 47, 8913–8916 CrossRef CAS.
- (a) J.-Y. Shin, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2001, 40, 619–621 CrossRef CAS; (b) S. Mori, J.-Y. Shin, S. Shimizu, F. Ishikawa, H. Furuta and A. Osuka, Chem.–Eur. J., 2005, 11, 2417–2425 CrossRef; (c) S. Shimizu, J.-Y. Shin, H. Furuta, R. Ismael and A. Osuka, Angew. Chem., Int. Ed., 2003, 42, 78–82 CrossRef CAS; (d) M. Suzuki, R. Taniguchi and A. Osuka, Chem. Commun., 2004, 2682–2683 RSC; (e) S. Saito and A. Osuka, Chem.–Eur. J., 2006, 12, 9095–9102 CrossRef CAS; (f) J. M. Lim, M. Inoue, Y. M. Sung, M. Suzuki, T. Higashino, A. Osuka and D. Kim, Chem. Commun., 2011, 47, 3960–3962 RSC; (g) C.-H. Hung, J.-P. Jong, M.-Y. Ho, G.-H. Lee and S.-M. Peng, Chem.–Eur. J., 2002, 8, 4542–4548 CrossRef CAS.
- (a) J. Li, L. R. Subramanian and M. Hanack, Eur. J. Org. Chem., 1998, 2759–2767 CrossRef CAS; (b) B. Ramdhanie, C. L. Stern and D. P. Goldberg, J. Am. Chem. Soc., 2001, 123, 9447–9448 CrossRef CAS; (c) A. Młodzianowska, L. Latos-Grażyński and L. Szterenberg, Inorg. Chem., 2008, 47, 6364–6374 CrossRef CAS; (d) E. Pacholska-Dudziak, F. Ulatowski, Z. Ciunik and L. Latos-Grażyński, Chem.–Eur. J., 2009, 15, 10924–10929 CrossRef CAS; (e) T. Miura, T. Higashino, S. Saito and A. Osuka, Chem.–Eur. J., 2010, 16, 55–59 CrossRef CAS; (f) T. Higashino, J. M. Lim, T. Miura, S. Saito, J.-Y. Shin, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 4950–4954 CrossRef CAS.
- (a) S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS; (b) S. Mori, K. S. Kim, Z. S. Yoon, S. B. Noh, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 11344–11345 CrossRef; (c) M.-C. Yoon, S. Cho, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2009, 131, 7360–7367 CrossRef CAS.
- (a) L. Horner and W. D. Balzer, Tetrahedron Lett., 1965, 6, 1157–1162 CrossRef; (b) S. E. Cremer and R. J. Chorvat, J. Org. Chem., 1967, 32, 4066–4070 CrossRef CAS; (c) K. Naumann, G. Zon and K. Mislow, J. Am. Chem. Soc., 1969, 91, 7012–7023 CrossRef CAS; (d) K. L. Marsi, J. Org. Chem., 1974, 39, 265–267 CrossRef; (e) H.-C. Wu, J.-Q. Yu and J. B. Spencer, Org. Lett., 2004, 6, 4675–4678 CrossRef CAS.
- (a) L. D. Field and M. P. Wilkinson, Tetrahedron Lett., 1997, 38, 2779–2782 CrossRef CAS; (b) L. Horner, H. Hoffman and P. Beck, Chem. Ber., 1958, 91, 1583–1588 CrossRef CAS; (c) S. R. Gilbertson and G. W. Starkey, J. Org. Chem., 1996, 61, 2922–2923 CrossRef CAS.
- (a) T. Imamoto, Pure Appl. Chem., 1993, 65, 655–660 CrossRef CAS; (b) Y. Dienes, S. Durben, T. Kárpáti, T. Neumann, U. Englert, L. Nyulászi and T. Baumgartner, Chem.–Eur. J., 2007, 13, 7487–7500 CrossRef CAS.
- (a) T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS; (b) S. R. Piettre, Tetrahedron Lett., 1996, 37, 4707–4710 CrossRef CAS.
- (a) J. K. M. Sanders, N. Bampos, Z. Clyde-Watson, S. L. Darling, J. C. Hawley, H.-J. Kim, C. C. Mak and S. J. Webb, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 1999, vol. 3, pp. 1–48 Search PubMed; (b) P. Sayer, M. Gouterman and C. R. Connell, J. Am. Chem. Soc., 1977, 99, 1082–1087 CrossRef CAS; (c) P. Sayer, M. Gouterman and C. R. Connell, Acc. Chem. Res., 1982, 15, 73–79 CrossRef CAS; (d) S. Mangani, E. F. Meyer, D. L. Cullen, M. Tsutsui and C. J. Carrano, Inorg. Chem., 1983, 22, 400–404 CrossRef CAS; (e) T. Barbour, W. J. Belcher, P. J. Brothers, C. E. F. Rickard and D. C. Ware, Inorg. Chem., 1992, 31, 746–754 CrossRef CAS; (f) Y. Yamamoto, R. Nadano, M. Itagaki and K. Akiba, J. Am. Chem. Soc., 1995, 117, 8287–8288 CrossRef CAS.
- Although some examples of porphyrinoid containing phosphorus atoms have been reported, a “phosphorus(III) complex” has not been reported so far. See the following references. P(III) phosphaporphyrins: (a) Y. Matano, T. Nakabuchi, T. Miyajima and H. Imahori, Organometallics, 2006, 25, 3105–3107 CrossRef CAS; (b) Y. Matano, T. Miyajima, T. Nakabuchi, H. Imahori, N. Ochi and S. Sakaki, J. Am. Chem. Soc., 2006, 128, 11760–11761 CrossRef CAS; (c) Y. Matano, T. Nakabuchi, T. Miyajima, H. Imahori and H. Nakano, Org. Lett., 2006, 8, 5713–5716 CrossRef CAS; (d) Y. Matano, T. Nakabuchi, S. Fujishige, H. Nakano and H. Imahori, J. Am. Chem. Soc., 2008, 130, 16446–16447 CrossRef CAS; (e) Y. Matano, M. Nakashima, T. Nakabuchi, H. Imahori, S. Fujishige and H. Nakano, Org. Lett., 2008, 10, 553–556 CrossRef CAS; (f) Y. Matano and H. Imahori, Acc. Chem. Res., 2009, 42, 1193–1204 CrossRef CAS. Phosphonium species of meso- or β-P(III) porphyrinoid: (g) K. M. Smith, G. H. Barnett, B. Evans and Z. Martynenko, J. Am. Chem. Soc., 1979, 101, 5953–5961 CrossRef CAS; (h) A. Małek, L. Latos-Grażyński, T. J. Bartczak and A. Żądło, Inorg. Chem., 1991, 30, 3222–3230 CrossRef CAS; (i) K. Youfu and A. Osuka, Org. Lett., 2005, 7, 4381–4384 CrossRef CAS. Meso-Phosphanylporphyrin: (j) Y. Matano, K. Matsumoto, Y. Nakao, H. Uno, S. Sasaki and H. Imahori, J. Am. Chem. Soc., 2008, 130, 4588–4589 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.2), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS; (b) Z. Chen, C. S. Wannerere, C. Corminboeuf, R. T. Puchta and P. von R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental methods, HR-ESI-TOF mass spectra, UV/Vis absorption spectra, NMR spectra, cyclic voltammograms, X-ray crystal structures and results of DFT calculations. CCDC reference numbers 827554-827556 and 842955. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00653c |
‡ Crystallographic data for 2: 4(C55H6F24N5OP)·13(C6H5Cl), Mr = 6421.63, monoclinic, space groupP21/a (No.14), a = 29.5322(5), b = 10.3705(2), c = 41.5808(8) Å, β = 99.1602(8)°, V = 12572.3(4) Å3, T = 93(2)K, ρc = 1.696 g cm−3, Z = 2, 114624 reflections measured, 19582 unique (Rint = 0.1338), R1 = 0.0993 [I > 2.0σ(I)], wR2 = 0.2961 (all data), GOF = 0.988; Crystallographic data for 3: C55H6F24N5PS·3(C7H8), Mr = 1532.09, monoclinic, space groupP21/c (No.14), a = 19.4591(5), b = 8.6913(2), c = 37.1609(11) Å, β = 94.5998(18)°, V = 6264.6(3) Å3, T = 93(2)K, ρc = 1.624 g cm−3, Z = 4, 60930 reflections measured, 11391 unique (Rint = 0.1115), R1 = 0.0675 [I > 2.0σ(I)], wR2 = 0.1500 (all data), GOF = 1.057; Crystallographic data for 4: 2(C55H11BF24N5P)·3(C6H14)·C7H8, Mr = 2829.59, monoclinic, space groupC2/c (No.15), a = 30.1073(6), b = 18.0820(3), c = 22.4608(4) Å, β = 106.2569(10)°, V = 11738.8(4) Å3, T = 93(2)K, ρc = 1.601 g cm−3, Z = 4, 55183 reflections measured, 10540 unique (Rint = 0.1381), R1 = 0.0759 [I > 2.0σ(I)], wR2 = 0.1619 (all data), GOF = 1.001. Crystallographic data for 5: C55H8F24N5OP·C2H3N·0.75(CCl4)·0.5(water), Mr = 1405.92, triclinic, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No.2), a = 10.1849(5), b = 14.9773(8), c = 17.6759(10) Å, α = 85.568(3), β = 88.271(3), γ = 76.766(3)°, V = 2616.7(2) Å3, T = 93(2)K, ρc = 1.784 g cm−3, Z = 2, 21103 reflections measured, 7529 unique (Rint = 0.1076), R1 = 0.1190 [I > 2.0σ(I)], wR2 = 0.3747 (all data), GOF = 1.076. (No.2), a = 10.1849(5), b = 14.9773(8), c = 17.6759(10) Å, α = 85.568(3), β = 88.271(3), γ = 76.766(3)°, V = 2616.7(2) Å3, T = 93(2)K, ρc = 1.784 g cm−3, Z = 2, 21103 reflections measured, 7529 unique (Rint = 0.1076), R1 = 0.1190 [I > 2.0σ(I)], wR2 = 0.3747 (all data), GOF = 1.076. |
| This journal is © The Royal Society of Chemistry 2012 |