Quantitative preparation of 3,4-di(methylene)tetrahydrothiophene-1,1-dioxide by Zn-induced 1,4-debromination. A valuable 6-C reactive diene in [4+2] cycloadditions with DMAD and [60]fullerene†
Marios S.
Markoulides
,
Charalambos P.
Ioannou
,
Manolis J.
Manos
and
Nikos
Chronakis
*
Department of Chemistry, University of Cyprus, P.O. Box 20537, 1678, Nicosia, Cyprus. E-mail: nchronak@ucy.ac.cy; Fax: +357 22892801; Tel: +357 22892781
First published on 18th October 2012
Abstract
Optimum reaction conditions for the quantitative preparation of the highly reactive 3,4-di(methylene)tetrahydrothiophene-1,1-dioxide are described. The method involves the zinc-induced 1,4-debromination of 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide in acetone solvent either by using conventional heating, microwave or ultrasonic irradiation. The [4+2] cycloaddition reaction of 3,4-di(methylene)tetrahydrothiophene-1,1-dioxide with dienophiles such as DMAD and C60 led to the efficient and clean formation of the corresponding Diels–Alder cycloadducts. Specifically for [60]fullerene, the short-chain C60 monoadduct was formed in a short reaction time and in high overall yield (56%). In contrast, the iodine-induced 1,4-debromination using KI in toluene, in the presence of 18-crown-6 as a phase transfer catalyst, failed to give the corresponding [4+2] C60 monoadduct at room temperature or in refluxing toluene and a low product yield (13%) was only obtained at a temperature of 45–50 °C.
Introduction
More than a century ago, the reaction between cyclopentadiene and quinones was carried out by Albrecht in 1906.1 The first reported Diels–Alder reaction was carried out in 19282 and the [4+2] cycloaddition is still one of the most important tools to construct cyclic molecular architectures.3 The dienophilic behaviour of [60]fullerene has been widely exploited in such reactions,4 as the Diels–Alder transformation is a powerful method for the covalent attachment of organic moieties on the surface of C60. This approach appears to be particularly attractive and has been fruitful in preparing rigidified donor–bridge–C60 electroactive systems for various applications in materials science.5 The advantage of the Diels–Alder strategy concerns the more rigid spatial orientation of the HOMO of the donor with respect to the LUMO of C60 due to the folded-boat conformation of the cyclohexene ring, therefore favouring through-space interactions in electron transfer processes. In most cases, the key step is the preparation of versatile bis- or tetrakis(bromomethylated) derivatives which can generate the corresponding transient o-quinodimethane analogues via reductive elimination.6 However, in almost all of these cases an aromatic core is covalently linked to the fullerene cage through a cyclohexene ring in order to prevent the retro Diels–Alder reaction. To date, only a limited number of short-chain fullerene derivatives have been reported in the literature, probably due to difficulties met in the synthesis and isolation of stable dienes required for the Diels–Alder reaction with C60.7More recently, Sambrook et al.8 reported the synthesis of a short-chain C60 monoadduct and a bis-fullerene adduct by reacting the tetrabromo precursor 1 with C60 in toluene solvent in an one-pot manner and by using KI in the presence of 18-crown-6 as a phase transfer catalyst. This is the only literature example reporting the utilisation of the transient intermediate 2,2′-bisallyl diradical 2 (Scheme 1) towards the synthesis of a [4+2] C60 derivative.
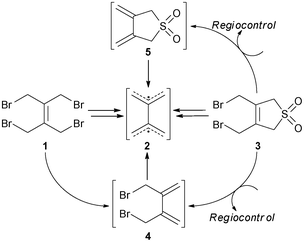 | ||
| Scheme 1 Precursors for the generation of the 2,2′-bisallyl diradical 2. | ||
Searching for an elegant method to access short-chain [4+2] C60 monoadducts bearing functional groups for further derivatization, it has been realised that despite the enormous body of literature reports dealing with the in situ generation and Diels–Alder reactions of mono-9 and bis-o-quinodimethanes10 and their derivatives, very few reports describe the synthesis of Diels–Alder reactions utilising the transient intermediate 2,2′-bisallyl diradical 2. The chemistry of the diradical 2 was originally reported by Trost and Shimizu11 and was subsequently extended by Hosomi et al.12 Presumably, the diene-masking possibilities available for o-quinodimethanes9,10 either cannot be applied for the generation of the 2,2′-bisallyl diradical 213 or require a multi-step synthesis.14,15 Easily accessible and stable masked bis-diene precursors for the generation of 2 are: (a) 1,4-dibromo-2,3-bis(bromomethyl)but-2-ene 1,16 which can undergo sequential 1,4-debromination15b and (b) 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3,17 which can undergo 1,4-debromination17b,c and SO2 extrusion either thermally18 or chemically19 (Scheme 1).
Although the diene intermediates 4 and 5 have been known for several decades, there are very few reports describing their preparation and their subsequent Diels–Alder reactions with dienophiles.15,17b,c For example, the only direct methods for the preparation of the diene-sulfone 5 were reported by Nasyrov et al.17b and Alder et al.17c In both cases, NaI was used as a de-brominating reagent and the reactions were carried out in acetone at room temperature. Specifically, in the report of Nasyrov et al.,17b 2 g of the starting material 3 were used at a concentration of 100 mg mL−1 and the product 5 was obtained after 2 h in 60% yield, following a standard work-up procedure with Na2S2O3 and recrystallization from benzene–Et2O. The authors did not mention any instability problems of the formed diene-sulfone 5. In the report by Alder et al.,17c 16 g of the starting material 3 were used at a concentration of 70 mg mL−1 and the product 5 was obtained after 18 h in 100% yield following a standard work-up with Na2S2O3 but with no further purification. In this case, the authors reported that compound 5 polymerised rapidly upon solvent removal and it was not explained how the yield of 5 was calculated.
An indirect method for the preparation of 5 was reported by Gaoni and Sadeh.15b The reaction of 1 with an excess of a Zn–Cu couple, in Et2O and in the presence of HMPT, gave the dibromo-diene 4 in 70–73% yield. Diene 4 was then reacted with sodium sulfide followed by oxidation of the resulting cyclic sulfide to afford 5 (25% overall yield from 1). Despite the multistep, low yielding sequence, the authors clearly stated the facile polymerisation of 5 either in the absence of solvents or in solution.
It is apparent that in all procedures where 1 is utilised for either sequential15b or tandem8 Diels–Alder reactions, chemo- or regioselective control over the diene formation is not feasible. In addition, the cycloaddition of 1 with C60 results in a mixture of fullerene adducts, namely the [4+2] C60 monoadduct and the bis-fullerene adduct with two fullerenes attached on the in situ formed bisallyl diradical 2. These products showed very similar polarity with unreacted C60 and therefore their separation could be achieved only by means of preparative HPLC.8 Furthermore, the disadvantage of using NaI or KI for the 1,4-debromination step lies in the fact that the presence of bromine in the reaction mixture leads to the formation of undesirable adducts, especially when C60 is employed as a dienophile. This process usually requires longer reaction times4a and repetitive chromatographic purification or preparative HPLC is necessary for the isolation of the formed adducts. The alternative method of metal-induced 1,4-debromination to access the desired diene is not compatible with C60 due to its pronounced electron accepting ability. It is well-established in the literature that C60 is reduced by metals including zinc,4b thus indicating that the pure diene intermediate (e.g., 4 or 5, Scheme 1) should be first isolated in a pure form and then subjected to the [4+2] cycloaddition reaction with C60.
Eliminating the problem of chemoselectivity of the tetrabromo precursor 1, a selective Diels–Alder reaction can be envisioned with the structure of 3 (Scheme 1) by taking advantage of either the chemoselective 1,4-debromination or the elimination of the sulfone moiety to form the corresponding dienes. Nevertheless, the incompatibility of C60 in either iodine- or metal-induced debromination conditions results in the inefficient preparation of [4+2] C60 monoadducts and is therefore a major drawback.
Probably due to instability problems, no attempts have been made in the past to optimise the conditions and therefore generalise the preparation of the highly valuable 6-C synthon 5. Another reason might arise from the fact that simple dienophiles were used in the subsequent [4+2] cycloadditions and therefore, the scale of the reaction or the purity of 5 were not a major problem.15,17b,c Finally, the one-pot reaction of 3 with simple dienophiles under the iodine-induced 1,4-debromination conditions usually results in the formation of fewer undesirable products that are easier to purify, giving higher overall yields compared with the usual low yielding reactions of C60.
Our interest in the synthesis of [60]fullerene derivatives derived from the Diels–Alder reaction,20 in combination with the limitations posed by the current methods which utilise synthon 2, prompted us to investigate the optimum reaction conditions for the preparation of the diene-sulfone 5 and the subsequent efficient synthesis of a short-chain [4+2] C60 monoadduct bearing a reactive site for further derivatization.
Results and discussion
The preparation of the dibromo-sulfone 3 was performed following modified literature procedures.17a,21 The reaction of SO2 with 2,3-dimethyl-1,3-butadiene 6 gave the cyclic sulfone 7 in 74% yield,21 which was then subjected to an allylic bromination with N-bromosuccinimide in dry dichloromethane to furnish the dibromo-sulfone 3 in 41% yield (Scheme 2).17a Product 3 showed a pronounced ability to crystallise and colourless single crystals were easily obtained by recrystallization from ethanol. The proposed structure was unambiguously confirmed by X-ray crystallography (see Supporting Information†).![A chemoselective approach towards the preparation of the short-chain dibromo [4+2] C60 monoadduct 8.](/image/article/2012/RA/c2ra22502f/c2ra22502f-s2.gif) | ||
| Scheme 2 A chemoselective approach towards the preparation of the short-chain dibromo [4+2] C60 monoadduct 8. | ||
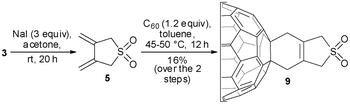
Our investigation was initiated by the reaction of C60 with dibromo-diene 4, generated in situ from 3via thermal SO2 extrusion (see ESI† for the DSC and TGA of 3). The dibromo [4+2] C60 monoadduct 8 (Scheme 2) was obtained as a brown solid contaminated with unreacted C60. Purification of 8 could not be achieved by column chromatography in all attempted eluent systems due to its similar polarity with C60. This was also confirmed in the report of Sambrook et al.,8 where preparative HPLC was required for the purification of 8 synthesised starting from 1. Nonetheless, the thermal SO2 extrusion of 3 followed by the Diels–Alder reaction with C60 is advantageous compared to the literature method8 due to the regiocontrol over the diene formation.
Next, we examined the Diels–Alder reaction of diene 5 derived from the 1,4-debromination of dibromo-sulfone 3 with C60 (Table 1). This transformation was carried out under iodine-induced 1,4-debromination conditions, previously reported for the preparation of o-quinodimethane derivatives.4,8 KI was used as the de-brominating reagent in toluene in the presence of 18-crown-6 as a phase transfer catalyst. In all reported examples, the reaction between C60 and the in situ formed o-quinodimethanes proceeds smoothly in refluxing toluene. In the case of 3, the one-pot cycloaddition reaction in refluxing toluene yielded only intractable baseline (TLC) material (Table 1, entry 1). At room temperature, the reaction gave no indication of product formation even after prolonged reaction periods (Table 1, entry 2) and the best result was obtained at a temperature of 45–50 °C (Table 1, entry 3). The purification of product 9 was easily achieved by column chromatography on silica gel (toluene–EtOAc = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) followed by precipitation from CHCl3–pentane. The increased polarity of the [4+2] C60 monoadduct 9 compared with 8 allowed its facile separation and purification and was isolated in 13% yield.
:1) followed by precipitation from CHCl3–pentane. The increased polarity of the [4+2] C60 monoadduct 9 compared with 8 allowed its facile separation and purification and was isolated in 13% yield.
|
|
|||
|---|---|---|---|
| Entry | Temp/°C | Time | Yield of 9 (%) |
| a Conditions: 3 (0.06 mmol), C60 (0.07 mmol), KI (0.15 mmol), 18-crown-6 (0.45 mmol), toluene (50 mL). Results based on an average of three runs. b Baseline material on TLC (inseparable). c Starting materials on TLC. d Isolated yield. | |||
| 1 | reflux | 2 h | —b |
| 2 | rt | 5 d | —c |
| 3 | 45–50 | 24 h | 13d |

The 1H NMR spectrum of 9 showed two broad singlets at 4.22 and 4.38 ppm which correspond to the methylenic hydrogens of the cyclohexene and the heterocyclic five-membered ring of the addend, respectively. In the 13C NMR spectrum, three signals at 40.81, 59.68 and 65.32 ppm were observed corresponding to the sp3 carbon atoms of the C60 core and the methylenic carbons of the organic addend. In the region between 133.11–155.14 ppm, 18 peaks were found, 17 of which are attributed to the fullerene sp2 carbons and one to the carbons of the double bond of the cyclohexene moiety. All the observed signals reflect nicely the C2v symmetry of the molecule, which arises from the fast ring flipping of the cyclohexene unit. Finally, the MALDI-TOF mass spectra (negative mode, DCTB as the matrix) of the fullerene monoadduct 9 showed the [M–SO2]− ion at m/z 800.0619 Da indicating that the extrusion of sulfur dioxide is facile under the mass spectrometric conditions.
The low yield of 9 obtained following the one-pot method with KI turned our efforts to the preparation of diene 5 prior to the cycloaddition reaction with C60. Following standard literature procedures,17b,c we investigated the 1,4-debromination reaction of 3 using NaI in acetone (Scheme 3). Despite extensive repetition of the procedure, in our hands, the isolation of 5 or pure solutions of it was not feasible. The reaction was repeated up to a 1.65 mmol scale of 3, as well as in dilute concentrations up to 6 mg mL−1. Monitoring was performed by means of thin-layer chromatography (EtOAc–hexane = 5:4) and upon disappearance of the starting material a standard work-up with Na2S2O3 was followed. Isolation of compound 5 or purification by recrystallization was never accomplished due to its rapid decomposition to a polymeric material that was not soluble in a wide range of solvents. Purification by column chromatography on silica gel (EtOAc–hexane = 5:4) using dry, de-acidified solvents and exchanging the solvent of the combined fractions with dry benzene was the best way to manipulate the highly reactive diene 5. However, impurities of similar polarity were still present and further purification attempts did not prove to be effective. By subjecting a benzene solution of 5, prepared by the aforementioned procedure, to a Diels–Alder reaction with C60 in toluene, at 45–50 °C for 12 h, we succeeded in isolating the adduct 9 in 16% yield. The yield and consequently the efficiency of this method are very similar to the one-pot procedure in which KI was used.
 | ||
| Scheme 3 Synthesis of the diene-sulfone 5 by the NaI procedure and its Diels–Alder reaction with C60. | ||
Realising the lack of an efficient and practical way for the clean preparation of 5, we started our investigations with the reductive elimination of 3 using activated Zn-dust. Due to the electron accepting ability of C60 with many metals,4b our initial optimisation strategy was focused on the reaction of the dibromo-sulfone 3 with activated Zn-dust in various solvents, in the absence of a dienophile in the reaction mixture (Table 2). Since diene 5 could not be isolated, TLC monitoring of the reaction reflected the stability of compound 5 in the solvents tested and at different concentrations. Of the solvents screened, acetone was the only solvent which facilitated the exclusive formation of product 5, at refluxing temperature for 16–18 h (Table 2, entry 9). Under these conditions, no decomposition was observed and the results were optimum at a concentration of 6 mg mL−13 in acetone, using 2.3 equiv of activated Zn-dust. In more concentrated solutions, fewer equivalents of zinc were required but decomposition material was the major result. When the reaction was carried out in refluxing Et2O or EtOAc (Table 2, entries 2 and 10), TLC revealed the formation of product 5, which was accompanied by decomposition material in substantial amounts. No indication of the product formation was observed when toluene, chloroform and acetonitrile (Table 2, entries 3, 5 and 13) were used as solvents, while in THF (Table 2, entries 11 and 12), the reaction failed to proceed at RT and at 55–60 °C; it afforded exclusively decomposition material.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp/°Cb | Time | TLC resultc |
|
a The reactions were performed up to 0.33 mmol scale of 3. Conditions: 3 (1 equiv), concentration 6 mg mL−1, activated Zn-dust (2.3 equiv). Reported results are based on an average of two runs on each scale.
b Conventional heating.
c TLC monitoring (EtOAc–hexane = 5:4).
|
||||
| 1 | Et2O | rt | 3 d | starting material |
| 2 | Et2O | reflux | 2 d | product + decomposition |
| 3 | toluene | 75–80 | 3 d | starting material |
| 4 | DMSO | rt | 2–3h | decomposition |
| 5 | CHCl3 | reflux | 3 d | starting material |
| 6 | DMF | rt | 3 d | starting material + decomposition |
| 7 | DMF | 45–50 | 2–3 h | decomposition |
| 8 | acetone | rt | 3 d | starting material |
| 9 | acetone | reflux | 16–18 h | product |
| 10 | EtOAc | 55–60 | 2 d | product + decomposition |
| 11 | THF | rt | 3 d | starting material |
| 12 | THF | 55–60 | 2–3 h | decomposition |
| 13 | acetonitrile | 55–60 | 2 d | starting material |

The clean transformation of dibromo-sulfone 3 to diene 5, under the optimum experimental conditions found (Table 2, entry 9), prompted us to attempt its NMR characterisation in an effort to confirm the results obtained by TLC analysis. For this purpose, the reaction was repeated under the conditions mentioned before but this time acetone-d6 was employed as a solvent. After completion (TLC), the reaction mixture was cooled to RT, filtered and the filtrate was directly subjected to NMR analysis. The observed absorptions in the 1H and 13C NMR spectra (Fig. 1) led to the unambiguous structural assignment of diene 5, which was the sole product of the Zn-induced 1,4-debromination reaction. In both spectra, the absence of any peaks corresponding to the starting material or to other by-products provided the ultimate proof for the remarkable efficiency of the applied method.
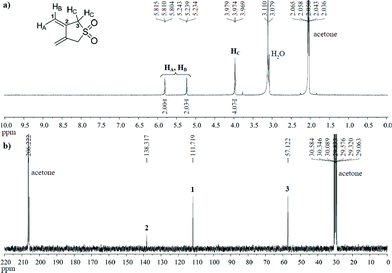 | ||
| Fig. 1 (a) 1H NMR (500 MHz, acetone-d6) and (b) 13C NMR (125 MHz, acetone-d6) of diene 5 formed by Zn-induced 1,4-debromination of 3. | ||
A minor disadvantage in the synthesis of diene 5 from 3 under the optimum experimental conditions (Table 2, entry 9) is the relatively long reaction time (16–18 h). In an effort to shorten the reaction time, we used non-conventional heating methods. The Zn-induced 1,4-debromination reaction of 3 under conventional heating, microwave irradiation and sonication was carried out in acetone-d6 at a concentration of 6 mg mL−13. In each case, the yield of diene 5 was calculated using an equimolar amount (with respect to the starting material 3) of 1,3,5-trimethylbenzene (mesitylene, bp 163–166 °C), as an internal standard. Control experiments confirmed that mesitylene is stable under the experimental conditions used in all three methods employed. The progress of the reactions was followed by TLC and upon completion the solutions were filtered and analysed by 1H NMR spectroscopy. The diene-sulfone 5 was the sole product independent of the heating method applied and by integrating the appropriate peaks in the 1H NMR spectra (see ESI†) the yields were calculated to be more than 97% in all cases (Table 3). Conventional heating afforded the product in 16–18 h (Table 3, entry 1), sonication in 2 h and 50 min (Table 3, entry 2) and microwave irradiation led to the quantitative formation of diene 5 in only 55 min (Table 3, entry 3). Most impressive was the fact that diene 5 remained stable under microwave irradiation and thus, this widely used method for accelerating reactions rates can be employed in the synthesis of the highly reactive diene 5. The acetone solutions of 5 can be used directly for subsequent Diels–Alder reactions without any purification. Furthermore, when the solvent was exchanged with dry benzene and stored in the freezer at −20 °C, diene 5 was stable for several weeks as indicated by TLC.
|
|
||||
|---|---|---|---|---|
| Entry | Heating method | Temp/°C | Time | Yield of 5 (%)b |
| a Conditions: 3 (1 equiv), concentration 6 mg mL−1, activated Zn-dust (2.3 equiv), mesitylene (1 equiv). Reported results are based on an average of two runs. b NMR yields using one equivalent of mesitylene (bp 163–166 °C) as an internal standard. c The reactions were carried out on a 0.038 mmol scale of 3. d CEM Discover microwave reactor at 250 W, 21 psi. | ||||
| 1 | conventionalc | reflux | 16–18 h | 99–100 |
| 2 | sonicationc | reflux | 2 h 50 min | 98–99 |
| 3 | microwavecd | 60 | 55 min | 97–98 |

In the next step, the Diels–Alder cycloaddition reaction of the diene-sulfone 5 with a zinc-compatible dienophile was investigated in solvents employed in our initial studies and are summarised in Table 2. The Zn-induced 1,4-debromination of 3 followed by the [4+2] cycloaddition with an excess of dimethyl acetylenedicarboxylate (DMAD) (10 equiv) was examined in a one-pot procedure for each solvent tested (Table 4). The yield of product 10 was calculated after its isolation and purification by column chromatography. Recrystallization from methanol afforded single crystals of 10 suitable for X-ray analysis, which further supported the proposed structure. It has to be mentioned here that except for acetone, which was the best solvent for the synthesis of diene 5via the Zn-induced 1,4-debromination of 3, we examined the cycloaddition reaction with DMAD in solvents where diene 5 was formed together with the decomposition material (Et2O, EtOAc) and solvents where the decomposition material was the only outcome of the reaction (DMSO, DMF, THF). Our purpose was to clarify if in certain solvents the [4+2] cycloaddition reaction of 5 with DMAD is faster than its decomposition due to polymerisation and consequently to determine whether they can be employed in reactions where the dienophile is used in situ. The trend observed was very similar to the initial experiments (Table 4). When the reaction was carried out in acetone, product 10 was isolated in 91–92% isolated yield (Table 4, entry 5). In Et2O, EtOAc and THF (Table 4, entries 1, 6 and 7) the isolated yields ranged from 50 to 75%, while the same reaction in either DMSO or DMF (Table 4, entries 2, 3 and 4) led only to decomposition. Finally, the one-pot Zn-induced 1,4-debromination of 3/[4+2] cycloaddition with DMAD was repeated under microwave irradiation or sonication giving product 10 in 90–92% isolated yield, in identical reaction times with those reported in Table 3 (entries 2 and 3).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp/°Cb | Time | Yield of 10 (%)c |
| a The reactions were performed up to 0.33 mmol scale of 3. Conditions: 3 (1 equiv), concentration 6 mg mL−1, activated Zn-dust (2.3 equiv), DMAD (10 equiv). Reported results are based on an average of two runs on each scale. b Conventional heating. c Isolated yields. | ||||
| 1 | Et2O | reflux | 3 d | 72–75 |
| 2 | DMSO | rt | 2–3 h | decomposition |
| 3 | DMF | 45–50 | 2–3 h | decomposition |
| 4 | DMF | rt | 2 d | starting material + decomposition |
| 5 | acetone | reflux | 16–18 h | 91–92 |
| 6 | EtOAc | 55–60 | 2.5 d | 59–62 |
| 7 | THF | 55–60 | 5 h | 50–52 |

In order to complete our investigation in finding the optimum conditions for the preparation of diene 5 and its Diels–Alder reaction with dienophiles, we turned our attention to the metal reagent used for the 1,4-debromination of dibromo-sulfone 3. Following a procedure reported by Smith and Simmons,22 the Zn–Cu couple was employed and compared with activated Zn-dust and NaI for the debromination of 3 (Table 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Equivs of reagent | Temp/°Cb | Time | Yield of 10 (%)c |
| a The reactions were carried out on a 0.076 mmol scale of 3. Conditions: 3 (1 equiv), concentration 6 mg mL−1, DMAD (10 equiv). Reported results are based on an average of two runs. b Conventional heating. c Isolated yields. | |||||
| 1 | Zn | 2.3 | reflux | 16–18 h | 91–92 |
| 2 | Zn–Cu | 16 | reflux | 5 d | 90–93 |
| 3 | NaI | 3 | 45–50 | 24 h | 50–54 |

All reactions were carried out in the presence of DMAD as the dienophile. Although the procedure with Zn–Cu gave cycloadduct 10 in an excellent yield (Table 5, entry 2), an excess of the reagent was used but more importantly, the reaction required 5 d to reach completion. A wasteful stoichiometry of the Zn–Cu couple and long reaction times were also observed in the report of Gaoni and Sadeh15b during the preparation of dibromo-diene 4 starting from 1 (Scheme 1). Finally, the inefficiency of NaI to promote the 1,4-debromination was confirmed by performing the reaction in the presence of DMAD (10 equiv) wherein the Diels–Alder adduct 10 was isolated in only 50–54% yield (Table 5, entry 3).
Having defined the optimum conditions for the synthesis of diene-sulfone 5 and its Diels–Alder reactions with dienophiles, we explored the versatility of the method to prepare the sulfone derivative 11, a useful precursor for the synthesis of unusual α-amino acid derivatives. We chose this example as 11 was previously prepared in a four step sequence starting from 6, in 27% overall yield.10b Following our approach (Scheme 4), sulfone 11 was prepared in a one-pot procedure consisting of three sequential steps, namely zinc-induced 1,4-debromination of 3, [4+2] cycloaddition of 5 with DMAD and finally oxidation of 10 with MnO2. By this procedure, product 11 was isolated in 90% overall yield.
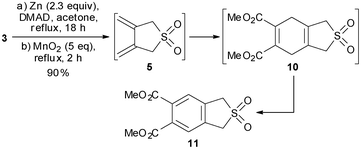 | ||
| Scheme 4 One-pot three-step synthesis of 11. | ||
To reach our initial target and establish an elegant and practical method to access short-chain [4+2] C60 monoadducts bearing functional groups for further derivatization, diene-sulfone 5 was subjected to a [4+2] cycloaddition reaction with C60 (Scheme 5).
![Synthesis of the Diels–Alder [60]fullerene monoadduct 9.](/image/article/2012/RA/c2ra22502f/c2ra22502f-s5.gif) | ||
| Scheme 5 Synthesis of the Diels–Alder [60]fullerene monoadduct 9. | ||
The diene was prepared quantitatively under our optimum conditions and used in acetone solution after the filtration of the zinc salts. The Diels–Alder reaction with C60 proceeded smoothly at 50 °C to successfully afford monoadduct 9. After purification by column chromatography on silica gel (toluene–EtOAc = 20:1) and precipitation from CHCl3–pentane, monoadduct 9 was isolated in 56% yield, which was a major improvement over the previous procedures involving either the preparation of 9 from 3 using KI/18-crown-6 in toluene or the preparation of 5 using the NaI procedure. By this way, a facile access to short-chain [4+2] C60 monoadducts was established utilising the valuable 6-C synthon 5 and eliminating the problems arising from the use of reagents which are incompatible with C60.
Conclusions
In summary, we have demonstrated a practical and efficient preparation of the reactive 3,4-di(methylene)tetrahydrothiophene-1,1-dioxide (5). Optimisation of the reaction parameters allowed the quantitative preparation of the diene-sulfone 5via a zinc-induced 1,4-debromination of 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide (3) with no signs of polymerisation. In particular, acetone was the solvent of choice for the preparation of 5 from 3 at a concentration of 6 mg mL−1. Activated Zn-dust efficiently promoted the 1,4-debromination step, while the shortest reaction time was achieved with microwave irradiation. The formed diene can be employed in Diels–Alder reactions with dienophiles either in acetone or after exchanging the solvent with dry benzene where the diene remains stable for several weeks at −20 °C. The overall yield of the one-pot procedure (formation of diene 5/Diels–Alder reaction) when DMAD was utilised as a dienophile was more than 90%. Zinc incompatible dienophiles can be treated with acetone solutions of 5 by simple filtration of the zinc salts prior to the Diels–Alder cycloaddition.The diene-sulfone 5 synthesised under the optimum conditions reported before was utilised for the synthesis of a [4+2] short-chain C60 monoadduct bearing a masked-diene moiety capable of further derivatization. The applied method led to the successful preparation of cycloadduct 9 which was isolated in 56% yield. Dienophiles with a wide range of chemical functionalities could be employed in order to provide a novel post-synthetic modification strategy via cheletropic extrusion of SO2 targeting novel C60 derivatives for various applications in materials science. Research in this direction is currently underway in our group and the results will be presented in the near future.
Experimental section
General information
All starting materials were purchased from commercial sources and used without further purification. The solvents were dried using appropriate standard procedures. Zinc dust was activated following a known procedure.23 Column chromatography was carried out using Merck silica gel 60H (40–60 nm, 230–300 mesh). Thin-layer chromatography (TLC) was carried out on aluminum plates coated with Merck HF254/366 silica gel. Visualisation was accomplished under a 254 nm ultraviolet (UV) light source and/or by immersion in potassium permanganate (KMnO4) or phosphomolybdic acid (PMA) solutions, followed by heating. 1H and 13C NMR spectra were recorded on Bruker Avance 300 (300 MHz) and Bruker Avance III 500 Ultrashield Plus (500 MHz) spectrometers. Residual non-deuterated solvent was used as the internal standard for 1H NMR spectra and a carbon signal of the solvent was used as the internal standard for 13C NMR spectra. Chemical shifts (δH and δC) are quoted in parts per million (ppm) downfield from tetramethylsilane (TMS). The resonance multiplicity patterns are described as singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), quintet (quin.), multiplet (m), or combinations of these. Coupling constants (J) are quoted in hertz (Hz). Peak assignments were aided by 1H–1H COSY, 1H–13C HMQC, DEPT-135 and/or DEPT-90, whenever necessary. High resolution mass spectra were recorded either on a MALDI TOF Bruker Autoflex III Smartbeam instrument using DCTB as a matrix or on a LTQ Orbitrap XL spectrometer. Infrared (IR) spectra were recorded on a Shimadzu FTIR-NIR Prestige-21 spectrometer with a Pike Miracle Ge ATR accessory and bands are quoted in cm−1. Differential scanning calorimetric (DSC) measurements were performed on a DSC TA Q1000 apparatus using a heating curve from 25 to 300 °C under argon atmosphere with a heating rate of 5 °C min−1. The samples (0.8–1.5 mg) were measured in hermetically sealed aluminium pans. Thermogravimetric analysis (TGA) measurements were performed on a TGA TA Q500 analyser under argon atmosphere with about 10 mg of the samples at a heating rate of 10 °C min−1 from 25 to 400 °C, using ceramic pans. A CEM Discover Microwave Reactor was used for microwave experiments and an Elmasonic S 30 (H) ultrasonic laboratory cleaner (220–240 V, 280 W, 50/60 Hz) was used for ultrasonic experiments. Melting points (mp) were determined on a Stuart Scientific SMP10 apparatus and were uncorrected.:1). Further purification by recrystallization from methanol afforded product 7 as colourless long needles (19.13 g, 74%). Rf 0.36 (EtOAc–hexane = 1:1, PMA). mp 136–137 °C (from MeOH) (lit.,21 134.5–135.5 °C). IR, νmax (ATR)/cm−1: 2985, 2954, 2922, 1444, 1402, 1386, 1288, 1263, 1176, 1109, 823. 1H NMR (300 MHz, CDCl3): δH 3.68 (4H, d, J = 1.13 Hz, 2 × CH2), 1.74 (6H, t, J = 1.13 Hz, 2 × CH3). 13C NMR (75 MHz, CDCl3): δC 125.61 (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 60.70 (CH2), 14.60 (CH3).
:4). Recrystallization from ethanol (the ethanol solution was stored in the fridge for one day) afforded product 3 as colourless needles (1.79 g, 41%). Rf 0.68 (EtOAc–hexane = 5:4, UV or KMnO4). mp 121–122 °C (from EtOH) (lit., 17a 118–120 °C). IR, νmax (ATR)/cm−1: 2976, 2968, 2922, 1454, 1427, 1391, 1313, 1302, 1250, 1229, 1198, 1130, 1112, 1101, 1087, 1005, 893, 864, 818. 1H NMR (300 MHz, CDCl3): δH 4.07 (4H, br s, 2 × BrCH2), 4.01 (4H, br s, 2 × CH2–S). 13C NMR (75 MHz, CDCl3): δC 131.18 (>C), 58.43 (CH2–S), 24.00 (Br–CH2).
:1). The reaction mixture was allowed to reach room temperature and was subjected to column chromatography on SiO2 (100% toluene) to remove the baseline material. Removal of the solvent under reduced pressure afforded product 8 as a brown solid which was contaminated with unreacted C60. Further purification of 8 could not be accomplished due to the similar Rf with C60 in all attempted eluent systems. The 1H and 13C NMR resonances were in good agreement with the data reported in the literature.8
:4). [1H NMR monitoring can also be performed using acetone-d6 instead of dry acetone and mesitylene (10.52 μL, 0.076 mmol) as an internal standard to calculate the yield of the reaction.] The reaction mixture was then filtered and the solid was washed thoroughly with dry acetone. The filtrate and washings were combined, the solvent was exchanged with dry benzene and the solution was stored in the freezer at −20 °C for subsequent reactions without further purification. Under these conditions the product was stable for several weeks giving a single spot on the TLC (EtOAc–hexane = 5:4). However, complete evaporation of the solvent resulted in rapid decomposition to a polymeric material that was not soluble in any solvent tested. The yield of product 5 was calculated by 1H NMR analysis of the reaction mixture using acetone-d6 and mesitylene (10.52 μL, 0.076 mmol) as an internal standard and was found to be quantitative (99–100% using conventional heating and 98–99% using sonication). Alternatively, the reaction can be carried out in a microwave reactor, on a 0.038 mmol scale with respect to 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3, in a sealed tube and by heating at 60 °C (250 W, 21 PSI) for 55 min. The yield was calculated by 1H NMR analysis of the reaction mixture using acetone-d6 and mesitylene (0.038 mmol) as an internal standard and was found to be 97–98%. Method 2 (iodine-induced 1,4-debromination of 3): In a dry 10 mL single-neck round-bottomed flask equipped with a magnetic stirrer, 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3 (23 mg, 0.076 mmol) was dissolved in dry acetone (4 mL) under an atmosphere of dry nitrogen. NaI (34 mg, 0.227 mmol) was then added and the resulting mixture was stirred for 20 h at room temperature. The consumption of the starting material was monitored by TLC (EtOAc–hexane = 5:4). The reaction mixture was then filtered and the solid was washed thoroughly with acetone. The filtrate and washings were combined, the solvent was evaporated down to 2/3 volume and 5 ml DCM was added. The organic layer was washed with a 5% sodium thiosulfate solution (3 × 4 mL), water (3 × 4 mL) and dried over Na2SO4. Complete evaporation of the solvent resulted in rapid decomposition to a polymeric material that was not soluble in any solvent tested. Therefore, the solution was subjected to column chromatography on SiO2 (100% DCM). The fractions containing the product were combined and the solvent was exchanged with benzene and stored in the freezer at −20 °C for subsequent reactions without further purification (impurities of similar polarity were still present). Rf 0.38 (100% DCM, UV or KMnO4), Rf 0.60 (EtOAc–hexane = 5:4, UV or KMnO4). 1H NMR (300 MHz, acetone-d6): δH 5.81 (2H, t, J = 1.70 Hz, 2 × CH), 5.24 (2H, t, J = 1.50 Hz, 2 × CH), 3.97 (4H, t, J = 1.50 Hz, 2 × CH2). 13C NMR (75 MHz, acetone-d6): δC 138.32 (>C), 111.72 (CH2), 57.12 (–CH2–).
:1). The reaction mixture was then allowed to cool at room temperature and the crude mixture was subjected to column chromatography on SiO2. Unreacted C60 and other impurities were eluted with toluene and the fullerene monoadduct 9 was then eluted with toluene–EtOAc = 20:1. Precipitation from CHCl3–pentane afforded product 9 as a brown solid (6.7 mg, 13%). Method 2 (stepwise procedure: zinc-induced 1,4-debromination of 3/[4+2] cycloaddition of 5 with C60): In a dry 10 mL single-neck round-bottomed flask fitted with a magnetic stirrer and a condenser, 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3 (23 mg, 0.076 mmol) was dissolved in dry acetone (4 mL) under an atmosphere of dry nitrogen. Activated Zn-dust (11.4 mg, 0.174 mmol) was then added and the resulting mixture was refluxed for 18 h. The consumption of the starting material was monitored by TLC (EtOAc–hexane = 5:4). The reaction mixture was then filtered and the solid was washed thoroughly with dry acetone (10 mL). The filtrate and washings were combined and the acetone solution was added in a 250 mL two-neck round-bottomed flask fitted with a magnetic stirrer, condenser and a thermometer, containing a stirred solution of C60 (65.4 mg, 0.091 mmol) in dry toluene (70 mL) under an atmosphere of dry nitrogen. The reaction mixture was heated at 45–50 °C for 12 h and the consumption of the starting material was monitored by TLC (toluene 100% then toluene–EtOAc = 20:1). The reaction mixture was allowed to cool at room temperature and the crude mixture was subjected to column chromatography on SiO2. Unreacted C60 and other impurities were eluted with toluene and the fullerene monoadduct 9 was then eluted with toluene–EtOAc = 20:1. Precipitation from CHCl3–pentane afforded product 9 as a brown solid (36.6 mg, 56%). Rf 0.39 (toluene–EtOAc = 20:1). 1H NMR (300 MHz, CS2–CDCl3, 1:1): δH 4.38 (4H, br s, 2 × CH2), 4.22 (4H, br s, 2 × CH2). 13C NMR (75 MHz, CS2–CDCl3, 1:1): δC 155.14, 147.60, 147.57, 146.48, 146.20, 145.60, 145.48, 145.43, 144.65, 144.52, 143.06, 142.57, 142.10, 141.95, 141.55, 140.17, 135.45, 133.11, 65.32 (sp3C of C60), 59.68 (CH2), 40.81 (CH2). (The 18 signals in the region 155.16–133.11 ppm correspond to sp2carbon atoms; 17 signals are attributed to sp2carbon atoms of C60.) HRMS (MALDI TOF, negative mode, DCTB): calculated for C66H8 [M–SO2]− requires 800.0620; found: 800.0619.
:4). The reaction mixture was then allowed to cool at room temperature, filtered and the solid was washed thoroughly with acetone (10 mL). The filtrate and washings were combined and the solvent was evaporated under reduced pressure to give a colourless solid which was purified by column chromatography on silica gel (EtOAc–hexane = 5:4). Product 10 was isolated as a colourless solid (19.9 mg, 92%). Alternatively, the reaction can be carried out in a microwave reactor, in a 0.038 mmol scale with respect to 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3, in a sealed tube and by heating at 60 °C (250 W, 21 PSI) for 55 min. Rf 0.39 (EtOAc–hexane = 5:4, UV or KMnO4). mp 170–171 °C (from MeOH) (lit.,15b 168–169 °C). IR, νmax (ATR)/cm−1: 2974, 2966, 1736, 1713, 1647, 1445, 1418, 1310, 1283, 1256, 1244, 1207, 1196, 1157, 1144, 1111, 1090, 1066, 1001, 941, 914, 852, 819, 783. 1H NMR (500 MHz, CDCl3): δH 3.73 (6H, br s, 2 × CO2CH3), 3.70 (4H, br s, 2 × CH2), 3.03 (4H, br s, 2 × CH2). 13C NMR (125 MHz, CDCl3): δC 167.13 (CO), 131.24 (>C), 124.31 (>C), 58.37 (CH2), 52.43 (CO2CH3), 28.57 (CH2). HRMS (APCI+): calculated for C12H15O6S [M+H]+ requires 287.0584; found: 287.0587.
:4). The reaction mixture was then allowed to cool at room temperature and MnO2 (19.5 mg, 0.224 mmol) was added. The mixture was refluxed for another 2 h, filtered and the solid was washed thoroughly with acetone (10 mL). The filtrate and washings were combined and the solvent was evaporated under reduced pressure to give a colourless solid which was purified by column chromatography on silica gel (EtOAc–hexane = 5:4). Product 11 was isolated as a colourless solid (11.4 mg, 90%). The 1H and 13C NMR resonances were in good agreement with the data reported in the literature.10b
), 60.70 (CH2), 14.60 (CH3).
:4). Recrystallization from ethanol (the ethanol solution was stored in the fridge for one day) afforded product 3 as colourless needles (1.79 g, 41%). Rf 0.68 (EtOAc–hexane = 5:4, UV or KMnO4). mp 121–122 °C (from EtOH) (lit., 17a 118–120 °C). IR, νmax (ATR)/cm−1: 2976, 2968, 2922, 1454, 1427, 1391, 1313, 1302, 1250, 1229, 1198, 1130, 1112, 1101, 1087, 1005, 893, 864, 818. 1H NMR (300 MHz, CDCl3): δH 4.07 (4H, br s, 2 × BrCH2), 4.01 (4H, br s, 2 × CH2–S). 13C NMR (75 MHz, CDCl3): δC 131.18 (>C), 58.43 (CH2–S), 24.00 (Br–CH2).
:1). The reaction mixture was allowed to reach room temperature and was subjected to column chromatography on SiO2 (100% toluene) to remove the baseline material. Removal of the solvent under reduced pressure afforded product 8 as a brown solid which was contaminated with unreacted C60. Further purification of 8 could not be accomplished due to the similar Rf with C60 in all attempted eluent systems. The 1H and 13C NMR resonances were in good agreement with the data reported in the literature.8
:4). [1H NMR monitoring can also be performed using acetone-d6 instead of dry acetone and mesitylene (10.52 μL, 0.076 mmol) as an internal standard to calculate the yield of the reaction.] The reaction mixture was then filtered and the solid was washed thoroughly with dry acetone. The filtrate and washings were combined, the solvent was exchanged with dry benzene and the solution was stored in the freezer at −20 °C for subsequent reactions without further purification. Under these conditions the product was stable for several weeks giving a single spot on the TLC (EtOAc–hexane = 5:4). However, complete evaporation of the solvent resulted in rapid decomposition to a polymeric material that was not soluble in any solvent tested. The yield of product 5 was calculated by 1H NMR analysis of the reaction mixture using acetone-d6 and mesitylene (10.52 μL, 0.076 mmol) as an internal standard and was found to be quantitative (99–100% using conventional heating and 98–99% using sonication). Alternatively, the reaction can be carried out in a microwave reactor, on a 0.038 mmol scale with respect to 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3, in a sealed tube and by heating at 60 °C (250 W, 21 PSI) for 55 min. The yield was calculated by 1H NMR analysis of the reaction mixture using acetone-d6 and mesitylene (0.038 mmol) as an internal standard and was found to be 97–98%. Method 2 (iodine-induced 1,4-debromination of 3): In a dry 10 mL single-neck round-bottomed flask equipped with a magnetic stirrer, 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3 (23 mg, 0.076 mmol) was dissolved in dry acetone (4 mL) under an atmosphere of dry nitrogen. NaI (34 mg, 0.227 mmol) was then added and the resulting mixture was stirred for 20 h at room temperature. The consumption of the starting material was monitored by TLC (EtOAc–hexane = 5:4). The reaction mixture was then filtered and the solid was washed thoroughly with acetone. The filtrate and washings were combined, the solvent was evaporated down to 2/3 volume and 5 ml DCM was added. The organic layer was washed with a 5% sodium thiosulfate solution (3 × 4 mL), water (3 × 4 mL) and dried over Na2SO4. Complete evaporation of the solvent resulted in rapid decomposition to a polymeric material that was not soluble in any solvent tested. Therefore, the solution was subjected to column chromatography on SiO2 (100% DCM). The fractions containing the product were combined and the solvent was exchanged with benzene and stored in the freezer at −20 °C for subsequent reactions without further purification (impurities of similar polarity were still present). Rf 0.38 (100% DCM, UV or KMnO4), Rf 0.60 (EtOAc–hexane = 5:4, UV or KMnO4). 1H NMR (300 MHz, acetone-d6): δH 5.81 (2H, t, J = 1.70 Hz, 2 × CH), 5.24 (2H, t, J = 1.50 Hz, 2 × CH), 3.97 (4H, t, J = 1.50 Hz, 2 × CH2). 13C NMR (75 MHz, acetone-d6): δC 138.32 (>C), 111.72 (CH2), 57.12 (–CH2–).
:1). The reaction mixture was then allowed to cool at room temperature and the crude mixture was subjected to column chromatography on SiO2. Unreacted C60 and other impurities were eluted with toluene and the fullerene monoadduct 9 was then eluted with toluene–EtOAc = 20:1. Precipitation from CHCl3–pentane afforded product 9 as a brown solid (6.7 mg, 13%). Method 2 (stepwise procedure: zinc-induced 1,4-debromination of 3/[4+2] cycloaddition of 5 with C60): In a dry 10 mL single-neck round-bottomed flask fitted with a magnetic stirrer and a condenser, 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3 (23 mg, 0.076 mmol) was dissolved in dry acetone (4 mL) under an atmosphere of dry nitrogen. Activated Zn-dust (11.4 mg, 0.174 mmol) was then added and the resulting mixture was refluxed for 18 h. The consumption of the starting material was monitored by TLC (EtOAc–hexane = 5:4). The reaction mixture was then filtered and the solid was washed thoroughly with dry acetone (10 mL). The filtrate and washings were combined and the acetone solution was added in a 250 mL two-neck round-bottomed flask fitted with a magnetic stirrer, condenser and a thermometer, containing a stirred solution of C60 (65.4 mg, 0.091 mmol) in dry toluene (70 mL) under an atmosphere of dry nitrogen. The reaction mixture was heated at 45–50 °C for 12 h and the consumption of the starting material was monitored by TLC (toluene 100% then toluene–EtOAc = 20:1). The reaction mixture was allowed to cool at room temperature and the crude mixture was subjected to column chromatography on SiO2. Unreacted C60 and other impurities were eluted with toluene and the fullerene monoadduct 9 was then eluted with toluene–EtOAc = 20:1. Precipitation from CHCl3–pentane afforded product 9 as a brown solid (36.6 mg, 56%). Rf 0.39 (toluene–EtOAc = 20:1). 1H NMR (300 MHz, CS2–CDCl3, 1:1): δH 4.38 (4H, br s, 2 × CH2), 4.22 (4H, br s, 2 × CH2). 13C NMR (75 MHz, CS2–CDCl3, 1:1): δC 155.14, 147.60, 147.57, 146.48, 146.20, 145.60, 145.48, 145.43, 144.65, 144.52, 143.06, 142.57, 142.10, 141.95, 141.55, 140.17, 135.45, 133.11, 65.32 (sp3C of C60), 59.68 (CH2), 40.81 (CH2). (The 18 signals in the region 155.16–133.11 ppm correspond to sp2carbon atoms; 17 signals are attributed to sp2carbon atoms of C60.) HRMS (MALDI TOF, negative mode, DCTB): calculated for C66H8 [M–SO2]− requires 800.0620; found: 800.0619.
:4). The reaction mixture was then allowed to cool at room temperature, filtered and the solid was washed thoroughly with acetone (10 mL). The filtrate and washings were combined and the solvent was evaporated under reduced pressure to give a colourless solid which was purified by column chromatography on silica gel (EtOAc–hexane = 5:4). Product 10 was isolated as a colourless solid (19.9 mg, 92%). Alternatively, the reaction can be carried out in a microwave reactor, in a 0.038 mmol scale with respect to 3,4-bis(bromomethyl)-2,5-dihydrothiophene-1,1-dioxide 3, in a sealed tube and by heating at 60 °C (250 W, 21 PSI) for 55 min. Rf 0.39 (EtOAc–hexane = 5:4, UV or KMnO4). mp 170–171 °C (from MeOH) (lit.,15b 168–169 °C). IR, νmax (ATR)/cm−1: 2974, 2966, 1736, 1713, 1647, 1445, 1418, 1310, 1283, 1256, 1244, 1207, 1196, 1157, 1144, 1111, 1090, 1066, 1001, 941, 914, 852, 819, 783. 1H NMR (500 MHz, CDCl3): δH 3.73 (6H, br s, 2 × CO2CH3), 3.70 (4H, br s, 2 × CH2), 3.03 (4H, br s, 2 × CH2). 13C NMR (125 MHz, CDCl3): δC 167.13 (CO), 131.24 (>C), 124.31 (>C), 58.37 (CH2), 52.43 (CO2CH3), 28.57 (CH2). HRMS (APCI+): calculated for C12H15O6S [M+H]+ requires 287.0584; found: 287.0587.
:4). The reaction mixture was then allowed to cool at room temperature and MnO2 (19.5 mg, 0.224 mmol) was added. The mixture was refluxed for another 2 h, filtered and the solid was washed thoroughly with acetone (10 mL). The filtrate and washings were combined and the solvent was evaporated under reduced pressure to give a colourless solid which was purified by column chromatography on silica gel (EtOAc–hexane = 5:4). Product 11 was isolated as a colourless solid (11.4 mg, 90%). The 1H and 13C NMR resonances were in good agreement with the data reported in the literature.10b
Acknowledgements
This work was financially supported by the Cyprus Research Promotion Foundation (Grant NEKYP/0308/02). We also thank Dr P. A. Koutentis (Department of Chemistry, University of Cyprus) for providing access to the TGA, DSC and the microwave reactor.References
- W. Albrecht, Justus Liebigs Ann. Chem., 1906, 348, 31–49 CrossRef CAS.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- For general examples, see: (a) G. Jayamurugan, J.-P. Gisselbrecht, C. Boudon, F. Schoenebeck, W. B. Schweizer, B. Berneta and F. Diederich, Chem. Commun., 2011, 47, 4520–4522 Search PubMed; (b) R. Meissner, X. Garcias, S. Mecozzi and J. Rebek, Jr., J. Am. Chem. Soc., 1997, 119, 77–85 CrossRef CAS; (c) S. Sarkar, E. Bekyarova, S. Niyogi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 3324–3327 CrossRef CAS; (d) A. Dag, H. Sahin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 886–892 CrossRef CAS.
- For examples of [4+2] cycloadditions with C60, see: (a) W. Sliwa, Fullerene Sci. Technol., 1997, 5, 1133–1175 CrossRef CAS; (b) A. Hirsch and M. Brettreich, In Fullerenes - Chemistry and Reactions; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2005 Search PubMed.
- For examples of donor–bridge–C60 electroactive systems, see (a) H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537–546 CrossRef CAS; (b) H. Imahori, Org. Biomol. Chem., 2004, 2, 1425–1433 RSC; (c) S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841–843 CrossRef CAS; (d) P. Hudhomme, C. R. Chim., 2006, 9, 881–891 CrossRef CAS.
- P. Belik, A. Gügel, J. Spickermann and K. Müllen, Angew. Chem., Int. Ed. Engl., 1993, 32, 78–80 CrossRef.
- For examples of short-chain fullerene derivatives, see (a) G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Nature, 1997, 387, 583–586 CrossRef CAS; (b) K. Komatsu, G.-W. Wang, Y. Murata, T. Tanaka, K. Fujiwara, K. Yamamoto and M. Saunders, J. Org. Chem., 1998, 63, 9358–9366 CrossRef CAS; (c) S. Lebedkin, S. Ballenweg, J. Gross, R. Taylor and W. Krätschmer, Tetrahedron Lett., 1995, 36, 4971–4974 CAS; (d) A. B. Smith, H. Tokuyama, R. M. Strongin, G. T. Furst and W. J. Romanow, J. Am. Chem. Soc., 1995, 117, 9359–9360 CrossRef CAS; (e) A. Gromov, S. Lebedkin, S. Ballenweg, A. G. Avent, R. Taylor and W. Krätschmer, Chem. Commun., 1997, 209–210 Search PubMed; (f) N. Dragoe, H. Shimotani, J. Wang, M. Iwaya, A. de Bettencourt-Dias, A. L. Balch and K. Kitazawa, J. Am. Chem. Soc., 2001, 123, 1294–1301 CrossRef CAS; (g) Y. Murata, A. Han and K. Komatsu, Tetrahedron Lett., 2003, 44, 8199–8201 CrossRef CAS; (h) K. Fujiwara and K. Komatsu, Org. Lett., 2002, 4, 1039–1041 CrossRef CAS; (i) C. Zhang, S. Chen, Z. Xiao, Q. Zuo and L. Ding, Org. Lett., 2012, 14, 1508–1511 CrossRef CAS.
- T. J. Hingston, M. R. Sambrook, K. Porfyrakis and G. A. D. Briggs, Tetrahedron Lett., 2006, 47, 7413–7415 CrossRef CAS.
- J. L. Charlton and M. M. Alauddin, Tetrahedron, 1987, 43, 2873–2889 CrossRef CAS.
- For recent examples of bis-o-quinodimethanes, see (a) S. Kotha and P. Khedkar, J. Org. Chem., 2009, 74, 5667–5670 CrossRef CAS; (b) S. Kotha and A. S. Chavan, J. Org. Chem., 2010, 75, 4319–4322 CrossRef CAS.
- B. M. Trost and M. Shimizu, J. Am. Chem. Soc., 1982, 104, 4299–4301 CrossRef CAS.
- A. Hosomi, K. Otaka and H. Sakurai, Tetrahedron Lett., 1986, 27, 2881–2884 CrossRef CAS.
- (a) F. Monnat and P. Vogel, Helv. Chim. Acta, 2002, 85, 712–732 CrossRef CAS; (b) E. Roversi, R. Scopelliti, E. Solari, R. Estoppey, P. Vogel, P. Brana, B. Menendez and J. A. Sordo, Chem.–Eur. J., 2002, 8, 1336–1355 CrossRef CAS.
- P. Dowd, J. Am. Chem. Soc., 1970, 92, 1066–1068 CrossRef CAS.
- (a) S. Sadeh and Y. Gaoni, Tetrahedron Lett., 1973, 14, 2365–2368 CrossRef; (b) Y. Gaoni and S. Sadeh, J. Org. Chem., 1980, 45, 870–881 CrossRef CAS.
- A. C. Cope and F. Kagan, J. Am. Chem. Soc., 1958, 80, 5499–5502 CrossRef CAS.
- (a) N. Watanabe, N. Kihara and T. Takata, Org. Lett., 2001, 3, 3519–3522 CrossRef CAS; (b) G. A. Tashbaev, L. Yu. Krivehikova and I. M. Nasyrov, Chem. Heterocycl. Compd., 1992, 28, 139–141 CrossRef; (c) R. W. Alder, P. R. Allen, L. S. Edwards, G. I. Fray, K. E. Fuller, P. M. Gore, N. M. Hext, M. H. Perry, A. R. Thomas and K. S. Turner, J. Chem. Soc., Perkin Trans. 1, 1994, 21, 3071–3077 RSC.
- For thermal SO2 extrusion, see selected examples (a) T. Suzuki, K. Kubomura, H. Fuchii and H. Takayama, J. Chem. Soc., Chem. Commun., 1990, 1687–1689 RSC; (b) K. Ando, M. Kankake, T. Suzuki and H. Takayama, J. Chem. Soc., Chem. Commun., 1992, 1100–1102 RSC.
- For examples of chemical SO2 extrusion, see (a) T.-S. Chou and M.-L. You, J. Org. Chem., 1987, 52, 2224–2226 CrossRef CAS; (b) Y. Gaoni, Tetrahedron Lett., 1977, 18, 947–950 CrossRef.
- (a) G. Vassilikogiannakis, N. Chronakis and M. Orfanopoulos, J. Am. Chem. Soc., 1998, 120, 9911–9920 CrossRef CAS; (b) N. Chronakis and M. Orfanopoulos, Org. Lett., 2001, 3, 545–548 CrossRef CAS; (c) N. Chronakis and M. Orfanopoulos, Tetrahedron Lett., 2001, 42, 1201–1204 CrossRef CAS; (d) N. Chronakis, G. Froudakis and M. Orfanopoulos, J. Org. Chem., 2002, 67, 3284–3289 CrossRef CAS.
- O. Grummitt and A. L. Endrey, J. Am. Chem. Soc., 1960, 82, 3614–3619 CrossRef CAS.
- R. D. Smith and H. E. Simmons, Org. Synth., 1973, 5, 855 Search PubMed.
- Vogel's Textbook of Practical Organic Chemistry, Longman Group UK Limited, 5th edn, 1989, ch. 4, pp. 395–469 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Characterisation data are available. CCDC reference numbers 868567 and 868568. See DOI: 10.1039/c2ra22502f |
| This journal is © The Royal Society of Chemistry 2012 |