Superparamagnetic nano-composite scaffolds for promoting bone cell proliferation and defect reparation without a magnetic field
Kuilin
Lai†
a,
Wen
Jiang†
a,
James. Z.
Tang
b,
Yao
Wu
*a,
Bin
He
a,
Gang
Wang
a and
Zhongwei
Gu
*a
aNational Engineering Research Center for Biomaterials, Sichuan University, Chengdu, P. R. China. E-mail: zwgu@scu.edu.cn; Yaowu_amanda@126.com; Tel: +86 28 8541 0336; Fax: +86 28 8541 0653
bDepartment of Pharmacy, School of Applied Sciences, University of Wolverhampton, Wulfruna Street, Wolverhampton, WV1 1SB, United Kingdom
First published on 8th October 2012
Abstract
Apart from chemical molecules, physical regulations also greatly determine the efficiency of healing in regenerating functional tissues. In this study, we fabricated superparamagnetic nano-composite scaffolds for tissue engineering and investigated their effects on different bone cells without an external magnetic field. Poly(lactic-co-glycolic acid) (PLGA) and hydrophobic superparamagnetic magnetite nanoparticles (MNPs) were combined together with different mass ratios in order to construct composite scaffolds using an electrospinning method for the first time. The diameters of the fibers were 400–600 nm with the MNPs uniformly dispersed in them, as shown by transmission (TEM) and scanning (SEM) electron microscopy observations. All composite scaffolds retained superparamagnetism at room temperature, but the saturation magnetization did not increase linearly as the magnetite content increased. The composite scaffolds with different MNP content showed excellent biocompatibility and significantly promoted cell proliferation compared with PLGA nanofibrous scaffold without an external magnetic field. Cell cycle analysis proved that the composite scaffolds decreased cell numbers in G0/G1 phase while increasing those in S phase, which resulted in positive effects on cell proliferation. However, the composite scaffolds had no effect on the differentiation of MC3T3-E1 cells because of the different impact mechanism between proliferation and differentiation. Therefore, the composite scaffolds composed of superparamagnetic MNPs could be considered as an ideal substrate for accelerating osteoblast cell proliferation and tissue repair.
1. Introduction
Tissue engineering has been developing continuously in recent years and is considered a promising regenerative medicine for the future. However, this technology is hindered in some respects, such as poor biocompatibility and long cell propagation time, which might cause inflammation and a longer tissue repair period. The scaffold substrate, in which cells are attached and cultured, is one of the major impact factors on the reconstruction of functional tissue in tissue engineering. There are various scaffold fabrication approaches, including fiber-spinning, leaching, microsphere, phase separating, printing and prototyping methods. Among these, electrospinning is the most convenient and versatile way of fabricating composite scaffolds with continuous and tunable nanofibers, ranging from nanometers to micrometers. A wide range of natural materials, such as chitosan, sodium hyaluronate, collagen, and biocompatible polymers like poly(L-lactide), polycaprolactone and poly(lactic-co-glycolic acid) (PLGA), have been used for electrospinning of tissue engineering scaffolds.1,2 The obtained porous and nanoscale structures mimic the natural extracellular matrix (ECM) with random or aligned fibers,3 which is essential for cell attachment, proliferation and differentiation.4,5During the past twenty years, magnets or static magnetic fields have been considered to be one of the most useful treatments for pain release and wound healing, especially for bone repair, due to their stimulating effects on cell proliferation.6–9 Wolsko et al. found that extremely weak static magnetic fields had significant biological and clinical effects.10 When the dimensions of a bulk magnet, such as magnetite, drop to less than 20 nanometers, its magnetization direction can flip randomly under the influence of temperature. In this circumstance, magnetite becomes superparamagnetic with only one magnetism domain.11 Superparamagnetic magnetite nanoparticles (MNPs) are widely used in biomedical fields, such as magnetic resonance imaging (MRI) contrast agents14 and bio-separation materials,12 for their high magnetism saturation and excellent biocompatibility.12,13 Similarly, a composite scaffold composed of polymer and magnetite could become an important component for tissue regeneration. Novel hydrogels, with MNPs as a substrate, could be used in hyperthermia cancer therapy.15 Some researchers have used MNPs combined with a magnetic force for constructing cell sheets in tissue engineering.16–18 Our previous research work showed that a composite scaffold composed of MNPs and porous hydroxyapatite could promote the proliferation of osteoblast cells.19 Gu’s group20 fabricated paramagnetic nanofibers with polylactide, hydroxyapatite and γ-Fe2O3 combined with a magnetic field for tissue regeneration. They also found that composite films significantly enhanced the proliferation, differentiation and ECM secretion of the osteoblast cells. However, these composite scaffolds were unable to solely explain the impact of magnetism property on the cell behaviors, due to the bio-inductive and osteogenic property of calcium phosphate ceramic.21 Moreover, the bio-function of MNPs in the composite scaffold was usually neglected due to the synergistic effect of an external magnetic field.22
Although some electrospun magnetic fibrous scaffolds have been fabricated, such as Fe3O4/PVP nanofibers,23 Fe3O4/PVA nanofibers,24 FePt/PCL nanofibers25 and CPT/Fe2O3-embedded PLGA composite fibers,26 the bioeffects of MNPs on cells, besides the biocompatibility of the magnetic composite, have not been considered and understood.27 Recently, Wei28 reported that magnetic biodegradable Fe3O4/CS/PVA nanofibrous membranes could promote MG63 proliferation. However, the mechanism by which magnetic scaffolds promote cell proliferation is still unclear. Based on the positive effect of MNPs in bone repair, the bioeffects and mechanism of MNPs in cells have strongly attracted our interest, especially when they are combined with biocompatible polymers without bio-inductive property and external magnetic fields. In this study, we have aimed to develop biocompatible superparamagnetic scaffolds with different iron content to investigate the reasons for the impact of MNPs in nano-composite scaffolds on the bio-behaviors of osteoblast cells. We chose the biocompatible polymer PLGA, which is approved by the Food and Drug Administration (FDA), and hydrophobic MNPs, synthesized via a thermal decomposition method, to fabricate composite scaffolds with controlled MNP contents using electrospinning technology. The physicochemical properties, such as size, morphology, dispersion of MNPs, thermogravimetric analysis (TGA) and superparamagnetism of obtained nano-fibers, were systematically studied. The interactions between composite scaffolds and two osteoblast cell lines, Ros17/2.8 and MC3T3-E1, including cell spread, proliferation, differentiation and cell cycle were investigated in order to explore the mechanism for the impact of MNPs on cells.
2. Materials and methods
2.1. Materials
CHCl3, CHCl2, dimethylformamide (DMF) and ethanol were obtained from Chengdu Kelong chemical company. Oleylamine, oleic acid, benzyl ether, diethanolamine and p-nitrophenyl phosphate disodium salt were obtained from Sigmal-Aldrich. Milli-Q water was used throughout the experiments.2.2. Synthesis of PLGA copolymers
L-Lactide (LA) and glycolide (GA) (Purac, Netherlands) monomers were polymerized in the presence of stannous octoate (Tin(II) 2-ethylhexanoate) catalyst to produce PLGA as follows. LA and GA monomers were recrystallized from ethyl acetate, dried under vacuum, and transferred to a 20 mL round-bottom reaction flask at 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 molar ratio. After purging with nitrogen and degassing, the flask was sealed and placed in an oil bath at 170 °C for 30 min and 150 °C for another 12 h. The product was dissolved in CHCl3, precipitated in ethanol and dried to obtain pure PLGA.
:25 molar ratio. After purging with nitrogen and degassing, the flask was sealed and placed in an oil bath at 170 °C for 30 min and 150 °C for another 12 h. The product was dissolved in CHCl3, precipitated in ethanol and dried to obtain pure PLGA.
1H NMR was used to characterize the polymerization reaction. Briefly, the sample was dissolved in chloroform-d (CDCl3, Cambridge Isotope Laboratories, MA) and analyzed with a Bruker Avance II NMR Spectrometer working at 400 MHz at room temperature (RT).
2.3. Synthesis of superparamagnetic magnetite nanoparticle
MNPs were prepared according to the method reported by Sun et al.29 Briefly, iron (III) acetylacetonate (2.0 mmol), 1,2-hexadecanediol (10.0 mmol), oleylamine (6.0 mmol) and oleic acid (6.0 mmol) were dissolved in 20 mL benzyl ether. The solution was heated to 200 °C for 2 h, refluxed at 300 °C for another 1 h, and finally cooled to RT. The black particles were precipitated via ethanol and collected by centrifugation at 8000 rpm for 10 min. Particles were reprecipitated and washed with ethanol for 3 times. Finally, the product was redispersed in n-hexane and stored at 4 °C.2.4. Characterization of MNPs
Hydrodynamic diameter and size distribution of MNPs were measured by dynamic light scattering (DLS, Malvern Nano-ZS, l = 632.8 nm) at RT. Samples were dispersed in n-hexane in a glass cuvette and tested at 25 °C. X-ray diffraction (XRD, X’ Pert Pro MPD, Philips, Netherlands) was employed to study the crystalline properties of samples with angle ranging from 10° to 90° at 0.1° s−1, using a copper anode. The FTIR analysis of MNPs was performed using a PE spectrometer. In brief, dry KBr crystals were ground with MNPs at a 300:1 weight ratio and pressed to form a thin film. The absorbance was collected at 4000–400 cm−1.
2.5. Preparation of electrospinning scaffolds
The electrospinning scaffolds were prepared on clean glass coverslips. 5 wt% PLGA in a mixed solvent of CH2Cl2:DMF (7:3 in v/v) was used for all experiments. MNPs were added into the PLGA solution at weight ratios of 1:9 or 1:1 to PLGA and electrospun to prepare PLGA-10% MNPs and PLGA-50% MNPs composite scaffolds, respectively. Similarly, quantum dots Cd/Se ZnO (QDs) (Jiayuan Quantum Dots Co.) were also added into the PLGA solution at a weight ratio of 1:9 to PLGA and electropun to fabricate PLGA–10% QDs scaffolds. Each solution was drawn into a 10 mL glass syringe connected with a 6-gauge needle. An electrode was mounted on the needle. During the electrospinning process, the voltage was kept constantly at 20 kV while the feeding rate was 1 mL h−1 and the ground metal plate was 20 cm away from the needle tip. Coverslips with a diameter of 20 mm were placed on the metal plate to collect electrospinning scaffolds for investigation of the morphology of scaffolds and for cell experiments. Additionally, ferrofluids in hexane was dripped onto pure PLGA scaffolds and dried at RT to produce surface deposited scaffolds (PLGA-deposit MNPs).
For cell culture studies, all the scaffolds were sterilized under ultraviolet light for 1 h on each side and subsequently immersed in 75% ethanol solution for 2 h. Finally, the scaffold was rinsed three times and subsequently prewetted in phosphate buffer saline (PBS, pH 7.4) overnight before use.
2.6. Microstructure characterization of scaffolds
Electrospinning scaffolds were coated with gold and characterized with a field emission SEM (LEO 1550) operating at 5 kV with a secondary electron detector. Hundreds of fibers per sample were measured to determine average fiber diameters. Additionally, TEM was used to observe the dispersity of the MNPs in the fibers by collecting them on a TEM copper net.2.7. Composition and magnetization of MNPs and scaffolds
The composition of MNPs and their contents in scaffolds were measured by TGA using a Netzsch STA499C thermal analyzer within an Al2O3 crucible. The mass losses of the samples (5–10 mg) were monitored from 35 °C to 1000 °C at a heating rate of 10 K min−1 in a dynamic nitrogen atmosphere. The magnetization of the MNPs and composite scaffolds was measured by a vibrating sample magnetometer (VSM, Model BHV-525, Riken Japanese Electronics Company) with the field from 0 to 15000 Oe at 300 K.
2.8. Cell culture
Ros17/2.8 and MC3T3-E1 cells were purchased from the Chinese Academy of Sciences Cell Bank in Shanghai. Ros17/2.8 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Invitrogen) supplemented with 10% fetal calf serum (FCS, Thermo Scientific), penicillin (100 U mL−1) (Gibco) and streptomycin (100 mg mL−1) (Gibco). MC3T3-E1 cells were cultured in alpha minimum essential medium (α-MEM, Invitrogen) supplemented with 10% FCS, 10% penicillin and streptomycin for proliferation at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. The medium was changed every other day.2.9. Fluorescence microscopy of cell morphology
At the time of cell seeding, cells were washed with PBS, lifted from culture dishes with trypsin/ethylenediaminetetraacetic acid (EDTA, Gibco). The cells were then cultured onto PLGA and PLGA-10% MNPs scaffolds at low density (2 × 105 cells per sample) and incubated at 5% CO2, 37 °C. After different culture time, the cellular scaffolds were rinsed with PBS to remove unattached cells and stained with 5 μg mL−1 fluorescein diacetate (FDA) in PBS at RT for 3–5 min. The samples were washed with PBS twice and photographed by confocal laser scanning microscopy (CLSM) (Leica).2.10. Proliferation assay
1 × 105 cells were cultured in each sample and analyzed for proliferation after 2, 4 and 6 days of culture. For the assay, cell proliferation was determined with Cell Counting Kit-8 solution (CCK-8, Dojindo) according to the recommendation of the manufacturer. Briefly, the supernatant was removed and 500 μL cell culture medium containing 10% CCK-8 solution was added into each well. The control group (cells cultured in well plates) and cells in the scaffolds were then incubated in the dark at 37 °C and 5% CO2 for 2 h to allow the formation of water-soluble formazan dye. After gently shaken, 100 μL culture medium per well was added into an enzyme label plate and the absorbance at 450 nm was measured using a microplate reader (Bio-Rad).2.11. Cell cycle study
Cell cycles were analyzed by flow cytometry. After culturing on scaffolds for several days, cells in logarithmic growth were trypsinized and washed twice with cold PBS. For each sample, about 1 × 106 cells were fixed in 1 mL of 75% ethanol solution and stored at −20 °C. At the time of analysis, the fixed cells were precipitated by centrifugation at 800 × g for 5 min and washed with PBS three times. Finally, the cell pellet was resuspended in 200 μL PBS and 100 μL RNase A (Beyotime Institute of Biotechnology) was added. After incubation for 30 min at 37 °C, 200 μL propidium iodide (PI) solution was added and incubated in the dark at 4 °C for 60 min. After filtered with 40 μm cell strainers, the DNA content was analyzed using standard techniques with 488 nm excitation of the PI, gating on FL-3 width and area, and FL-3 at 610 ± 20 nm was collected on a Beckman-Coulter Cytomics F.C. 500 flow cytometer.2.12. Differentiation assay
The MC3T3-E1 cells were cultured on composite scaffolds in ascorbic acid free α-MEM for 2, 4 and 14 days. Alkaline phosphatase activity (ALP), osteocalcin (OC) and collagenase I (Col I) contents were measured separately to assess cell differentiation. Cellular scaffolds were washed with PBS and 200 μL protein extraction reagent (M-PER, thermo scientific) was added for each sample. After shaking for 5 min at RT, the cell lysates were collected and centrifuged at 13000 × g for 10 min and the supernatant was collected and stored at −20 °C. Prior to analysis, cell lysates were thawed and agitated for 1 min. Protein concentration was determined by BCA Protein Assay Kit (Thermo Scientific). Alkaline phosphatase activity was measured by the method described by Walter et al.30 Briefly, 50 μL cell lysate per sample was mixed with 49.5 μL of buffer solution at pH 9.8 (0.5 mM MgCl2 and 1 M diethanolamine) and 0.5 μL of substrate solution (450 mM p-nitrophenyl phosphate disodium salt) and incubated at 37 °C for 30 min. The reaction was stopped with the addition of 50 μL NaOH (1 M) and the absorbance at 405 nm was measured using a microplate reader. Alkaline phosphatase activity was normalized by μg of protein. Col I and OC contents were measured using an enzyme-linked immunosorbent assay kit (R&D) according to the recommendation of the manufacturer.
2.13. Statistics
For each group, data were collected from 4 to 6 samples and expressed as mean ± standard deviation. Statistical analysis was performed using the Student’s t test, and significance was determined at p < 0.05.3. Results
3.1. Characterization of the MNPs
The FTIR results in Fig. 1a confirmed the surface properties of synthesized MNPs. Compared with pure oleic acid, MNPs showed new strong Fe–O vibrations at 599 cm−1. The C–H stretching of alkyl chains at 2923 and 2853 cm−1 were clearly retained. Two carboxylate stretchings at 1533 and 1424 cm−1 revealed a bidentate ligand between the iron oxide core and oleate,31,32 which indicated the presence of oleate on the surface and provided the MNPs with excellent monodispersity in non-polar solvents. The narrow size distribution of the MNPs in hexane, as determined by DLS in Fig. 1b, corresponded to an average hydrodynamic diameter of 8.47 ± 2.12 nm. XRD results showed that the scattering angle of the MNPs was the same as that of pure magnetite from the standard Powder Diffraction File (PDF#19-0629), and corresponded to different crystal faces (Fig. 1c).33 From the Scherrer formula D = 0.89λ/Bcosθ,34 the crystal size of the MNPs could be calculated as ∼6.4 nm, a little smaller than that given by the DLS measurement because of the oleic acid surface shell. The magnetization curve for the MNPs was characterized by VSM measurement in Fig. 1d. The magnetic loops illustrated the typical superparamagnetic behavior of the MNPs at RT, as the curve passed through 0 Oe and no magnetic residue remained without a magnetic field.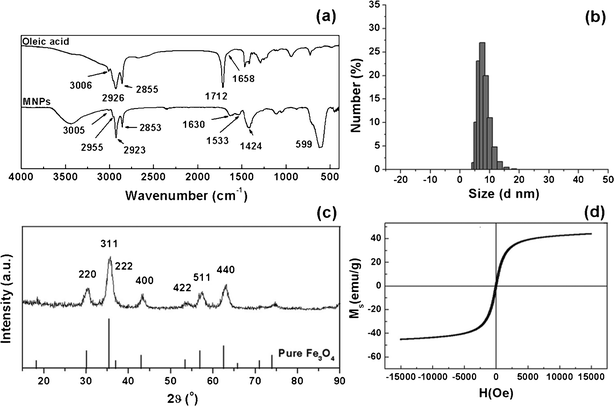 | ||
| Fig. 1 Characterization of the MNPs by (a) FTIR: the positions of characteristic oleic acid and MNPs peaks are indicated; (b) DLS: the hydrodynamic diameter; (c) XRD: the scattering angles are indicated, as are those of pure magnetite from standard Powder Diffraction File (PDF#19-0629); (d) VSM: the magnetization curve. | ||
3.2. Electrospining scaffolds
The chemical structure of PLGA was determined by the chemical shifts of lactide methyl protons at 1.5 ppm and glycolide methylene protons at 4.75 ppm. The results showed that the molar ratio of lactide to glicolide in PLGA was 8:2. Fig. 2 shows the experimental procedures of fabricating different scaffolds for cell experiments. Different kinds of electrospinning scaffolds were prepared on glass coverslips (Fig. 3, insets). It can be clearly observed from the SEM images that the fibers of PLGA-10% QDs (Fig. 3b) and PLGA-10% MNPs (Fig. 3c) scaffolds were both as smooth as those of PLGA scaffolds (Fig. 3a). However, for PLGA-50% MNPs, there were some magnetite aggregates in the subsurface, which can be observed in Fig. 3d. Furthermore, for PLGA-deposit MNPs scaffolds (Fig. 3e), some fibers were knotted by the aggregated MNPs compared with PLGA scaffolds (Fig. 3a). The color of the composite scaffolds turned from brown to black as the concentration of MNPs increased (Fig. 3c, d, e, insets). After analysis of the SEM images, it could be seen that different scaffolds had similar fiber sizes ranging from 400 to 600 nm with narrow statistical distributions as shown in Table 1. TEM images (Fig. 4) confirmed the distribution of MNPs in the composite scaffolds, which could be observed as black particles. For PLGA-10% MNPs scaffolds in Fig. 4a, the particles were uniformly monodisperse in the fiber. But for PLGA-50% MNPs in Fig. 4b, MNPs tended to form small aggregates in the fiber.
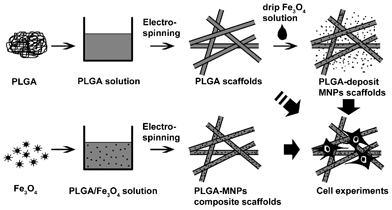 | ||
| Fig. 2 Schemes of fabricating different scaffolds for cell experiements. PLGA with/without MNPs in CH2Cl2/DMF solvent was used to fabricate PLGA scaffolds and PLGA-MNPs composite scaffolds. Some PLGA scaffolds were subsequently used to make PLGA-deposit MNPs scaffolds by dripping MNP solution onto their surface and evaporating the solvent. Finally, these scaffolds were cultured with osteoblast cells on the surface for cell experiment studies. | ||
 | ||
| Fig. 3 SEM images of various scaffolds: (a) PLGA, (b) PLGA-10% QDs, (c) PLGA-10% MNPs, (d) PLGA-50% MNPs and (e) PLGA-deposit MNPs. Photographs of each kind of scaffold are added as insets in the corresponding SEM images. | ||
 | ||
| Fig. 4 TEM images of (a) PLGA-10% MNPs and (b) PLGA-50% MNPs scaffolds with part of each image amplified for clarity. Fibers were collected on TEM copper net and the dispersion of MNPs in the fibers was measured. | ||
3.3. Composition and magnetization of composite scaffolds
To quantify the content of MNPs in composite scaffolds, thermal decomposition processes were measured by TGA and are presented in Fig. 5 with the corresponding data summarized in Table 1. For PLGA scaffolds (Fig. 5a), there was only one decomposition step for the polymer, from 280 to 350 °C. In contrast, composite scaffolds exhibited multiple decomposition steps during the heating process (Fig. 5b, c, d), because of the different decomposition steps of PLGA and MNPs. MNPs showed a strong primary mass loss at 350 °C followed by a second mass loss peak at the higher temperature of 650 °C (Fig. 5e). With MNP content increased in different composite scaffolds (Fig. 5b, c, d), a decreased primary mass loss at 250–300 °C, followed by a significantly increased second mass loss at 300–400 °C could be observed, which corresponded to the decompositions of PLGA and the outer layer oleic acid of the MNPs respectively. In addition, weight loss over 600 °C originated from the inner layer oleic acid of the MNPs, due to the greater bonding strength of the ligand to the surface of the MNPs. These desorption and decomposition patterns were in good agreement with that for pure oleic-acid-stabilized MNPs (Fig. 5e). After measurement, the remained mass weight of iron oxide increased according to increasing MNP content in the composite scaffold. However, for PLGA-10% MNPs and PLGA-50% MNPs scaffolds, the calculated mass fractions of MNPs to PLGA were lower than the theoretical value (Table 1). PLGA-deposit MNPs scaffolds possessed the largest MNP content, greater than 30%, among the composite scaffolds.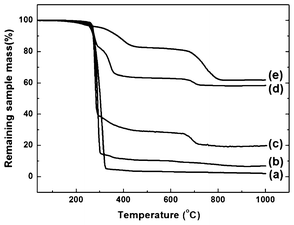 | ||
| Fig. 5 TGA curves of (a) PLGA, (b) PLGA-10% MNPs, (c) PLGA-50% MNPs, (d) PLGA-deposit MNPs scaffolds and (e) MNPs. | ||
Fig. 6 shows the magnetization curves of composite scaffolds. The corresponding saturation magnetization (Ms) values are summarized in Table 1. Similarly to the hysteresis loop of the MNPs in Fig. 1d, no remanence or concavity was observed, because Ms became 0 emu g−1 when the magnetic field came down to 0 Oe. The magnetization curves of the scaffolds were reversible S-shaped increasing functions, meaning that the superparamagnetic property was successfully retained for different composite scaffolds. The Ms value increased from 4 to 10 emu g−1 as the MNP content increased, which was in good agreement with TGA results.
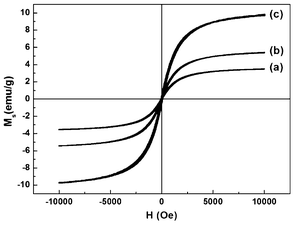 | ||
| Fig. 6 VSM measurements of (a) PLGA-10% MNPs, (b) PLGA-50% MNPs and (c) PLGA-deposit MNPs scaffolds at room temperature. | ||
3.4. Cell morphology
Cell morphology was observed using fluorescence microscopy, as shown in Fig. 7. The projected cell area is presented in Fig. 8. From the fluorescence microscopy observation of Ros17/2.8 cells (Fig. 7a), cell number and projected area were similar in all groups after 12 h culture, and the cells did not spread much. After 24 h culture, the cells spread much more than before. The semi-quantification results showed that the control group had the biggest projected cell area, and the PLGA-10% MNPs group possessed a bigger one than the PLGA group (Fig. 8a). For MC3T3-E1 (Fig. 7b), the cells spread better in PLGA-10% MNPs group and control groups than PLGA scaffolds after the initial 4 h. After 24 h culture, there was no significant difference between the PLGA and PLGA-10% MNPs groups, while the control group showed the biggest projected cell area (Fig. 8b). In summary, the control group had the largest projected cell area in both kinds of cells, and the composite scaffold promoted faster and better cell attachments compared with the PLGA group, especially at the early incubation time of less than 12 h.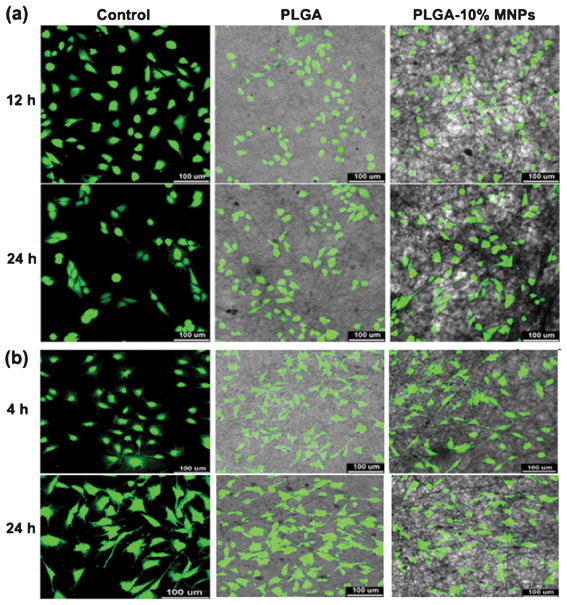 | ||
| Fig. 7 Fluorescence microscopy images of (a) Ros17/2.8 and (b) MC3T3-E1 cells cultured in well plates (control), PLGA and PLGA-10% MNPs scaffolds. The cells were stained with FDA after 12 and 24 h culture for Ros17/2.8 and 4 and 24 h for MC3T3-E1, and were photographed by CLSM. | ||
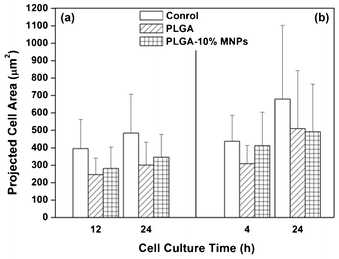 | ||
| Fig. 8 Projected cell area in control (white column), PLGA (strip column) and PLGA-10% MNPs scaffolds (grid column) groups for (a) Ros17/2.8 cells and (b) MC3T3-E1 cells. Osteoblast cells cultured on different groups were stained by FDA at different culture times and the projected cell areas were measured using software (Image-Pro Plus Version 6.0.0.260). | ||
3.5. Cell proliferation
Cell proliferation is an important factor for assessing the influence of composite scaffolds on osteoblast cells. Cell viability was determined using a CCK-8 kit after 2, 4 and 6 days culture, and the values of different groups were normalized with that of the control (Fig. 9). Impressively, both Ros17/2.8 and MC3T3-E1 cells proliferated significantly faster in PLGA-10% MNPs than in PLGA scaffold groups and this difference became significant after 4 days of culture (Fig. 9a, p < 0.05).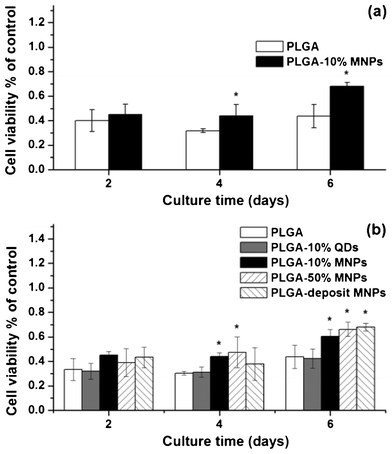 | ||
| Fig. 9 Cell proliferation of (a) Ros17/2.8 and (b) MC3T3-E1 cells in PLGA (white column), PLGA-10% QDs (gray column), PLGA-10% MNPs (black column), PLGA-50% MNPs (left twill column) and PLGA-deposit MNPs (right twill column) scaffold groups after 2, 4 and 6 days culture. Cell viability was measured using a CCK-8 kit and the values of the above composite scaffold groups were normalized with that of the control group. (* p< 0.05) | ||
To assess the effect of MNP content on cell viability, MC3T3-E1 cells were further used to investigate cell proliferation on different scaffolds (Fig. 9b). PLGA-10% QDs scaffolds without magnetic response were used as comparison, because the QDs had similar dimensions and surface oleic acid to those of the MNPs, but had neglegible magnetic response. PLGA-50% MNPs and PLGA-deposit MNPs scaffolds, which had much higher MNP content compared with PLGA-10% MNPs scaffolds, were also used in the cell proliferation investigation. Interestingly, the osteoblast cells proliferated significantly faster on all magnetic composite scaffolds than PLGA-10% QDs and PLGA scaffolds after 4 and 6 days culture. PLGA-10% QDs and PLGA scaffolds showed the lowest and similar cell viability. After 6 days culture, the cell numbers increased in accordance with the increasing MNP content in composite scaffolds as follows: PLGA ≈ PLGA-10% QDs < PLGA-10% MNPs < PLGA-50% MNPs < PLGA-deposit MNPs.
3.6. Cell cycle
Compared with the control group, both PLGA and PLGA-10% MNPs scaffold groups increased the percentage of Ros17/2.8 and MC3T3-E1 cells in G1 phase (Fig. 10) and these data are presented in Table 2. Interestingly, the percentage of cells in G1 phase on PLGA-10% MNPs scaffolds was significantly less than that of PLGA scaffolds. Simultaneously, the percentage of cells in S phase on PLGA-10% MNPs scaffolds was significantly more than that of PLGA scaffolds. There was no apoptosis peak for all groups, indicating that the scaffolds affected the cell viability state without leading to cell apoptosis.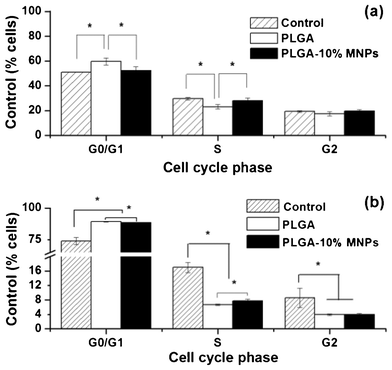 | ||
| Fig. 10 Cell cycle of (a) Ros17/2.8 and (b) MC3T3-E1 in control (stripe column), PLGA (white column) and PLGA-10% MNPs (black column) scaffolds groups. Tens of thousands of cells were measured in each group. (* p < 0.005) | ||
| Cell cycle phase | Ros17/2.8 (%) | MC3T3-E1 (%) | ||||
|---|---|---|---|---|---|---|
| Control | PLGA | PLGA-10% MNPs | Control | PLGA | PLGA-10% MNPs | |
| G0/G1 | 51.1 | 59.7 | 52.3 | 74.4 | 89.4 | 88.3 |
| S | 29.6 | 23.1 | 28.1 | 17 | 6.7 | 7.7 |
| G2 | 19.3 | 17.2 | 19.6 | 8.6 | 3.9 | 4 |
3.7. Cell differentiation
MC3T3-E1 cells could differentiate under the influence of ascorbic acid and dexamethasone, presenting proliferation, differentiation and maturation stages successively. In the proliferation stage, the cells synthesize little ALP, OC and Col I, but the cells enter the differentiation stage after 14 days culture and begin to synthesize more bone characteristic proteins. To assess the differentiation potential of MC3T3-E1 cells on composite scaffolds, characteristic protein contents of osteoblast cells (ALP, OC and Col I) were measured and normalized with the amounts of total protein in cells (Fig. 11). Results showed that characteristic protein contents were very low in all groups, indicating unsuccessful differentiation of MC3T3-E1 cells in all groups. At the same time, there were no significant differences in the characteristic proteins contents among control, PLGA and PLGA-10% MNPs scaffolds groups.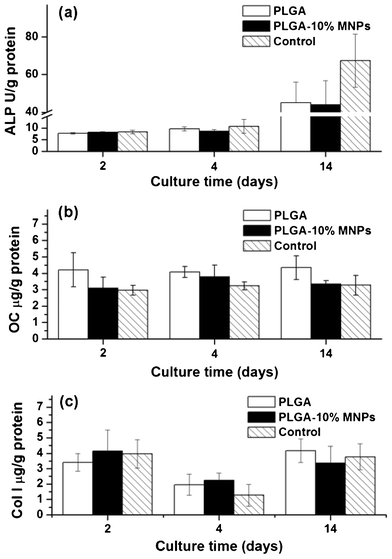 | ||
| Fig. 11 Characteristic protein contents of MC3T3-E1 cells in PLGA scaffolds (white colume), PLGA-10% MNPs scaffolds (black colume) and control (stripe colume) groups. After 2, 4 and 14 days culture, the cells in these groups were lysed and the characteristic proteins: (a) ALP, (b) OC and (c) Col I were analyzed using ELISA method. | ||
4. Discussion
The magnetic field has proved to be quite effective and is widely used in bone repair.35 Some researchers have found that a magnetic field could promote the proliferation of osteoblast cells.36,37 This study attempted to combine superparamagnetic MNPs and PLGA bulk materials to fabricate composite scaffolds via electrospinning method, and to investigate the effects of superparamagnetic scaffolds on the morphology, proliferation, differentiation and cell cycle of Ros17/2.8 and MC3T3-E1 osteoblast cells without an external magnetic field.There are several methods for the synthesis of MNPs, such as thermal decomposition, co-precipitation and hydrothermal methods.13 Herein, we chose a thermal decomposition method to synthesize 6 nm hydrophobic superparamagnetic MNPs. In this way, surface oleic acid contributed excellent monodispersibility to the MNPs in a non-polar solvent, CH2Cl2, which was a preferred solvent for PLGA. The small diameter and narrow size distribution of the MNPs not only afforded them excellent superparamagnatism and reproducibility in the following work, but also had little effect on the smoothness of the fibers at low MNP contents.
Through optimization of electrospinning parameters (e.g. solvent, solution concentration, voltage), we used 5 wt% PLGA in CH2Cl2/DMF (7/3 in v/v) for all electrospining experiments. The working voltage was 20 kV while the feeding rate was 1 mL h−1 and the collecting plate was 20 cm away from the needle tip. The diameters of the fibers ranged from 400 to 600 nm, which could be easily reproduced in order to exclude the influence of fiber size on cell proliferation.38 For the fabrication of composite scaffolds, we firstly used a physical blend method to make PLGA-10% MNPs, PLGA-50% MNPs and PLGA-10% QDs scaffolds. The hydrophobic nanoparticles could homogeneously disperse in the PLGA solution at low content. But it was difficult to produce composite scaffolds with a theoretical feed greater than 50% MNPs in weight. One reason is because oleic-acid-coated MNPs tended to agglomerate in polar solvent DMF above this high content and precipitate before electrospinning. Another one is that the solution of high MNP content was too viscous to be electrospun. TEM images proved that some MNP aggregates had formed in PLGA-50% MNPs but not in PLGA-10% MNPs scaffolds (Fig. 4). In fact, the actual weight percentage of MNPs in PLGA-50% MNPs scaffolds was only 30.2%, which was lower than the theoretical feed of 50% (Table 1). In order to make composite scaffolds with higher MNP content, we fabricated PLGA-deposit MNPs scaffold by surface depositing ferrofluid and successfully increased their MNP content up to ∼60% (Table 1). Both blend and deposit methods facilitated maintenance of superparamagnetism for the MNPs in the composite scaffolds. Higher MNP content led to a higher Ms value of the scaffolds, but the Ms values did not increase proportionally because of the different physical properties and measurement manipulation between powders and nanofilms.
In the cell experiment, we first used fluorescence microscopy to investigate cell morphology and attachment on composite scaffolds. Considering the slower adherence of Ros17/2.8 than MC3T3-E1 cells to the culture plate, different culture times were introduced for both types of cells. Results showed that attachments of both cells in the scaffolds were slower than those of the control (culture plate), but appeared similar after a longer culture time (Fig. 7 and 8). Notably, the projected cell area of the composite scaffolds group was larger than that of the PLGA scaffolds group at the early culture time, indicating that composite scaffolds could enhance cell attachment during the initial period of cultivation. Previous studies found that a magnetic field could affect the nucleation and growth of protein crystals and intramembrane protein distribution in cell cultures.39,40 In this research, one reason for the influence of the composite scaffolds on the cell attachment might be the change of the protein species and configuration on the scaffolds.
According to previous studies, an alternative magnetic field could affect cell proliferation while a static magnetic field only accelerated the cell proliferation during the first 4 days culture.41 In our study without a magnetic field, the cells proliferated significantly faster merely on the composite scaffolds composed of MNPs. For PLGA-10% QDs scaffold, although QDs had the same surface properties and particle size as the MNPs, the scaffold showed a similar impact on cells as did the pure PLGA scaffold, and did not have the capability of accelerating osteoblast cell proliferation (Fig. 9). Therefore, the introduction of MNPs into PLGA scaffolds was the key factor in increasing cell proliferation. However, composite scaffolds with different MNP contents exhibited slightly different bio-effects. After 6 days culture, the cell numbers increased in accordance with the increasing MNP contents in composite scaffolds as follows: PLGA ≈ PLGA-10% QDs < PLGA-10% MNPs < PLGA-50% MNPs < PLGA-deposit MNPs (Fig. 9b). We used atomic absorption spectrophotometry to analyze culture media and cell lysates during the cell proliferation experiment. The Fe content was similar to fresh culture media and did not change with incubation time, indicating that no MNPs were released from the composite scaffolds (data not shown). Meanwhile, the degradation of PLGA could be neglected in the experiment, according to previous reports.42 It is the magnetism property of scaffolds rather than cellular endocytosis of the MNPs that affects cell proliferation. This result indicated that the magnetic effect of the composite scaffolds on cells was different from previous researches using a static magnetic field,41 because each superparamagnetic MNP in the scaffold was a single magnetism domain with its magnetic direction changing constantly. The bulk material PLGA could be considered as glue, which combined lots of tiny magnets into nanometer fibers, as shown in Fig. 2.
The control of cell growth is complex, because not only is it stimulated by growth factors, but it is also regulated by the energy state of the cell and numerous nutrients, such as amino acids. Cell cycle signaling is responsible for the control of a sequence of events that occur when a cell is activated to grow and divide. Cell cycle investigation would provide deep understanding of how the composite scaffolds affect cell proliferation. Up-regulation of cyclin-dependent kinase (CDK) is a key factor in cell propagation. According to previous studies, a magnetic field could influence the Ca2+ influent into cells, and subsequently affected the Ca2+/Calmodulin (CaM) function and CDK activity.43 Meanwhile, even an extremely small magnetic field could affect protein phosphorylation related to Ca2+/CaM.44,45 For PLGA scaffolds in this research, there was a G1 phase arrest as indicated by the increase of cell number in G1 phase for both cell lines, which accounted for the decrease in cell proliferation. But the composite scaffolds decreased the percentage of cells in G1 phase, which accounted for the increase in cell growth compared with PLGA scaffolds (Fig. 9 and Fig. 10). This effect was more noticeable in Ros17/2.8 than in MC3T3-E1 cells. Some researchers found that a magnetic field could stimulate cells to enter G1 phase, and the effects differentiated in different cell lines.46,47 Our results indicated that the composite scaffolds could induce osteoblast cells to depart from G1 phase and enter S phase. The reason for this might be the effect of the scaffolds’ magnetism on the culture medium protein or cell membrane proteins, which is still under investigation.
Although a composite fiber could significantly speed proliferation of osteoblast cells, it was not capable of promoting MC3T3-E1 cell differentiation. Cells can switch from proliferation to differentiation through the combined effect of various genes and transcription factors. Cell cycle change was one reason for the cell proliferation accelerating. For the induction of differentiation in MC3T3-E1 cells, combining the composite scaffold with other stimulates would be a better idea. Therefore, the composite scaffolds composed of MNPs could be considered an ideal substrate for accelerating tissue repair and osteoblast cell proliferation. Further research on in vivo experiments and molecular studies are still in progress in our group.
5. Conclusions
In this study, we combined hydrophobic MNPs and PLGA to fabricate composite scaffolds by electrospinning method and explored their potential usage in tissue engineering without an external magnetic field. The composite scaffolds composed of PLGA and different contents of 6 nm superparamagnetic MNPs (10%, 50% and 60%) were synthesized and investigated for their morphology and magnetism property. The fiber diameters of the scaffolds were uniformly between 400 to 600 nm. The MNPs were well dispersed in the composite scaffold at low content (PLGA-10% MNPs), but showed a little aggregation at higher MNP content (PLGA-50% MNPs). All composite scaffolds successfully retained superparamagnetism at room temperature. Their saturation magnetizations were 3–10 emu g−1 and increased with increasing MNP content. Cell experiment results indicated that the composite scaffolds were biocompatible and enhanced osteoblast cell attachment (Ros17/2.8 and MC3T3-E1) at an early culture time compared with PLGA scaffolds. More importantly, combining MNPs into composite scaffolds could significantly promote cell proliferation, and the effects varied slightly with different MNP contents. Compared with PLGA scaffolds, the composite scaffolds could effectively decrease the percentage of cells in G0/G1 phase and increase that in S phase, which led to the acceleration of cell proliferation. However, the composite scaffolds did not induce differentiation of MC3T3-E1 cells. In conclusion, the composite scaffolds, which had a fibrous and porous structure and superparamagnetic property, could promote bone cell attachment and accelerate cell proliferation, showing greatly promising usage in repair of defects.Acknowledgements
The authors would like to thank National Basic Research Program of China (National 973 program, No. 2011CB606206), the National Natural Science Foundation of China (31070849, 50830105, 51133004), the Department of Science and Technology of Sichuan Province (2009HH0001, 2009SZ0137), the Ministry of Science and Technology (2010DFA51550), and European Commission Research and Innovation (PIRSES-GA-2011-295218)References
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- H. A. von Recum and T. J. Sill, Biomaterials, 2008, 29, 1989–2006 CrossRef.
- S. Ramakrishna, F. Yang, R. Murugan and S. Wang, Biomaterials, 2005, 26, 2603–2610 CrossRef.
- P. X. Ma and X. H. Liu, Ann. Biomed. Eng., 2004, 32, 477–486 CrossRef.
- J. H. Elisseeff and B. Sharma, Ann. Biomed. Eng., 2004, 32, 148–159 CrossRef.
- C. Vallbona, C. F. Hazlewood and G. Jurida, Arch. Phys. Med. Rehabil., 1997, 78, 1200–1203 CrossRef CAS.
- C. S. Brown, F. W. Ling, J. Y. Wan and A. A. Pilla, Am. J. Obstet. Gynecol., 2002, 187, 1581–1587 CrossRef.
- D. Man, B. Man and H. Plosker, Plast. Reconstr. Surg., 1999, 104, 2261–2266 CAS.
- M. I. Weintraub, G. I. Wolfe, R. A. Barohn, S. P. Cole, G. J. Parry, G. Hayat, J. A. Cohen, J. C. Page, M. B. Bromberg, S. L. Schwartz and M. R. Grp, Arch. Phys. Med. Rehabil., 2003, 84, 736–746 CrossRef.
- P. M. Wolsko, D. M. Eisenberg, L. S. Simon, R. B. Davis, J. Walleczek, M. Mayo-Smith and T. J. Kaptchuk, Altern Ther Health M, 2004, 10, 36–43 Search PubMed.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120–129 CrossRef.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- R. N. Muller, S. Laurent, D. Forge, M. Port, A. Roch, C. Robic and L. V. Elst, Chem. Rev., 2008, 108, 2064–2110 CrossRef.
- D. Patel, A. Kell, B. Simard, B. Xiang, H. Y. Lin and G. H. Tian, Biomaterials, 2011, 32, 1167–1176 CrossRef CAS.
- S. A. Meenach, J. Z. Hilt and K. W. Anderson, Acta Biomater., 2010, 6, 1039–1046 CrossRef CAS.
- K. Ino, M. Okochi and H. Honda, Biotechnol. Bioeng., 2009, 102, 882–890 CrossRef CAS.
- A. Ito, K. Ino, T. Kobayashi and H. Honda, Biomaterials, 2005, 26, 6185–6193 CrossRef CAS.
- K. Shimizu, A. Ito, M. Arinobe, Y. Murase, Y. Iwata, Y. Narita, H. Kagami, M. Ueda and H. Honda, J. Biosci. Bioeng., 2007, 103, 472–478 CrossRef CAS.
- W. Yao, J. Wen, W. Xiantao, H. Bin, Z. Xiaobo, W. Gang and G. Zhongwei, Biomed. Mater., 2010, 5, 015001 CrossRef.
- J. Meng, Y. Zhang, X. Qi, H. Kong, C. Wang, Z. Xu, S. Xie, N. Gu and H. Xu, Nanoscale, 2010, 2, 2565–2569 RSC.
- Z. J. Yang, H. P. Yuan, W. D. Tong, P. Zou, W. Q. Chen and X. D. Zhang, Biomaterials, 1996, 17, 2131–2137 CrossRef CAS.
- X. B. Zeng, H. Hu, L. Q. Xie, F. Lan, W. Jiang, Y. Wu and Z. W. Gu, Int. J. Nanomed., 2012, 7, 3365–3378 CrossRef CAS.
- H. Wang, H. Tang, J. He and Q. Wang, Mater. Res. Bull., 2009, 44, 1676–1680 CrossRef CAS.
- S. Wang, C. Wang, B. Zhang, Z. Sun, Z. Li, X. Jiang and X. Bai, Mater. Lett., 2010, 64, 9–11 CrossRef CAS.
- T. Song, Y. Zhang, T. Zhou, C. T. Lim, S. Ramakrishna and B. Liu, Chem. Phys. Lett., 2005, 415, 317–322 CrossRef CAS.
- T. Amna, M. S. Hassan, K. T. Nam, Y. Y. Bing, N. A. Barakat, M. S. Khil and H. Y. Kim, Int. J. Nanomed., 2012, 7, 1659–1670 CrossRef CAS.
- O. Jordan, P. E. Le Renard, A. Faes, A. Petri-Fink, H. Hofmann, D. Rufenacht, F. Bosman, F. Buchegger and E. Doelker, Biomaterials, 2010, 31, 691–705 CrossRef.
- W. Yan, Z. Xuehui, S. Yu, H. Bing, H. Xiaoyang, W. Xinzhi, L. Yuanhua and D. Xuliang, Biomed. Mater., 2011, 6, 055008 CrossRef.
- S. H. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. X. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- H. U. Bergmeyer and K. Gawehn, Methods of enzymatic analysis, Verlag Chemie; New York : Academic Press, Weinheim, 1974 Search PubMed.
- L. M. Bronstein, X. L. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- H. C. Gu, L. Zhang and R. He, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef.
- R. Y. Hong, S. Z. Zhang, Y. P. Han, H. Z. Li, J. Ding and Y. Zheng, Powder Technol., 2006, 170, 1–11 CrossRef CAS.
- F. H. Lin, S. Y. H. Wu and C. L. Tseng, J. Nanopart. Res., 2010, 12, 1173–1185 CrossRef.
- A. A. Pilla, in Handbook of Biological Effects of Electromagnetic Fields, ed. F. S. B. a. B. Greenebaum, Taylor & Francis Group, Third edn, 2007 Search PubMed.
- B. D. Boyan, C. H. Lohmann, Z. Schwartz, Y. Liu, Z. Li, B. J. Simon, V. L. Sylvia, D. D. Dean, L. F. Bonewald and H. J. Donahue, J. Orthop. Res., 2003, 21, 326–334 CrossRef.
- G. Ito, P. Diniz, K. Shomura and K. Soejima, Bioelectromagnetics, 2002, 23, 398–405 CrossRef.
- A. S. Goldstein, A. S. Badami, M. R. Kreke, M. S. Thompson and J. S. Riffle, Biomaterials, 2006, 27, 596–606 CrossRef.
- F. Bersani, F. Marinelli, A. Ognibene, A. Matteucci, S. Cecchi, S. Santi, S. Squarzoni and N. M. Maraldi, Bioelectromagnetics, 1997, 18, 463–469 CrossRef CAS.
- G. Sazaki, E. Yoshida, H. Komatsu, T. Nakada, S. Miyashita and K. Watanabe, J. Cryst. Growth, 1997, 173, 231–234 CrossRef CAS.
- T. Eremenko, C. Esposito, A. Pasquarelli, E. Pasquali and P. Volpe, Bioelectromagnetics, 1997, 18, 58–66 CrossRef CAS.
- J. A. Hunt and Z. G. Tang, Biomaterials, 2006, 27, 4409–4418 CrossRef.
- A. R. Means and C. R. Kahl, Endocr. Rev., 2003, 24, 719–736 CrossRef.
- S. Engstrom, M. S. Markov, M. J. McLean, R. R. Holcomb and J. M. Markov, Bioelectromagnetics, 2002, 23, 475–479 CrossRef CAS.
- M. S. Markov and A. A. Pilla, Bioelectrochem. Bioenerg., 1997, 43, 233–238 CrossRef CAS.
- A. Xu, G. P. Zhao, S. P. Chen, Y. Zhao, L. Y. Zhu, P. Huang, L. Z. Bao, J. Wang, L. Wang, L. J. Wu and Y. J. Wu, Plasma Sci. Technol., 2010, 12, 123–128 CrossRef.
- L. H. Qiu, X. N. Tang, M. Zhong and Z. Y. Wang, Shanghai Kou Qiang Yi Xue, 2004, 13, 469–470 Search PubMed.
Footnote |
| † These authors made an equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2012 |