Laser dye doped nanoparticles for highly photostable optical nanoamplifiers†
Angelo
Monguzzi
*a,
Michel
Frigoli
b,
Chantal
Larpent
b and
Francesco
Meinardi
a
aDipartimento di Scienza dei Materiali, Università Milano Bicocca, via R. Cozzi 53, 20125 Milano, Italy. E-mail: angelo.monguzzi@mater.unimib.it; Fax: +39 02 64485400; Tel: +39 02 64485173
bInstitut Lavoisier UMR-CNRS 8180, Université de Versailles, Versailles-Saint Quentin en Yvelines 45, Avenue des Etats-Unis, 78035 Versailles Cedex, France
First published on 2nd October 2012
Abstract
Much research on organic advanced materials is currently focused on reducing the size of optical devices and fluorescent probes, and, moreover, on avoiding the photobleaching processes which hinder their extensive use for durable devices. To fulfil both goals, we propose the encapsulation of the laser dye pyrromethene 567 in small (<30 nm) polymeric nanoparticles. These ultra-stable nanoshells are used as vessels which efficiently protect included compounds from the external environment, and especially from O2, by transforming the doped NPs themselves into a durable optically active gain material. An improved resistance to photobleaching has been observed, while the unbeaten gain properties of the liquid lasing system are fully preserved after encapsulation. Their small size and their amplification efficiency make fluorescent NPs particularly suitable for biological imaging and drug delivery applications when the tissue porosity requires nanometre size vectors, such as in solid tumors.
Introduction
The attractive properties of organic semiconductors such as high photoluminescence quantum yield (PLQY), broad emission spectra across the visible range and ease of fabrication make them attractive gain media for exploitation in solid state devices for lasers, optical communication technology and sensing, which is also thanks to the rapid growth of nanophotonics.1–13 Moreover, the application of nanoparticles (NPs) for the delivery and targeting of organic pharmaceutical, therapeutic and diagnostic agents has received significant attention in recent years. NPs may be constructed from a wide range of materials and used to encapsulate or solubilize chemotherapeutic agents for improved delivery in vivo or to provide unique optical, magnetic and electrical properties for imaging and therapy. In particular, dye loaded NPs have achieved great popularity in cancer imaging in recent years because of their attractive optical properties. As a matter of fact, compared to traditional organic dyes and fluorescent proteins, the large family of dye loaded NPs offers i) an improved photostability (suitable for imaging studies at cellular, subcellular, and even molecular level, and needed for implementation in photonic devices), ii) improved sensitivity (as NPs can be optical amplifiers), iii) multiplexed imaging (multiple markers can be imaged using different color dye-loaded NPs), iv) targetability (NP surfaces can be functionalized to attach to targeting molecules), and v) biocompatibility (by selecting suitable polymers or by modifying the NP surface with biocompatible molecules/polymers).While great progress has been made in reducing the size of optically active materials and in implementing the long desired electrical pumping,14 the photobleaching (photoinduced degradation) of organic species hinders their extensive use for the fabrication of endurable, cheap photonic devices and/or long term stable optical probes. Generally, photobleaching occurs from both photothermal decomposition (i.e. it depends on the ability of the dye to dissipate the thermal energy accumulated during the high intensity excitation required for lasing) and photochemical degradation.15,16 Although the latter is not a simple mechanism, it is known that singlet oxygen plays a major role in the photodegradation of organic molecule based optoelectronics.17 The sealed packaging of devices or the use of antioxidants improves the photostability,18 but more efficient protection is demanded to develop solid devices which do not require the commonly used dye solution to be refreshed to eliminate bleached emitters.19 This lack of photostability is a key point even for laser dyes belonging to the pyrromethene (Pyr) family, which receives particular attention owing to its high lasing efficiency.17,20,21 Pyr photobleaching depends on the host composition and particularly on the degree of environmental O2 pollution. The main photobleaching mechanisms include self-sensitized and radical-sensitized photo-oxidation20 in which photo-sensitized singlet oxygen reacts with the Pyr ground state leading to permanent fluorophore bleaching.22
To improve organic dye photostability, here we propose the encapsulation of the laser dye pyrromethene 567 (Pyr567) in a polymeric nanoparticle (NP). Particles are used as nano-vessels, each one containing several luminescent molecules entrapped in fixed positions within the rigid and disordered host matrix. These nanoshells have been recently exploited to host different chromophore species in order to avoid quenching by O2 and allow multi-photon processes in solid matrixes. The results obtained demonstrated that the electronic properties of the chromophores are preserved upon the inclusion which fully protect the dyes from the external environment and from O2 by transforming the doped NP itself into a photoactive material.23 Herein, optical absorption, photoluminescence (PL) and optical gain properties of a series of Pyr567 doped NPs (Pyr567–NP) in an aqueous suspension and in films have been studied and compared with the unbeaten performance of Pyr567 in ethanol. A much improved resistance to photobleaching has been observed while the good gain properties of the liquid lasing system are completely preserved within the NP environment. It will be shown that the encapsulation technique proposed avoids dye aggregation, thus allowing high concentrations of ∼1 × 1019 dye molecules cm−3 to be reached without any detrimental quenching affecting the PL and light amplification properties. Each NP behaves as a high-efficiency single molecule emitter, which can be dispersed in a liquid or used as a dopant for polymeric matrices allowing the tuning of the host optical density to fit technological requirements without limits on the active dye content.
In addition to their excellent fluorescence and lasing performance, the small size of the NPs (<30 nm) makes them suitable for exploitation in biological imaging and drug delivery for cancer therapy because only objects with dimensions lower than ∼100 nm show a strong tendency to enter and be retained in solid tumors according to the tissue porosity. We want to underline that the idea of including the active species in an easy to handle rigid and non-toxic nano-vessel is general and adaptable to a large number of O2 sensitive molecules exploited in photonics, and for in vivo imaging and drug delivery applications which require affordable and reliable small vectors.24–26
Materials and methods
1. NP preparation and characterization
An aqueous suspension of NPs was prepared by copolymerization in a microemulsion stabilized with the cationic surfactant DTAB (15 wt%) using a procedure described previously.27–29 Two microemulsions were prepared by the progressive addition of a mixture of the monomers styrene/divinylbenzene: 92/3 mmol (56/1.5 mmol) and 2,2-dimethoxy-2-phenylacetophenone (radical photoinitiator, 1.3 mmol (0.065 mmol)) to 220 g of a 15 wt% solution of DTAB in demineralized water, under gentle magnetic stirring. The resulting microemulsion was gently degassed with nitrogen for 30 min and the polymerization was then carried out under white-light irradiation using two 60 W lamps, at room temperature under nitrogen, for 24 h. Chromatographic analysis indicated complete polymerization of all of the monomers. Stable, bluish translucent, aqueous suspensions of polystyrene nanoparticles were obtained. The average particle diameters were deduced from dynamic light scattering (DLS, Fig. 1). Polymeric nanoparticles with an average diameter of 18 nm with a polydispersity index (PDI) of 0.047 and a diameter of 27 nm with a PDI of 0.046 were obtained. The particle contents in the suspensions were 2 wt% and 4 wt% for NPs of 18 nm and 27 nm respectively. Recently, we have shown that the size of the polymeric NPs determined by DLS is perfectly in agreement with the size determined by TEM.30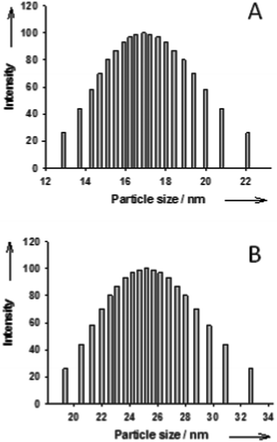 | ||
| Fig. 1 Particle size distribution of NPs with an average particle size of 18 nm (A) and NPs with an average particle size of 27 nm (B). | ||
2. NP doping
The doping (Fig. 2, panel A) was carried out with a particle content in the suspensions of 2 wt%. Therefore, prior doping, the suspension with NPs of 27 nm diameter was diluted twice with a 15 wt% aqueous solution of DTAB. 40 μL of a DCM solution containing Py567 at different concentrations was added to 2 mL of an aqueous suspension of the nanoparticles (2 wt%; 15% DTAB). The resulting mixtures were stirred at room temperature for 3 h in a capped sample tube. The tube was then uncapped and the solution was stirred for an additional 12 h to allow the evaporation of the DCM. The suspension was filtered through a 0.2 μm MF-MilliporeTM filter before use. The volumes and concentrations of the solutions of PM567 used for the loadings as well as the amount of PM567 encapsulated in the NP deduced from absorption spectra are detailed in the ESI,† Table S1.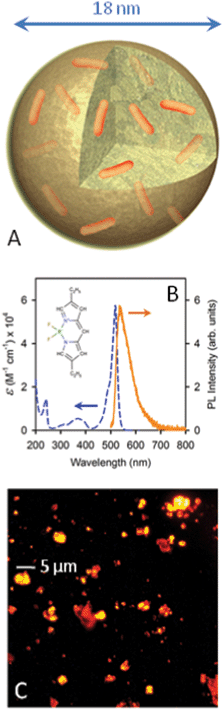 | ||
| Fig. 2 (A) Sketch of a Pyr567 (orange sticks) doped NP. (B) Molar extinction coefficient (dashed line) and PL spectrum (solid line) of a Pyr567 solution in ethanol (1.65 × 10−5 M) under 473 nm CW excitation. Inset: Pyr567 molecular structure. (C) Fluorescence image of a diluted Pyr567–NP cast film under 532 nm excitation. | ||
3. Photophysical studies
Optical absorption measurements were carried out using a Cary Varian 50 Spectrometer with a bandwidth of 1 nm and by using quartz cuvettes with an optical path of d = 0.1 cm. For steady-state PL measurements, a Coherent Verdi TEM00 CW laser at 532 nm (2.33 eV) and Roithner LaserTechnick RLTMBL-473-10 SSDL at 473 nm (2.62 nm) were used as excitation source. A nitrogen-cooled Spex 2000 charge coupled device (CCD) coupled to a polychromator (Triax 190 from J-Horiba) was used for the signal detection, with a bandwidth of 0.5 nm. For high power pulsed excitation and time-resolved (TRPL) measurements, we employed the II harmonic of a Nd:YAG laser at 532 nm (2.33 eV, Minilite Continuum) as light source and a Edimburgh LP920 Flash Photolysis System with time resolution <2 ns for signal decay detection. Laser stray-light was reduced by using a Edmund Optics 532 nm notch filter. All measurements were performed at room temperature (RT), and were corrected for the instrumental spectral response. PL quantum yield (PLQY) has been measured by using Rhodamine 6G (R6G) as fluorescence standard.31Results and discussion
Fig. 2B shows the Pyr567 molar extinction coefficient ε measured in ethanol solution and its PL spectrum under 473 nm CW excitation and Fig. 2C reports an image of highly fluorescent Pyr567–NP aggregates obtained by drop casting a diluted NP aqueous dispersion. Dyes have been embedded within cross-linked polystyrene-based NPs of 18 or 27 nm in diameter, obtained as stable aqueous suspensions by polymerization in an oil–water microemulsion stabilized with a cationic surfactant (dodecyltrimethylammonium bromide, DTAB).29 The embedding process within the NP core was achieved by swelling the NP with a dichloromethane (DCM) solution of Py567 at different initial concentrations followed by the evaporation of the solvent.27,28 The loading of each NP was calculated by considering that the ε value of Py567 in toluene solution is the same as that in the NP core. (ESI,† Table S1). Fig. 3A reports the Pyr567–NP absorption spectra. Centred in the green region of the visible spectrum, the absorption band presents its maximum at 524 nm, with an edge at 300 nm due to the NPs.32 The maximum is slightly red shifted by about 4 nm with respect to the ethanol solution, indicating that the NP environment does not significantly affect the vibronic ground state of the dye. PLQYs of the doped particles have also been measured to verify possible quenching effects due to the inclusion. Fig. 3B reports the PL spectra of the Pyr567–NP series, of Pyr567 in ethanol solution and of R6G in ethanol solution used as a standard, with 473 nm CW excitation. The data show the typical yellowish emission of Py567 with a slight red shift of the PL maxima due to self-absorption (ESI,† Fig. S2–S3). For the Pyr567–NP series, the PLQY doesn’t change significantly with the number of Pyr per particle ranging from 0.81 to 0.84 (±5%) and is very close to the PLQY value obtained in ethanol solution (0.84 ± 5%).31 These results demonstrate that NPs are suitable hosts for visible light emitting dyes because i) they are optically transparent in the spectral range considered and ii) the PL properties of the included chromophores are preserved. Moreover, iii) the inclusion protocol allows access to high local concentrations over the limits of the chromophore solubility in common organic solvents and polymers. Indeed, within Pyr567–NPs, a dye concentration as high as 8.29 × 1018 dye molecules cm−3 has been reached without any significant evidence of aggregation or concentration dependent quenching. The PL lifetime at 567 nm (inset of Fig. 3B) is the same for the whole sample series, demonstrating that no concentration related phenomenon occurs. These results open the way to the fabrication of photoactive polymeric materials based on high chromophore content thus promoting material waste reduction and a further miniaturization of photonic devices.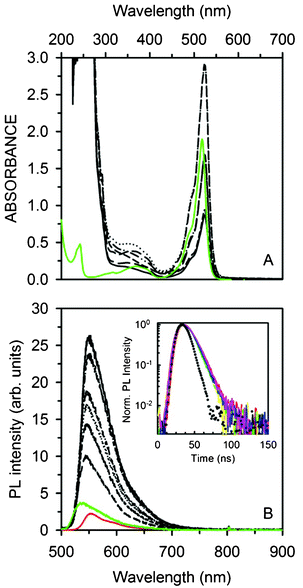 | ||
| Fig. 3 (A) Absorption spectra of the NP series (black lines) and Pyr567 ethanol solution (3.30 × 10−4 M, green solid line). (B) PL spectra of the NP series (black lines) compared to Pyr567 (1.65 × 10−5 M, green line) and R6G (3.20 × 10−4 M, red line) ethanol solutions under 473 nm CW excitation (normal incidence, 0.63 mW cm−2). Inset: NP luminescence decay at 567 nm (solid lines). The dotted curve is the laser pulse at 532 nm used for TRPL measurements. | ||
It is of course necessary to assess the stability of the proposed material under O2 exposure to estimate the photo-oxidation effectiveness of the Pyr567. PL intensities of all the samples have been monitored for 20 h under uninterrupted intense CW excitation at 532 nm in air (31.25 mW cm−2). None of the Pyr567–NPs undergo degradation or show a loss in PL efficiency (ESI,† Fig. S4). To unambiguously demonstrate the protection from O2 provided by the NP shell and to show the versatility of our strategy, rubrene doped NPs have been prepared as a reference system for photo-oxidation. In fact, rubrene can be used as an oxygen probe because it forms a colourless peroxide by reacting rapidly with singlet oxygen, thus losing its PL when excited at 532 nm.33,34 Moreover, in a DTAB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O dispersion rubrene forms stable nanocrystals within micellae of DTAB, which cannot be removed from the mother solution and which can be used as a model system of unprotected dyes.35 Despite the fact that the PL spectra of doped NPs, nanocrystals and rubrene in tetrahydrofuran (THF) solution are almost identical (Fig. 4B), the PL decay time of rubrene in the NPs (τnp = 20 ns) is longer than that obtained for the rubrene nanocrystals (τnc = 15 ns) or rubrene in THF (τsol = 12 ns) (Fig. 4A). This evidence suggests that all rubrenes are included within NPs. This hypothesis is completely confirmed since the rubrene doped NPs show no sign of degradation upon continuous irradiation for 20 h (Fig. 4C) while, in contrast, the PL intensity of the nanocrystals quickly decreases. Lastly, in ethanol solution rubrene photodegradation is 10 times quicker than for nanocrystals (this might be partially related to the fact that O2 solubility in ethanol is almost 5 times better than in water).36 These results demonstrate that encapsulation in polymeric NPs constitutes a straightforward strategy to efficiently protect dyes from O2.
:H2O dispersion rubrene forms stable nanocrystals within micellae of DTAB, which cannot be removed from the mother solution and which can be used as a model system of unprotected dyes.35 Despite the fact that the PL spectra of doped NPs, nanocrystals and rubrene in tetrahydrofuran (THF) solution are almost identical (Fig. 4B), the PL decay time of rubrene in the NPs (τnp = 20 ns) is longer than that obtained for the rubrene nanocrystals (τnc = 15 ns) or rubrene in THF (τsol = 12 ns) (Fig. 4A). This evidence suggests that all rubrenes are included within NPs. This hypothesis is completely confirmed since the rubrene doped NPs show no sign of degradation upon continuous irradiation for 20 h (Fig. 4C) while, in contrast, the PL intensity of the nanocrystals quickly decreases. Lastly, in ethanol solution rubrene photodegradation is 10 times quicker than for nanocrystals (this might be partially related to the fact that O2 solubility in ethanol is almost 5 times better than in water).36 These results demonstrate that encapsulation in polymeric NPs constitutes a straightforward strategy to efficiently protect dyes from O2.
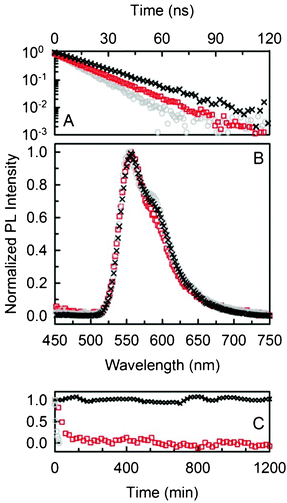 | ||
| Fig. 4 (A) PL decay of rubrene doped NPs (black crosses), nano-crystals (red squares) and THF solution (grey circles). (B) PL spectra of the same samples. (C) Normalized PL intensity as a function of time of exposure to air. | ||
Once we had assessed the effectiveness of the protection of NPs from O2, we verified the optical gain properties of the Pyr567–NPs by comparing their amplification efficiency to the standard Pyr567 in ethanol solution. Fig. 5A reports the PL intensity of the Pyr567 in solution and in the NP dispersion versus the pulse intensity in a quartz cuvette of optical path d = 1 cm, under 532 nm pulsed excitation at 15 Hz (pulse width 10 ns). The quartz edges provide the necessary reflectance to build up a simple Fabry–Perot oscillator. When the gain within the cavity overcomes the losses, the emission efficiency strongly increases as can be seen by monitoring the amplification threshold excitation intensity Ith.19 We also estimated the slope efficiency S of each system considered, here defined as the slope of the straight line of the amplified emission output vs. the pumping intensity (straight lines in Fig. 5A). Both the solution and the NP dispersion show Ith,sol ≈ Ith,disp = 0.8 mJ cm−2 (dashed line) and similarly Ssol ≈ Sdips = 0.54, as expected since the dye emitting properties are not affected by inclusion or by concentration quenching. A better result has been obtained by depositing a 275 μm film of water soluble poly-ethylene oxide (PEO) doped with Pyr567–NP sandwiched between quartz slides to create a cavity with the same reflecting edges as the cuvette. In this case, Ifilm = 0.5 mJ cm−2 (dotted line) is slightly lower than in the previous case, but the ratio Sfilm/Sdisp ∝ 1.4 demonstrates a better amplification efficiency for the film. This can be related to a reduction in the optical losses caused by the configuration or to an enhanced gain given by light trapping within the opaque film which increases the effective optical path L across the active medium.13,37 It is worthwhile to note that, again, the material gain properties are preserved despite the ultra-high dye concentration in the film. A further improvement of the light amplification performance has been achieved by closing the oscillator cavity with a 99% reflectivity aluminum mirror (film–Al). The threshold has been straightforwardly lowered to Ifilm–Al = 6 μJ cm−2 (Fig. 5A, empty green circles, dotted-dashed line, see also ESI† Fig. S6) with Sfilm–Al = 2.6, approximately 5 times higher than Sdisp. The improvement in the cavity quality is so marked that with excitation pulse energies of >1 mJ cm−2 it is possible to reach the saturation excitation fluency of the amplifier, with a reduction of the system gain properties.19 The material’s photodegradation during the time under the lasing working condition can be assessed by using an excitation intensity I ≫ Ith. Fig. 5B shows the emission efficiency as a function of 2 mJ cm−2 excitation pulse numbers. After some thousands of pulses, the PL intensity of the ethanol solution decreases by about 30% while the Pyr567–NP dispersion shows an emission decrease of only 10%.
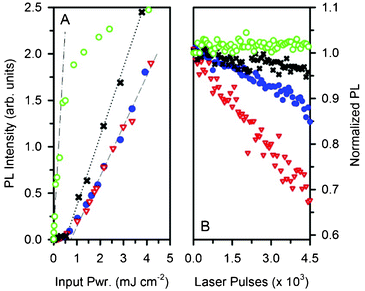 | ||
| Fig. 5 (A) PL intensity as a function of the excitation pulse energy of Pyr567 ethanol solution (red triangles), Pyr567–NP aqueous dispersion (blue dots), PEO:Pyr567–NP film (black crosses) and film–Al (green circles). (B) Normalized PL intensity of the samples shown as a function of the number of pumping cycles. | ||
Excluding photo-oxidation within NPs, we impute this slight degradation to the thermal decomposition of the dye, because of the lower thermal conductivity of the NPs (0.036 W m−1 K−1) with respect to ethanol (0.17 W m−1 K−1).38 In fact, the photodegradation of the sample with the PEO film is reduced to a mere 5% (black crosses), because the use of a less intense energy pulse of 1.3 mJ cm−2 to achieve the same output of light results in less heating of the sample. Finally, in the film–Al sample the dye stability is 100% when using an excitation energy pulse of 50 μJ cm−2.
Conclusions
To summarize, polystyrene based NPs have been successfully loaded with a laser dye in order to develop high performance solid state active photonic devices based on cheap organic materials. The results presented show that i) the electronic and gain properties of the dye are 100% preserved within the NP framework; ii) nano-encapsulation is proposed as a good method to overcome one of the major drawbacks which limits the wide application of organic material, namely photobleaching due to the permanent degradation of the emitting species by photo-oxidation. The NP shell efficiently protects the included moieties from O2 pollution thus eliminating the photo-oxidation mechanism; iii) the inclusion allows high concentrations of the gain medium to be achieved in polymeric films avoiding the detrimental PL quenching due to aggregation or a high density of excited states. Since each NP doped with dye acts as an independent emitting unit, it is possible to control the optical density of the material in order to fulfil the technological requirements, regardless of the limitation imposed by the solubility of common dyes in polymers and organic solvents. These results demonstrate that NPs are an efficient protection shell for a large number of dyes and organic chromophores for photonic and electro-optic applications. Moreover, from a more general point of view, small NPs with tuneable dimensions appear to be affordable water compatible vessels for O2 sensitive compounds and fluorescent probes used in drug delivery, imaging and sensing applications in biological systems where tissue porosity requires ultra-stable vectors with dimensions lower than 100 nm.Acknowledgements
This work was gratefully supported by Fondazione Cariplo. A. M. thanks the fund “Dote Ricercatori”: FSE, Regione Lombardia.References
- D. Moses, Appl. Phys. Lett., 1992, 60, 3215–3216 CrossRef CAS.
- N. Tessler, G. J. Denton and R. H. Friend, Nature, 1996, 382, 695–697 CrossRef CAS.
- F. Hide, M. A. DiazGarcia, B. J. Schwartz, M. R. Andersson, Q. B. Pei and A. J. Heeger, Science, 1996, 273, 1833–1836 CrossRef CAS.
- N. Tessler, Adv. Mater., 1999, 11, 363–370 CrossRef CAS.
- M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS.
- Y. Yang, I. D. W. Samuel and G. A. Turnbull, Adv. Mater., 2009, 21, 3205 CrossRef CAS.
- Y. Yang, G. A. Turnbull and I. D. W. Samuel, Adv. Funct. Mater., 2010, 20, 2093–2097 CrossRef CAS.
- J. Clark and G. Lanzani, Nat. Photonics, 2010, 4, 438–446 CrossRef CAS.
- A. Monguzzi, F. Scotognella, F. Meinardi and R. Tubino, Phys. Chem. Chem. Phys., 2010, 12, 12947–12950 RSC.
- F. Scotognella, A. Monguzzi, F. Meinardi and R. Tubino, Phys. Chem. Chem. Phys., 2010, 12, 337–340 RSC.
- F. Scotognella, D. P. Puzzo, A. Monguzzi, D. S. Wiersma, D. Maschke, R. Tubino and G. A. Ozin, Small, 2009, 5, 2048–2052 CrossRef CAS.
- P. N. Prasad, Nanophotonics, Wiley, Hoboken, NJ, 2004 Search PubMed.
- Y. Yang, G. A. Turnbull and I. D. W. Samuel, Appl. Phys. Lett., 2008, 92, 163306 CrossRef.
- A. N. Fletcher, Appl. Phys., 1978, 16, 93–97 CrossRef CAS.
- A. Otomo, S. Otomo, S. Yokoyama and S. Mashiko, Opt. Lett., 2002, 27, 891–893 CrossRef CAS.
- M. D. Rahn, T. A. King, A. A. Gorman and I. Hamblett, Appl. Opt., 1997, 36, 5862–5871 CrossRef CAS.
- A. Galvan-Gonzalez, K. D. Belfield, G. I. Stegeman, M. Canva, K. P. Chan, K. Park, L. Sukhomlinova and R. J. Twieg, Appl. Phys. Lett., 2000, 77, 2083–2085 CrossRef CAS.
- R. S. Quimby, Photonics and lasers : an introduction, Wiley-Interscience, Hoboken, N.J., 2006 Search PubMed.
- Y. Yang, M. Q. Wang, G. D. Qian, Z. Y. Wang and X. P. Fan, Opt. Mater., 2004, 24, 621–628 CrossRef CAS.
- A. Costela, I. Garcia-Moreno and R. Sastre, Phys. Chem. Chem. Phys., 2003, 5, 4745–4763 RSC.
- Y. Yang, G. D. Qian, D. L. Su and M. Q. Wang, Opt. Commun., 2004, 239, 415–420 CrossRef CAS.
- A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139–143 CrossRef CAS.
- B. E. Cohen, Nature, 2010, 467, 407–408 CrossRef CAS.
- D. J. Irvine, Nat. Mater., 2011, 10, 342–343 CrossRef CAS.
- S. Santra and A. Malhotra, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 501–510 CAS.
- M. Frigoli, K. Ouadahi and C. Larpent, Chem.–Eur. J., 2009, 15, 8319–8330 CrossRef CAS.
- F. Gouanve, T. Schuster, E. Allard, R. Meallet-Renault and C. Larpent, Adv. Funct. Mater., 2007, 17, 2746–2756 CrossRef CAS.
- R. Meallet-Renault, R. Pansu, S. Amigoni-Gerbier and C. Larpent, Chem. Commun., 2004, 2344–2345 RSC.
- C. Larpent, C. Cannizzo, A. Delgado, F. Gouanvé, P. Sanghvi, C. Gaillard and G. Bacquet, Small, 2008, 4, 833–840 CrossRef CAS.
- M. Montalti and S. L. Murov, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, 2006 Search PubMed.
- Y. Assor, Z. Burshtein and S. Rosenwaks, Appl. Opt., 1998, 37, 4914–4920 CrossRef CAS.
- M. Kytka, A. Gerlach, F. Schreiber and J. Kovac, Appl. Phys. Lett., 2007, 90, 131911 CrossRef.
- S. Uttiya, L. Raimondo, M. Campione, L. Miozzo, A. Yassar, M. Moret, E. Fumagalli, A. Borghesi and A. Sassella, Synth. Met., 2012, 161, 2603–2606 CrossRef.
- G. Laurent N. T. Ha-Duong R. Méallet-Renault and R. B. Pansu, in Handai Nanophotonics, ed. M. Hiroshi and K. Satoshi, Elsevier, 2004, 89–101 Search PubMed.
- I. B. Golovanov and S. M. Zhenodarova, Russ. J. Gen. Chem., 2005, 75, 73–78 CrossRef CAS.
- F. Scotognella, D. P. Puzzo, A. Monguzzi, D. S. Wiersma, D. Maschke, R. Tubino and G. A. Ozin, Small, 2009, 5, 2048–2052 CrossRef CAS.
- L. Song, Y. F. Chen and J. W. Evans, J. Electrochem. Soc., 1997, 144, 3797–3800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: additional data on NP synthesis, sizing, doping and PL characterization. See DOI: 10.1039/c2ra22314g |
| This journal is © The Royal Society of Chemistry 2012 |