Ferrocenyl BODIPYs: synthesis, structure and properties†
Prabhat
Gautam
,
Bhausaheb
Dhokale
,
Shaikh M.
Mobin
and
Rajneesh
Misra
*
Department of Chemistry, Indian Institute of Technology Indore, MP 452017, India. E-mail: rajneeshmisra@iiti.ac.in; Fax: +91 7312361482; Tel: +91 7312438710
First published on 29th October 2012
Abstract
A set of donor–π-acceptor–π-donor type ferrocenyl substituted BODIPYs were designed and synthesized by the Sonogashira coupling reaction. These compounds exhibit red shifted absorption and poor fluorescence. The electrochemical properties of these compounds exhibit strong donor–acceptor interactions. The crystal structure of 3b exhibits an extensive hydrogen bonded 2-D network.
Boron-dipyrromethane (BODIPY) dyes have attracted considerable attention because of their distinguished properties, such as high absorption coefficients, high quantum yields and high photostability.1 These dyes are used for a variety of applications in light harvesting molecular arrays, photodynamic therapy, sensors, energy transfer cassettes and photovoltaic devices.2 The electronic properties of the BODIPYs can be tuned by functionalization at the meso-position, as well as at the pyrrolic position. Functionalization of the BODIPY dye at the pyrrolic position perturbs the electronic properties more significantly compared to the meso-position because the pyrrolic site favors a coplanar geometry for the attached group, which maximizes the electronic coupling, whereas the meso-site favors an orthogonal geometry which minimizes the electronic communication.3 The BODIPY unit acts as an electron acceptor.4 It has been established that the structural motifs of D–A–D type compounds show promising nonlinear optical (NLO) behavior.5 Therefore, we were interested in incorporating the donor groups at both ends of the BODIPY dye and to explore its properties. There are many reports where donor groups are attached at the pyrrolic position of the BODIPY.6 Ferrocene is a strong donor and is highly stable.7 In this work, we have incorporated a ferrocenyl group on both the pyrroles of the BODIPY and generated a D–π-A–π-D type molecular system. Here, our aim was to explore the effect of the ferrocene unit on the photophysical and electrochemical behavior of BODIPY on enhancement of the π-conjugation length.
The synthetic route for ferrocenyl substituted BODIPYs 3a–3d is shown in Scheme 1. BODIPY 1 was synthesized following earlier reports.8 The 2,6-diiodo BODIPY 2 was synthesized by the iodination reaction of BODIPY 1.9 The ferrocenyl substituted BODIPYs 3a–3d were synthesised by Sonogashira coupling reactions of diiodo BODIPY 2 with the corresponding ferrocenyl derivatives. Diiodo substituted BODIPY 2 was reacted with ethynylferrocene, 4-ferrocenylphenylacetylene, 3-ferrocenylphenylacetylene and 4-ethynyl-phenylethynylferrocene, which resulted in compounds 3a, 3b, 3c and 3d in 60%, 40%, 50% and 55% yields respectively.
 | ||
| Scheme 1 General procedure for the synthesis of 3a–3d. | ||
The Sonogashira coupling reaction of 2,6-dibromo BODIPY with the corresponding ferrocenyl alkyne resulted in multiple products which were difficult to purify. On the other hand, the Sonogashira coupling reactions of 2,6-diiodo BODIPY 2 with the ferrocenyl derivatives were straight forward and the purification was achieved by column chromatography followed by repeated recrystallization of the product in a mixture of chloroform and ethanol. The isolation of ferrocenyl substituted BODIPY 3b was difficult because of its poor solubility. It is sparingly soluble in chloroform, dichloromethane and toluene and due to this we were able to record the 1H NMR spectrum only. The ferrocenyl BODIPYs 3a–3d were well characterized by 1H, 13C NMR and HRMS techniques. Compounds 3a–3b were also characterized by single crystal X-ray diffraction.
UV-visible absorption spectra of all the compounds in toluene were recorded on a Carry-100 Bio UV-visible Spectrophotometer with a concentration of 1.5 × 10−4 M at room temperature. Cyclic voltammetric (CV) studies for BODIPYs 3a–3b were carried out with an electrochemical system utilizing a three-electrode configuration consisting of a glassy carbon (working) electrode, platinum wire (auxiliary) electrode and a saturated calomel (reference) electrode. The experiments were performed in dry dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte at a concentration of 1.5 × 10−4 M . Crystals of BODIPYs 3a and 3b suitable for X-ray analysis were obtained by the slow diffusion of ethanol into their chloroform solutions. The vial containing this solution was loosely capped to promote crystallization upon ethanol diffusion. The structures were solved by direct methods using SHELXS-97 and were refined by full matrix least-squares with SHELXL-97, refining on F2.
The BODIPYs exhibit a strong absorption band at around 500 nm along with a broad transition in the higher energy region.10 The BODIPYs 3a–3d exhibit a strong absorption band between 575–590 nm, corresponding to the S0 → S1 (π → π*) transition, and a weak absorption band between 400–410 nm due to the S0 → S2 (π → π*) transition (Fig. 1). The substitution of the ferrocenyl group on the BODIPY results in a substantial red shift in the absorption maxima. The absorption maxima of 3a–3d is considerably red shifted compared to the unsubstituted BODIPY 1, which reflects the strong electronic interaction between the ferrocene and the BODIPY.11 The meta branching in compound 3c disrupts the extended π-conjugation compared to the other phenylacetylene spacers.12 Thus, a lower red shift is observed for compound 3c compared with the other ferrocenyl BODIPYs. The absorption maxima are broad in 3a–3d when compared to the sharp absorption of unsubstituted BODIPY 1 and this may be due to the overlap of the charge-transfer absorption.13 Compounds 3a–3d are poorly emissive in nature compared to BODIPY 1 due to the fast non-radiative deactivation of the exited state via intramolecular charge transfer.14,16
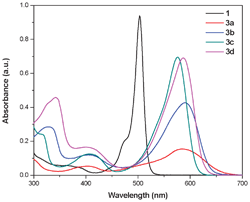 | ||
| Fig. 1 UV-visible absorption spectra of ferrocenyl substituted BODIPYs 1 and 3a–3d in toluene. The concentration used was 10−5 M. | ||
The electrochemical data of BODIPYs 3a–3b is listed in Table 1 and a representative cyclic voltammogram is presented in Fig. 2. The BODIPYs exhibit one oxidation and one reduction wave corresponding to the formation of a mono π-radical cation and mono π-radical anion respectively.15 The ferrocenyl substituted BODIPYs 3a–3d show one reversible reduction wave in the region of −1.08 to −1.05 V. The reduction potential of 3a–3d is shifted to lower values compared to unsubstituted BODIPY 1, indicating that the boron–dipyrromethane unit is easier to reduce.16 Oxidation peaks corresponding to the oxidation of ferrocene to ferrocenium and the formation of a BODIPY π-radical cation are observed for compounds 3a–3d. The ferrocenyl BODIPYs 3a–3d show one irreversible oxidation wave in the 1.24–1.20 V region. The trend in the oxidation potential of the ferrocenyl moiety in the ferrocenyl substituted BODIPYs 3a–3d follows the order 3a > 3b > 3d > 3c. Compared to the oxidation potential of free ferrocene, the oxidation of the ferrocenyl moiety became harder, which reflects the strong electronic communication between the ferrocenyl unit and the BODIPY core in 3a–3d.17
 | ||
| Fig. 2 Cyclic voltammogram of BODIPY 3a in DCM containing 0.1 M Bu4NPF6 as the supporting electrolyte, recorded at a scan speed of 100 mVs−1. | ||
| Compound | Photophysical dataa | Electrochemical datad | |||
|---|---|---|---|---|---|
| λ abs (nm)b | ε (M−1cm−1)c | E 1 ox (V) | E 2 oxid (V)e | E 1 red (V) | |
| a Measured in toluene. b λ abs (nm): absorption wavelength of the first absorption maximum. c ε: extinction coefficient. d Recorded by cyclic voltammetry in a 0.1 M solution of Bu4NPF6 in DCM (1.5 × 10−4 M) at a scan rate of 100 mV s−1, vs. a saturated calomel electrode. e For the irreversible redox process, the peak potential is quoted. | |||||
| Ferrocene | — | — | 0.38 | — | — |
| 1 | 503 | 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 989 989 |
— | 1.16 | −1.25 |
| 3a | 583 | 15500 |
0.53 | 1.24 | −1.05 |
| 3b | 590 | 42800 |
0.52 | 1.21 | −1.06 |
| 3c | 576 | 69600 |
0.44 | 1.20 | −1.08 |
| 3d | 586 | 69200 |
0.51 | 1.21 | −1.06 |
BODIPY 3a crystallizes in the monoclinic space group P21/c and BODIPY 3b crystallizes in the monoclinic space group C2/c, with a crystallographic two-fold axis which passes through atoms B1, C3 and C6–H6. Fig. 3 shows the single crystal X-ray structure of 3a and 3b.
 | ||
| Fig. 3 Single crystal X-ray structures of BODIPY 3a and 3b. (i) Front view and (ii) side view. | ||
The BODIPY core, B(N2F2), exhibits an F–B–F angle of 109.0(5)° for 3a and 110.2(4)° for 3b. The average bond length of the B–N bond for BODIPYs 3a and 3b lies in the range of 1.537(4) Å–1.557(7) Å and the average bond length of the B–F bond lies in the range of 1.372(7) Å–1.407(7) Å. The dihedral angles between the meso-substituted phenyl group and the BODIPY core are 89.15° and 79.79° for BODIPYs 3a and 3b respectively. This may be attributed to the steric hindrance from the 1,7-dimethyl-substituents of the BODIPY core. The meso-phenyl mean plane is tilted by 20.11° with respect to the BF2 mean plane in 3b, whereas they are almost parallel in 3a (2.61°). The two cyclopentadienyl rings of the ferrocene moiety are parallel with the syn-periplanar eclipsed conformation in both 3a and 3b. The tilt between the BODIPY and the ferrocene units is more prominent in 3b, with a dihedral angle of 35°, while for 3a it is 3.70°. The ferrocene unit in both 3a and 3b lies on the opposite sides of the BODIPY mean plane. The important bond lengths and bond angles are listed in Table S1 (see the ESI for details†). Moreover, in the case of 3a, the chloroform solvation is linked to F1 via C–H⋯F hydrogen bonding of 2.46(3) Å.
The packing diagram of 3b (Fig. 4) reveals intermolecular C–H⋯F and C–H⋯π interactions. The C–H⋯F interaction involves the meso-phenyl hydrogen (H6) of one molecule with both the F-atoms of a neighboring molecule along the b-axis, forming a 1D-linear chain. The other phenyl hydrogen atoms (two H4 atoms) are involved in C–H⋯π interactions with adjacent molecules along the a-axis, leading to the formation of a 2D-network.
 | ||
| Fig. 4 Packing diagram of 3b forming a 2D-network along the a-axis. | ||
In summary, we have synthesized a series of ferrocenyl substituted BODIPYs via a Pd catalyzed Sonogashira coupling reaction. The UV-visible absorption and electrochemical properties of these molecules show strong donor–acceptor interactions. The crystal structure of 3b shows a 2D-network.
References
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS.
- (a) T. Cheng, T. Wang, W. Zhu, X. Chen, Y. Yang, Y. Xu and X. Qian, Org. Lett., 2011, 13, 3656 CrossRef CAS; (b) O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Selcuk, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, J. Am. Chem. Soc., 2010, 132, 8029 CrossRef CAS; (c) H. S. Kim, T. C. T. Pham and K. B. Yoon, Chem. Commun., 2012, 48, 4659 RSC; (d) M. Benstead, G. H. Mehl and R. W. Boyle, Tetrahedron, 2011, 67, 3573 CrossRef CAS; (e) Y. Zhou, Y. Xiao, S. Chi and X. Qian, Org. Lett., 2008, 10, 633 CrossRef CAS; (f) S. J. Lord, N. R. Conley, H. I. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204 CrossRef CAS; (g) R. West, C. Panagabko and J. Atkinson, J. Org. Chem., 2010, 75, 2883 CrossRef CAS; (h) A. B. Descalzo, H. J. Xu, Z. L. Xue, K. Hoffmann, Z. Shen, M. G. Weller, X. Z. You and K. Rurack, Org. Lett., 2008, 10, 1581 CrossRef CAS; (i) S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghammbarian, Nature, 1997, 388, 845–851 CrossRef CAS; (j) S. M. LeCours, H. W. Guan, S. G. DiMagno, C. H. Wang and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 1497 CrossRef.
- (a) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS; (b) V. Lakshmi and M. Ravikanth, J. Org. Chem., 2011, 76, 8466 CrossRef CAS.
- F. Algi and A. Cihaner, Org. Electron., 2009, 10, 453 CrossRef CAS.
- (a) Y. Wang, D. Zhang, H. Zhou, J. Ding, Q. Chen, Y. Xiao and S. Qian, J. Appl. Phys., 2010, 108, 033520 CrossRef; (b) K. D. Belfield, A. R. Morales, B. S. Kang, J. M. Hales, D. J. Hagan, E. W. V. Stryland, V. M. Chapela and J. Percino, Chem. Mater., 2004, 16, 4634 CrossRef CAS; (c) V. Hrobarikova, P. Hrobarik, P. Gajdos, I. Fitilis, M. Fakis, P. Persephonis and P. Zahradnik, J. Org. Chem., 2010, 75, 3053 CrossRef CAS; (d) P. Gautam, B. Dhokale, V. Shukla, C. P. Singh, K. S. Bindra and R. Misra, J. Photochem. Photobiol., A, 2012, 239, 24 CrossRef CAS.
- D. K. Zhang, Y. C. Wang, Y. S. Xiao, X. Qian and X. H. Qian, Tetrahedron, 2009, 65, 8099 CrossRef CAS.
- M. L. H. Green, S. R. Marder, M. E. Thompson, J. A. Bandy, D. Bloor, P. V. Kolinsky and R. J. Jones, Nature, 1987, 330, 360 CrossRef CAS.
- (a) W. Yang, Y. Li, H. Liu, L. Chi and Y. Li, Small, 2012, 8, 504 CrossRef CAS; (b) X. Yin, Y. Li, Y. Li, Y. Zhu, X. Tang, H. Zheng and D. Zhu, Tetrahedron, 2009, 65, 8373 CrossRef CAS; (c) M. Yuan, Y. Li, J. Li, C. Li, X. Liu, J. Lv, J. Xu, H. Liu, S. Wang and D. Zhu, Org. Lett., 2007, 9, 2313 CrossRef CAS; (d) M. Yuan, X. Yin, H. Zheng, C. Ouyang, Z. Zuo, H. Liu and Y. Li, Chem.–Asian J., 2009, 4, 707 CrossRef CAS; (e) M. Zhu, L. Jiang, M. Yuan, X. Liu, C. Ouyang, H. Zheng, X. Yin, Z. Zuo, H. Liu and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7401 CrossRef CAS.
- J. Godoy, G. Vives and J. M. Tour, Org. Lett., 2010, 12, 1464 CrossRef CAS.
- Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192 CrossRef CAS.
- T. K. Khan, R. R. S. Pissurlenkar, M. S. Shaikh and M. Ravikanth, J. Organomet. Chem., 2012, 697, 65 CrossRef CAS.
- (a) J. S. Melinger, Y. Pan, V. D. Kleiman, Z. Peng, B. L. Davis, D. McMorrow and M. Lu, J. Am. Chem. Soc., 2002, 124, 12002 CrossRef CAS; (b) R. Misra, R. Kumar, T. K. Chandrashekar, C. H. Suresh, A. Nag and D. Goswami, J. Am. Chem. Soc., 2006, 128, 16083 CrossRef CAS.
- R. Ziessel, P. Retailleau, K. J. Elliott and A. Harriman, Chem.–Eur. J., 2009, 15, 10369 CrossRef CAS.
- (a) S. Fery-Forgues and B. Delavaux-Nicot, J. Photochem. Photobiol., A, 2000, 132, 137 CrossRef CAS; (b) B. Dhokale, R. Misra and P. Gautam, Tetrahedron Lett., 2012, 53, 2352 CrossRef CAS; (c) V. A. Nadtochenko, N. N. Denisov, V. Y. Gak, N. V. Abramova and N. M. Loim, Russ. Chem. Bull., 1999, 148, 1900 CrossRef; (d) S. Barlow and S. R. Marder, Chem. Commun., 2000, 1555 RSC.
- X. Yin, Y. Li, Y. Zhu, X. Jing, Y. Li and D. Zhu, Dalton Trans., 2010, 39, 9929 RSC.
- M. R. Rao, K. V. Pavan Kumar and M. Ravikanth, J. Organomet. Chem., 2010, 695, 863 CrossRef CAS.
- R. Maragani, T. Jadhav, S. M. Mobin and R. Misra, Tetrahedron, 2012, 68, 7302–7308 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectroscopic characterizations for new compounds 3a–3d and crystallographic data for 3a and 3b. CCDC 891093 and 891094. For ESI and crystallographic data in CIF or other electronic format details of any supplementary information available. See DOI: 10.1039/c2ra21964f |
| This journal is © The Royal Society of Chemistry 2012 |