Nitrogen supported solvent evaporation using continuous-flow microfluidics
Benjamin Z.
Cvetković
a,
Oliver
Lade
b,
Lucia
Marra
c,
Valentina
Arima
c,
Ross
Rinaldi
cd and
Petra S.
Dittrich
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Wolfgang-Pauli-Str. 10, CH-8093 Zurich, Switzerland. E-mail: dittrich@org.chem.ethz.ch; Tel: +41 44 633 68 93
bSiemens AG, Berlin, Germany
cNational Nanotechnology Laboratory (NNL), CNR–Institute of Nanoscience, U.O.S. Lecce, Italy
dUniversità del Salento, Dipartimento di Matematica e Fisica “E. De Giorgi”, ex Collegio Fiorini Campus extraurbano, Lecce, Italy
First published on 19th September 2012
Abstract
In this work we demonstrate a continuously operating microfluidic device for solvent removal and exchange for chemical syntheses by means of a supporting gas. The glass device consists of three sections: (i) three merging microchannels to create an annular gas–liquid stream; (ii) a serpentine channel with a heater underneath to allow efficient evaporation of the volatile solvent; (iii) a section with side capillaries to separate the liquid from the gas phase. We demonstrate the performance of the device for the removal of acetonitrile from an acetonitrile–water mixture. We achieved efficient removal of acetonitrile within a few seconds for flow rates of 20–30 μL min−1 and a nitrogen pressure of 1.2 bar. In three steps, acetonitrile was reduced from 50 wt% in the feed solution to 1 wt% in the final solution. We believe that the device can be easily applied to other solvent mixtures.
Introduction
In recent years microfluidic devices have gained great attraction in the field of chemical synthesis. The use of microreactors for chemical processes offers several unique advantages compared to conventional batch processes. Most importantly, rapid heat transfer inside microdevices allows for precise control of the reactor temperature. Additionally, mixing is reproducible due to the laminar flow regime present in the microreactors and can be fast due to the small dimensions. Many examples have demonstrated that the product yield and specificity of the reactions were significantly increased. Together with the short reaction times and the efficient use of chemicals, microreactors enable an easy and safe route to dangerous or sensitive compounds, whilst keeping the produced chemical waste to a minimum.1However, one of the remaining challenges is the realization of continuous operation, allowing multistep reactions to be conducted. In particular, simple solutions for the integration of standard processes such as solvent evaporation and exchange are required. Additionally, an integrated work-up of products would expand the applicability of microdevices to routine use in chemistry. Despite this importance, only a few approaches have been presented in recent years that demonstrate the continuous evaporation or distillation of solvents in a continuous flow reactor. Often, membrane supports2 or micro/nanosized pillar structures3 were implemented in a microfluidic device. Furthermore, the use of auxiliary gases was shown, where laminar streams of the liquid and the gas phases4 or segmented flow were realized.
In this paper we show a new microfluidic approach for nitrogen supported continuous solvent evaporation without the use of a membrane material. The device can serve as a powerful and robust tool for solvent exchange in continuous chemical synthesis processes. The channel material (glass), design of the microchip and the flow conditions ensured an annular flow regime and hence, provided a large interface between the gas and liquid phases for efficient solvent removal. To perform the removal of the solvent enriched gas phase from the remaining liquid phase we implemented small capillaries at the end of the separation channel (Fig. 1).
 | ||
| Fig. 1 Photograph (a) and schematic drawings (b–e) illustrating the channel geometry of the evaporation device. (a) The glass microdevice consists of three sections for (i) gas/liquid introduction, (ii) evaporation and (iii) gas–liquid phase separation. Scale bar 5 mm. (b) Schematic of the first section, where the liquid mixture is introduced into the middle channel (blue) and nitrogen is introduced from either side. The cross section (c) depicts the annular flow present in the microchannels. In the third section, the liquid phase is removed through side capillaries. The cross section (d) illustrates the geometry where the high channels are etched into the upper glass plate and the capillaries are etched into the bottom plate. (e) Removal of the liquid phase through the capillaries, while the gas phase remains in the main channel. | ||
The device is applicable for various syntheses and water–solvent systems. However, our ultimate goal was the development of a continuous flow reactor for radiotracers, which carry short-lived radionuclides such as 18F. On demand production in a continuous flow reactor promises a safe and reproducible synthesis as well as a simplified quality control.5
The first examples of radiotracer production in microreactors were presented recently,5a,5e–h,6 however, all of these approaches were discontinuous. Here, we demonstrate the operation of the device with the removal of acetonitrile from a water–acetonitrile mixture, which is required during the synthesis of the radiotracer 2-deoxy-2-(18F)-fluoro-glucose (18F-FDG).
Materials and methods
Device fabrication
The planar glass microdevice was designed using CAD software (AutoCAD 2010, Autodesk) and fabricated using standard lithography techniques and wet chemical etching.7 Microchannels and capillaries were etched into two B270 (Telic, Valencia, CA) glass plates, which were subsequently bonded together.The base plate contained the capillaries for separation of the liquid and gas phases, which had a height of 5 μm and a width of 10 μm. The input channel and the separation channel were fabricated into the top plate with a height of 100 μm and a width of 400 μm (Fig. 1a). Inlet and outlet holes (1/16′′ outer diameter (OD)) were drilled into the top glass plates, then both plates were aligned and thermally bonded. About 25 mm of standard PEEK tubing (1/16′′ OD × 0.030′′ inner diameter (ID) from IDEX Health and Science, USA) was glued (Torr Seal, Varian, USA) to the device enabling the connection of additional tubing for fluid supply. For the fluid supply chemically stable FEP tubing (1/16′′ OD × 0.25 mm ID, BGB Analytik AG, Switzerland) was used. Furthermore, the device was equipped with a resistive microheater, which heated one side of the device, guaranteeing isothermal conditions in the channel. The serpentine geometry compatible with the microfluidic module was used in order to reach high temperatures with low voltages (up to 15 V) and to heat locally. The heater was fabricated using a standard lithographic process, followed by the thermal evaporation of Cr (5 nm)/Au (300 nm) metals. A few nanometres of chromium were deposited in order to improve the adhesion of the gold layer onto the glass substrate.8 The heater was connected to a laboratory voltage source (DC power supply, DF 1730 SB). The meandering gold structure was used for resistive heating and a second structure for direct temperature sensing was patterned on the glass substrate. The thermal response of the heater was determined by I/V measurements (Keithley electrometer) and the temperature was measured by means of a micro-thermocouple. Temperatures up to 150 °C inside the channel at 15 V could be achieved with this configuration.
Peripheral instruments
An in-house nitrogen line was used to adjust the pressure of the gaseous nitrogen stream (0.8–1.6 bar). A syringe pump (Nexus 3000, Chemyx) was used to control the liquid flow rates, which typically ranged from 10–100 μL min−1. Liquid feed solutions were prepared using deionised water and acetonitrile (puriss p.a. quality, Sigma-Aldrich) and contained a maximum of 50 wt% acetonitrile in water.A laboratory source meter (U1271B, Agilent) was used to control the resistive microheater and the resulting temperature was determined by measuring the resistance change of an implemented resistor. The temperature was always kept below the boiling point (82 °C) of acetonitrile to prevent uncontrolled evaporation.
Optical imaging was performed on an inverted microscope (Olympus IX 70) equipped with a CCD camera (UK1117, ABS).
Determination of acetonitrile concentration
To determine the concentration of acetonitrile, we exploited the change of polarity for varying concentrations of acetonitrile and water, which could be visualized by the addition of the solvatochromic dye 1-methyl-4-[(oxocyclohexadienylidene) ethylidene]-1,4-dihydropyridine (MOED). Briefly, MOED was synthesized as described elsewhere9 and added to the sample solution (2.5 μM). The dye was excited by the 488 nm line of an Ar-ion laser (Laserlight), which was focused by a microscope objective (Olympus, 40×) and the fluorescence was detected using a fibre-coupled fluorescence spectrometer (USB-2000, Ocean Optics Inc.) (Fig. 2). The recorded fluorescence spectra are shifted to a longer wavelength for an increasing polarity, i.e. decreasing acetonitrile concentration. The shift in the fluorescence maximum was determined either online at the end of the microchannels, or off-chip in the collected solution.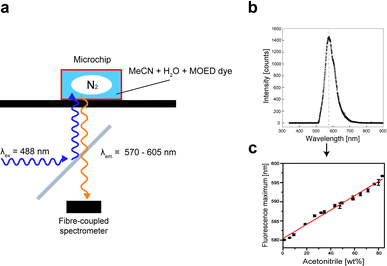 | ||
| Fig. 2 The concentration of acetonitrile in water was measured by fluorescence spectroscopy by adding a fluorescent dye (MOED) with a strong solvatochromic effect. (a) Schematic representation of the optical setup to measure the concentration online (in the microchip). A dichroic mirror is used to reflect the 488 nm line of a laser into the microscope. The fluorescence is recorded using a fibre-coupled spectrometer. (b) A fluorescence spectrum recorded from a pure water solution. (c) Calibration curve for various acetonitrile concentrations. | ||
Results and discussion
Organic syntheses frequently require the transfer of an intermediate or product from one solvent into another. The aim was to improve the formation of the extensively used radiotracer [18F]FDG and realize a continuous flow reaction in acetonitrile, followed by the hydrolysis of the product in water. Therefore, we focused on the removal of acetonitrile from an acetonitrile–water mixture. Due to the complete miscibility of these solvents, they cannot be separated by simple phase separation. Instead, we used nitrogen as an auxiliary gas to uptake the volatile organic solvent from the water. Finally, the gas phase is separated from the water stream.A schematic representation of the device is shown in Fig. 1. The device consists of three sections. In the first section (Fig. 1a (i)) gaseous and liquids streams are introduced, and an annular flow of a gas core and a fluid corona is created at the point of confluence. The second section consists of a serpentine channel (Fig. 1a (ii)) where the evaporation process takes place. The last section is where the liquid is separated from the gas phase (Fig. 1a (iii)). The separation of the vapour enriched gas phase from the liquid phase is achieved by small, hydrophilic capillaries (5 × 10 μm cross section), which allow the aqueous liquid phase to pass due to capillary forces while the gas phase remains in the main channel and flows towards the outlet. In the following, the operation of the device is described for each section and the performance is evaluated for various conditions.
Introduction of liquid and gas phases
When the liquid and gas phases are merged into a single stream, various flow regimes can occur depending on the flow conditions. For flow rates between 10–40 μL min−1 and <200 mbar nitrogen pressure, segmented flow (nitrogen bubbles in the liquid stream) is produced. The segmented flow regime provides a large contact surface for gas and liquid, which is advantageous for fast phase transfer. However, equilibrium conditions were quickly obtained resulting in saturation of the limited volume of the nitrogen bubble according to Henry's law. Furthermore, on reaching saturation, the segmented flow became unstable.Annular streams were created for liquid flow rates of 10–100 μL min−1 and nitrogen pressures of >400 mbar. At temperatures up to 60 °C, stable flow was achieved for nitrogen pressures of 800 to 1200 mbar. The hydrophilic surface of the chip material (glass) supports the annular flow regime, where the liquid phase contacts the channel walls, while nitrogen is flushed through the central channel at the same time (Fig. 1b and c). We chose these flow conditions with a dynamic headspace for further experiments as we expected less instability and fewer saturation effects at higher concentrations of acetonitrile. Once the annular flow regime is created, the volatile compound (acetonitrile) evaporates into the nitrogen gas. The thickness of the liquid stream was between 70–80 μm, hence the overall surface area along the entire evaporation channel was 70 mm2.
Gas–liquid separation
In order to implement the evaporation device in a continuous flow reactor, it is necessary to separate the vapour-enriched gas stream from the remaining liquid phase.We achieved this by adding capillaries into the third section of the glass device. Due the hydrophilic surface of the glass, the hydrophilic (aqueous) liquid phase enters the capillaries, while the gas stream is rejected and remains in the wider main channel. The performance of the separation is dependent on the capillary cross section. In contrast to previously discussed devices for oil–water phase separation with a width and height of 15 × 15 μm, respectively,10 we had to significantly reduce the capillary dimensions in order to prevent a breakthrough of the gas phase. To achieve this, a double layer fabrication technique was used, enabling independent variations of geometries and heights in the top and bottom glass plates (Fig. 1 d and e). For the evaporation device described herein, the main channels are etched into the upper glass plates with a height of 100 μm and a width of 400 μm, while the capillaries with a lower height of 5 μm and a width of 10 μm are etched into the other (bottom) plate. This device facilitated the complete separation of the gas and liquid phases for all relevant flow conditions in the annular flow regime. This is illustrated in the fluorescence image shown in Fig. 3. It is noteworthy to mention that the gas–liquid separation section also functions under segmented flow conditions.
 | ||
| Fig. 3 Fluorescence micrograph of the gas–liquid separation region with a high main channel and low narrow side capillaries. The fluid passing this separation region flows into the capillaries due to the capillary forces and finally into the liquid outlet channels. The gas phase is not able to enter the capillaries and remains in the main channel. Liquid and gas phase are collected through separate outlets. The aqueous solution was stained with fluorescein for better visualisation. Scale bar 200 μm. | ||
Removal of the volatile organic solvent
Finally, the performance of the device is demonstrated by the removal of acetonitrile from a water–acetonitrile mixture (with a maximum of 50 wt% acetonitrile) depending on the applied temperature, nitrogen pressure, and the flow rate of the liquid mixture.We first studied the evaporation process for three different starting feed concentrations (50, 35 and 18 wt% acetonitrile) at increasing temperatures, starting at 45 °C and heating up to 70 °C using the heating plate as described in the Materials and Methods section.
The nitrogen pressure and liquid flow rate were kept constant at 1.2 bar and 30 μL min−1. As expected, increasing the temperature had a strong effect on the evaporation processes. At 60 °C, approximately half of the original concentration of acetonitrile was removed from the liquid solution for all feed solutions (e.g. from initially 35 wt% to 18 wt%, see Fig. 4a). A further increase of the temperature (up to 70 °C) was counterproductive, because increased amounts of the water evaporate at these elevated temperatures.
 | ||
| Fig. 4 Acetonitrile concentration in the liquid phase after evaporation and phase separation in the microdevice. All experiments were performed under annular flow conditions. (a) Temperature dependence. Different temperatures between 45 and 70 °C were applied at a fixed feed flow rate of 30 μL min−1 and nitrogen pressure of 1.2 bar. Three feed solutions were investigated (red, green, blue columns: 50, 35, 18 wt% acetonitrile, respectively). (b) Dependence on the gas flow. Nitrogen pressures were varied from 1.0 to 1.4 bar at a temperature of 60 °C and a fixed feed flow rate of 30 μL min−1 (red, green, blue columns: 50, 35, 18 wt% acetonitrile, respectively). (c) Influence of the feed flow rates at a temperature of 60 °C and nitrogen pressure of 1.2 bar. (d) Three evaporation steps were performed sequentially with a feed flow rate of 20 μL min−1, nitrogen pressure of 1.2 bar and 50 wt% acetonitrile. | ||
Next, we varied the applied nitrogen pressure from 1.0 to 1.4 bar (Fig. 4b), corresponding to increasing gas flow rates. The liquid feed flow rate (30 μL min−1) and the temperature (60 °C) were kept constant. A faster gas stream should enable a better uptake of the vaporized solvent and prevent saturation effects of the nitrogen stream. Furthermore, a minor effect is the increase in the contact surface area of gas and liquid since the liquid phase is squeezed into a thinner film with an increased gas pressure. However, the amount of removed acetonitrile did not significantly improve for the flow and pressure conditions (under which the annular flow regime was stable). Moreover, at the highest tested nitrogen pressure (1.4 bar) instabilities in the downstream separation of the gas and liquid fractions were observed (see below).
Finally, the influence of the feed flow rate on the evaporation was studied. Flow rates between 10 and 100 μL min−1 were applied at a nitrogen pressure of 1.2 bar and at 60 °C (Fig. 4c), hence varying the residence time of the liquid. The results show that the most efficient removal of acetonitrile occurs at concentrations down to 20 wt% for the slower flow rates below 20 μL min−1. Further decrease of the liquid flow rate had no effect, indicating the saturation of nitrogen with vaporised acetonitrile.
Overall, the best conditions were found to be at flow rates of 20–30 μL min−1, nitrogen pressure of 1.2 bar and a temperature of 60 °C. This resulted in the largest decrease of acetonitrile concentration in one step, e.g. from 50 to 20 wt%. It should be emphasised that this process occurs within 5 s, i.e. the residence time of the liquid in the device.
Although the capacity of the device to remove volatile organic solvents has been demonstrated, complete removal of organic solvent was not possible in one step. Since we observed saturation effects of the gas for slow liquid flow rates, we did not use microchips with a longer (serpentine) channel for evaporation (see Fig. 1a, section (ii)). Also higher nitrogen pressures above 1.4 bar could not be applied due to the gas breakthrough in the liquid–gas separation section. Instead, we connected the evaporation modules serially, i.e. the collected liquid is reinjected into another module together with fresh (dry) nitrogen to further remove acetonitrile. Fig. 4d shows the result after passing through the evaporation modules three times. Finally, this resulted in the desired, nearly complete removal of acetonitrile with 1 wt% residual in the aqueous phase.
Conclusions
We presented a continuously operating microdevice for the removal of acetonitrile from water–acetonitrile mixtures within seconds by means of an auxiliary gas stream. Using annular flow conditions, a large contact area between the liquid and the gas phase allowed the efficient uptake of the volatile acetonitrile into the gas stream even at temperatures below the boiling point of acetonitrile. To achieve nearly complete solvent removal, several microdevices could simply be connected in a row. A higher throughput can be achieved by increasing the overall flow rates of the liquid and the gas phases and by modifications of the separation section, i.e. by integration of more capillaries with smaller dimensions.The device can be employed for many mixtures of organic solvents and water, provided that the organic solvent has a significantly higher volatility than water.
We believe that the device solves a bottleneck in the realization of continuous flow chemistry, where solvent removal or exchange is frequently required in multistep chemical reactions. Furthermore, by connecting a suitable detector at the gas outlet, the microdevice could be easily applied for the stripping of volatile organic compounds (in low concentrations), for water quality control.
Acknowledgements
The authors acknowledge the EU project Radiochemistry on chip “ROC”, grant agreement no. 213803 for financial support. We thank Tom Robinson for careful proofreading of the manuscript.References
- (a) A. Odedra and P. H. Seeberger, 5-(Pyrrolidin-2-yl) tetrazole-Catalyzed Aldol and Mannich Reactions: Acceleration and Lower Catalyst Loading in a Continuous-Flow Reactor, Angew. Chem., Int. Ed., 2009, 48(15), 2699–2702 CrossRef CAS; (b) R. L. Hartman, J. P. McMullen and K. F. Jensen, Deciding whether to go with the flow: evaluating the merits of flow reactors for synthesis, Angew. Chem., Int. Ed., 2011, 50(33), 7502–7519 CrossRef CAS; (c) P. Watts and C. Wiles, Recent advances in synthetic micro reaction technology, Chem. Commun., 2007,(5), 443–467 RSC; (d) S. J. Haswell and P. Watts, Green chemistry: synthesis in micro reactors, Green Chem., 2003, 5(2), 240–249 RSC; (e) C. Wiles and P. Watts, Continuous Flow Reactors, a Tool for the Modern Synthetic Chemist, Eur. J. Org. Chem., 2008,(10), 1655–1671 CrossRef CAS; (f) C. Wiles and P. Watts, Recent advances in micro reaction technology, Chem. Commun., 2011, 47(23), 6512–6535 RSC; (g) K. Jensen, Microreaction engineering-is small better?, Chem. Eng. Sci., 2001, 56, 293–303 CrossRef CAS; (h) P. Fletcher, S. Haswell, E. Pombo-Villar, B. Warrington, P. Watts, S. Wong and X. Zhang, Micro reactors: principles and applications in organic synthesis, Tetrahedron, 2002, 58(24), 4735–4757 CrossRef CAS; (i) S. J. Haswell, R. J. Middleton, B. O'Sullivan, V. Skelton, P. Watts and P. Styring, The application of micro reactors to synthetic chemistry, Chem. Commun., 2001,(5), 391–398 RSC; (j) A. Jahn, J. E. Reiner, W. N. Vreeland, D. L. DeVoe, L. E. Locascio and M. Gaitan, Preparation of nanoparticles by continuous-flow microfluidics, J. Nanopart. Res., 2008, 10(6), 925–934 CrossRef CAS; (k) R. L. Hartman and K. F. Jensen, Microchemical systems for continuous-flow synthesis, Lab Chip, 2009, 9(17), 2495 RSC; (l) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Chemistry in Microstructured Reactors, Angew. Chem., Int. Ed., 2004, 43(4), 406–446 CrossRef; (m) H. Pennemann, V. Hessel and H. Löwe, Chemical microprocess technology—from laboratory-scale to production, Chem. Eng. Sci., 2004, 59(22–23), 4789–4794 CrossRef CAS.
- (a) B. H. Timmer, K. M. van Delft, W. Olthuis, P. Bergveld and A. van den Berg, Micro-evaporation electrolyte concentrator, Sens. Actuators, B, 2003, 91(1–3), 342–346 CrossRef; (b) Y. Zhang, S. Kato and T. Anazawa, Vacuum membrane distillation by microchip with temperature gradient, Lab Chip, 2010, 10(7), 899–908 RSC.
- (a) A. Hibara, K. Toshin, T. Tsukahara, K. Mawatari and T. Kitamori, Microfluidic distillation utilizing micro–nano combined structure, Chem. Lett., 2008, 37(10), 1064–1065 CrossRef CAS; (b) K. F. Lam, E. Cao, E. Sorensen and A. Gavriilidis, Development of multistage distillation in a microfluidic chip, Lab Chip, 2011, 11(7), 1311 RSC.
- R. C. R. Wootton and A. J. deMello, Continuous laminar evaporation: micron-scale distillation, Chem. Commun., 2004,(3), 266–267 RSC.
- (a) C. C. Lee, Multistep Synthesis of a Radiolabeled Imaging Probe Using Integrated Microfluidics, Science, 2005, 310(5755), 1793–1796 CrossRef CAS; (b) A. M. Elizarov, Microreactors for radiopharmaceutical synthesis, Lab Chip, 2009, 9(10), 1326 RSC; (c) J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, B. A. Dekker and J. Zweit, Microfluidic technology for PET radiochemistry, Applied radiation and isotopes : including data, instrumentation and methods for use in agriculture, industry and medicine, 2006, 64(3), 333–336 CAS; (d) K. Liu, M.-W. Wang, W.-Y. Lin, D. L. Phung, M. D. Girgis, A. M. Wu, J. S. Tomlinson and C. K.-F. Shen, Molecular Imaging Probe Development Using Microfluidics, Curr. Org. Synth., 2011, 8(4), 473–487 CrossRef CAS; (e) A. M. Elizarov, C. Meinhart, R. Miraghaie, R. M. Dam, J. Huang, A. Daridon, J. R. Heath and H. C. Kolb, Flow optimization study of a batch microfluidics PET tracer synthesizing device, Biomed. Microdevices, 2010, 13(1), 231–242 CrossRef; (f) H.-J. Wester, B. W. Schoultz, C. Hultsch and G. Henriksen, Fast and repetitive in-capillary production of [18F]FDG, Eur. J. Nucl. Med. Mol. Imaging, 2008, 36(4), 653–658 CrossRef; (g) A. M. Elizarov, R. M. Van Dam, Y. S. Shin, H. C. Kolb, H. C. Padgett, D. Stout, J. Shu, J. Huang, A. Daridon and J. R. Heath, Design and optimization of coin-shaped microreactor chips for PET radiopharmaceutical synthesis, Journal of nuclear medicine: official publication, Society of Nuclear Medicine, 2010, 51(2), 282–287 CAS; (h) S. Sadeghi, J. Ly, Y. Deng and R. M. van Dam, A robust platinum-based electrochemical micro flow cell for drying of [18F] fluoride for PET tracer synthesis, Proceedings of the Fourteenth International Conference on Miniaturized Systems for Chemistry and Life Sciences, 2010, 318–320 Search PubMed; (i) C. J. Steel, A. T. O'Brien, S. K. Luthra and F. Brady, Automated PET radiosyntheses using microfluidic devices, J. Labelled Compd. Radiopharm., 2007, 50(5–6), 308–311 CrossRef CAS.
- (a) C. R. Buchanan, H. C. Padgett and T. L. Collier, Microfluidic apparatus and method for synthesis of molecular imaging probes including FDG, Patent application US 2005/0232861 A1 Search PubMed; (b) S.-Y. Lu, P. Watts, F. T. Chin, J. Hong, J. L. Musachio, E. Briard and V. W. Pike, Syntheses of 11C- and 18F-labeled carboxylic esters within a hydrodynamically-driven micro-reactor, Lab Chip, 2004, 4(6), 523–525 RSC; (c) H. Saiki, R. Iwata, H. Nakanishi, R. Wong, Y. Ishikawa, S. Furumoto, R. Yamahara, K. Sakamoto and E. Ozeki, Electrochemical concentration of no-carrier-added [18F]fluoride from [18O]water in a disposable microfluidic cell for radiosynthesis of 18F-labeled radiopharmaceuticals, Appl. Radiat. Isot., 2010, 68(9), 1703–1708 CrossRef CAS; (d) J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, W. Perrie, I. Hamblett, B. Dekker and J. Zweit, Microfluidic reactor for the radiosynthesis of PET radiotracers, Applied radiation and isotopes: including data, instrumentation and methods for use in agriculture, industry and medicine, 2006, 64(3), 325–332 CrossRef CAS.
- P. C. H. Li, Microfluidic Lab-on-a-Chip for Chemical and Biological Analysis and Discovery, CRC Press, 2005 Search PubMed.
- S. Kim, K. Kim and K. Kihm, Near-Field Thermometry Sensor Based on the Thermal Resonance of a Microcantilever in Aqueous Medium, Sensors, 2007, 7(12), 3156–3165 CrossRef.
- M. Minch, Merocyanin dye preparation for the introductory organic laboratory, J. Chem. Educ., 1977, 54, 709 CrossRef CAS.
- D. E. Angelescu, B. Mercier, D. Siess and R. Schroeder, Microfluidic Capillary Separation and Real-Time Spectroscopic Analysis of Specific Components from Multiphase Mixtures, Anal. Chem., 2010, 82(6), 2412–2420 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |