Changes in the hierarchical protein polymer structure: urea and temperature effects on wheat gluten films†
Ramune
Kuktaite
*a,
Tomás S.
Plivelic
b,
Hasan
Türe
c,
Mikael S.
Hedenqvist
c,
Mikael
Gällstedt
d,
Salla
Marttila
e and
Eva
Johansson
a
aDepartment of Agrosystems, Swedish University of Agricultural Sciences, SE-230 53 Alnarp, Sweden. E-mail: Ramune.Kuktaite@slu.se; Fax: +46 40 415119; Tel: +46 40 415337
bMAX IV Laboratory, Lund University, SE-221 00 Lund, Sweden
cFibre and Polymer Technology, Royal Institute of Technology, SE-100 44 Stockholm, Sweden
dInnventia, Box 5604, SE-11486 Stockholm, Sweden
eDepartment of Plant Protection Biology, Swedish University of Agricultural Sciences, SE-230 53 Alnarp, Sweden
First published on 2nd October 2012
Abstract
Understanding bio-based protein polymer structures is important when designing new materials with desirable properties. Here the effect of urea on the wheat gluten (WG) protein structure in WG–urea films was investigated. Small-angle X-ray scattering indicated the formation of a hexagonal close-packed (HCP) hierarchical structure in the WG–urea materials. The HCP structure was influenced significantly by the urea concentration and processing conditions. The interdomain distance dI between the HCP scattering objects increased with increasing content of urea and the objects seemed to be oriented in the extrusion direction. Additionally, the effect of temperature on the HCP structure was studied and it was shown that at ≥55 °C the HCP structure disappeared. Transmission electron microscopy revealed a rather denatured pattern of both HMW-glutenins and gliadins in the WG–urea films. The molecular packing of the WG protein polymer can be highly affected by an additive and the processing method used.
Introduction
Polymers from renewable resources, as an alternative for the production of plastic materials, have gained interest due to the future depletion of petroleum and the increasing environmental pollution by the use of petroleum-based plastics. Wheat gluten (WG), a relatively cheap by-product from the starch and bio-ethanol industries, is an attractive biopolymer with potential for the production of plastic material. The viscoelastic, thermoplastic, as well as excellent film forming and gas barrier properties of the WG protein contribute to the promising use of this biopolymer as packaging films,1 coatings on paperboards2 and foams for the insulation of houses.3Indeed, recent investigations on bio-based protein materials have revealed that the structure and functional behaviour of the material are closely related.4–6 For example, natural materials such as, spider silks, have shown outstanding mechanical properties7,8 and also specific structural geometries. For WG protein the structure and shape of extracted individual glutenins and gliadins have been described in a number of studies. Glutenins are considered as extended rod shaped molecules in solution,9 while gliadins (of α-, γ- and ω- types) are showed as being prolate ellipsoids of different axial ratios by small angle X-ray scattering.10 In addition, gliadin (in its hydrated solid state) showed a structure with dimensions of roughly 16 × 3 nm2.11 The protein polymerization, blend composition12 and thermal processing, e.g. extrusion, influence the performance of WG films.5 Supramolecular structures of the WG films with alkaline/alkaline-acidic additives i.e., sodium hydroxide (NaOH), ammonium hydroxide (NH4OH) and salicylic acid (SA)13,14 have also been associated with especially attractive mechanical and oxygen barrier properties. However, the WG protein morphology of the WG films with additives was found rather complex and only partially understood.5 Crystal structures of the large mixture of proteins in gluten are not available due to the protein lack of solubility in alcohol–water mixtures and also due to the proteins being present in mixtures.
During processing, high- and low-molecular weight glutenin subunits (HMW-gs and LMW-gs) form polymeric aggregates, which can either “trap” or crosslink monomeric gliadins.15,16 In fact, crosslinking of the gliadins, primarily through disulphide bonds with the glutenins was one of the explanations for the supramolecular structure found in the WG films.5 However, it still remains unclear why and how the supramolecular structures are formed during processing, and how these structures assist the formation of the WG polymer matrix and the functional properties of the material. For both petroleum-based polymers and biopolymers,17 good physical properties are also related to their thermal stability. Therefore, the thermal effect on the WG protein materials structure is of interest to investigate. The biological significance of studying wheat gluten protein is primarily based on collecting knowledge on the wheat gluten macropolymer formation as well as improving understanding on the protein structure-function relationship. Understanding the precise physical and chemical mechanisms of protein interactions is of importance.
Recent results have shown that the WG protein solubility can be manipulated by the use of a protein denaturant, urea, which also acts as a plasticiser, and as a consequence the aggregated/polymerized proteins formed have attractive tensile properties.18 Urea alters the protein structure and may exert its effect directly by binding to the WG protein, or indirectly by altering the protein “environment” (exposing of residues in the hydrophobic protein core).19 However, the molecular basis for urea’s effect on the WG protein structure remains not fully explained. The aim of this study was to reveal the supramolecular structure of the WG protein in WG–urea films with different concentrations of urea, and relate the structure to the functional properties of the materials. Additionally, the aim was to investigate the temperature effect on the protein structure. To assess the structure of the WG–urea films small-angle (SAXS) and wide-angle (WAXS) X-ray spectroscopy and transmission electron microscopy (TEM) were used.
Experimental
Materials
The WG powder was kindly provided by Lantmännen Reppe AB, Sweden. According to the supplier, the gluten powder contained 77.7% protein (according to modified NMKL nr. 6, Kjeltec, N × 5.7). Glycerol (≥99.5 wt%) was supplied by Karlshamns Tefac AB, Sweden. Urea (≥99.5 wt%) was purchased from Merck (Darmstadt, Germany).Sample preparation
The WG powder was placed at room temperature and 20% relative humidity (RH) in order to adjust its moisture content to approximately 6 wt% prior to extrusion. The WG powder was blended with glycerol (ratio 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, w/w), known to be a suitable plasticizer in the production of wheat gluten films20,21 and 5, 7, 9, 10, 15 and 20 wt% of urea was added to the blend at a concentration relative to the total mixture of WG–glycerol–urea (500 g), under ambient conditions. The mixture was consequently blended using a food processor (WATT; DUKA AB, Sweden) at “speed 2” (150 rpm) for 5 min. The samples were abbreviated with WGG followed by the amount of urea added, e.g. WGG + 5U corresponded to a sample with WG/glycerol with a weight ratio of 70/30 containing 5 wt% urea.
:30, w/w), known to be a suitable plasticizer in the production of wheat gluten films20,21 and 5, 7, 9, 10, 15 and 20 wt% of urea was added to the blend at a concentration relative to the total mixture of WG–glycerol–urea (500 g), under ambient conditions. The mixture was consequently blended using a food processor (WATT; DUKA AB, Sweden) at “speed 2” (150 rpm) for 5 min. The samples were abbreviated with WGG followed by the amount of urea added, e.g. WGG + 5U corresponded to a sample with WG/glycerol with a weight ratio of 70/30 containing 5 wt% urea.
Extrusion was performed according to Türe et al. (2011) using a single-screw extruder (BX18, Axon, Sweden).18 The extruder’s screw speed was 60 rpm and the die temperature was set to 130 °C as in Türe et al. (2011). Extruded films were cooled to room temperature and stored at −20 °C until further analysis.
Small angle X-ray scattering
Small angle X-ray scattering (SAXS) measurements were carried out at the beamlines I71122 and I911-423 of the MAX-lab Synchrotron, Lund University, Sweden. A monochromatic beam of 1.2 and 0.91 Å wavelengths was used at the respective beamlines. The range of the scattering vector q = (4π/λ)sin(θ) (where 2θ is scattering angle) was around 0.008 < q < 0.35 Å−1 for both cases. Two dimensional scattering pictures were obtained using an area-CCD detector (165 mm in diameter active area, from Marresearch, GmbH). WG–urea films of ca. 12 × 12 × 1 mm3 were measured with the incoming X-ray beam perpendicular to the film plane and only higher concentration WG–urea films i.e. WGG + 9U, WGG + 10U, WGG + 15U and WGG + 20U, were also scanned across the film plane using either 5 min or 30 s exposure times. The X-ray scattering data were processed using the graphical user interfaces bli911-423 and FIT2D.24 For comparison of the scattering intensities from different samples, the data were normalized by the integrated intensity incident on the sample during the exposure, and corrected for sample absorption, parasitic scattering and detector background.For the samples WGG, WGG + 10U, WGG + 15U and WGG + 20U, additional SAXS measurements at different temperatures e.g. 15, 20, 35, 45, 55, 60, 75, 80 and 90 °C were performed. For each temperature step 5 min thermalization was done before collecting the data. The exposure time was 60 s.
Wide angle X-ray scattering
Wide-angle X-ray scattering (WAXS) measurements were performed at the 911-2 beamline of the MAX-lab Synchrotron, Lund University, Sweden.25 The wavelength was 1.04 Å, and the sample-to-detector distance was 149.8 mm. Silicon powder was used as a calibration standard. Two-dimensional images were registered using an area-CCD detector (165 mm in a diameter active area, from Marresearch, GmbH) with 5 min of data acquisition. Parasitic scattering was subtracted from all diffractograms. The WG–urea films were analysed only with the incoming X-ray beam perpendicular to the film plane.TEM and double immunolocalization
Pieces of the WGG film (control), WGG + 10U and WGG + 20U urea films, size of approximately 2 × 2 × 0.5 mm3, were processed unfixed, dehydrated in graded series of ethanol followed by infiltration with London Resin White (LR White, TAAB, UK). Sample blocks were polymerized 48 h at 58 °C and post-polymerized 72 h at 40 °C. Ultrathin cross-sections (100–150 nm; perpendicular to thickness direction) were cut with an ultra-microtome and placed on Formvar-coated nickel grids (200 mesh). For double immunolocalization, section grids were treated with blocking solution of 5% goat normal serum and 1% bovine serum albumine (BSA) in phosphate buffered saline (PBS) (100 mM Na-phosphate, 150 mM NaCl, pH 7.2) for 30 min at RT followed by a mixture of two primary antibodies (rabbit anti-gliadin 1:2 and mouse anti-HMW glutenins 1:50 diluted with 1% BSA in PBS) overnight at 4 °C. Control grids were treated with buffer instead of primary antibodies. Sample grids were washed 4 × 5 min with PBS and incubated with a mixture of two secondary antibodies (goat anti-rabbit 2 nm gold 1:50 and goat anti-mouse 10 nm gold 1:20; British Biocell International, Cardiff, UK) for 2 h at RT. Grids were then washed 4 × 5 min with PBS and 6 × 2 min with distilled H2O, and routinely counterstained with uranyl acetate and lead citrate for analysis in a JEM-1010 transmission electron microscope (Jeol, Tokyo, Japan) at 60 kV.
Results and discussion
SAXS: X-ray beam perpendicular to the film plane
SAXS curves of the WG films containing 5, 7, 9, 10, 15 and 20% urea showed three well-defined Bragg peaks, which were distributed in the positional ratio (Fig. 1). This ratio indicated a two dimensional (2D) hexagonal close packed (HCP) arrangement of the scattering objects in the urea containing WG films. In contrast, for the WGG control film a broad peak (Fig. 1a) and for urea powder no clearly developed peak (Fig. 1b), were observed. For the WGG + 20U film at q = 0.116 Å−1 a small peak was noted (Fig. 1b, indicated by a star). The small peak seemed not to be correlated with the HCP structure (Fig. S1, ESI†) and may be possibly attributed to the greater amount of hydrogen bonds between the amine groups of the protein and urea.26
(Fig. 1). This ratio indicated a two dimensional (2D) hexagonal close packed (HCP) arrangement of the scattering objects in the urea containing WG films. In contrast, for the WGG control film a broad peak (Fig. 1a) and for urea powder no clearly developed peak (Fig. 1b), were observed. For the WGG + 20U film at q = 0.116 Å−1 a small peak was noted (Fig. 1b, indicated by a star). The small peak seemed not to be correlated with the HCP structure (Fig. S1, ESI†) and may be possibly attributed to the greater amount of hydrogen bonds between the amine groups of the protein and urea.26
 | ||
| Fig. 1 SAXS curves of control and urea containing WG films with the X-ray beam perpendicular to the film plane; Bragg peaks are indicated by arrows; * – indicates a small peak for WGG + 20U film. | ||
From the HCP symmetry presented in the urea containing WG films the interdomain distance dI (distance between the centres of the scattering objects) was calculated from the position of the first peak q1 with the equation:
 | (1) |
The unit cell parameter dI obtained for the urea containing WG films is reported in Table 1. A clear increase of dI with increasing concentration of urea was found (Table 1). Evidently urea, which is known to plasticise WG, swelled the HCP structure. In addition, urea is known to affect the protein in several ways, e.g. by chemical denaturation and promoting unfolding,27 decreasing the hydrophobicity, solvation effects,28 as well as by disruption of the secondary structure.29
| Material | d I interdomain distance [Å] |
|---|---|
| a The error bar is 0.2 Å, based on fitting of the first Bragg peak. | |
| WGG + 5U | 68.2 |
| WGG + 7U | 69.4 |
| WGG + 9U | 71.7 |
| WGG + 10U | 71.4 |
| WGG + 15U | 73.0 |
| WGG + 20U | 74.8 |
In a previous study, we have also observed hierarchical molecular superstructures in extruded films with specific additives, such as sodium hydroxide (NaOH), ammonium hydroxide (NH4OH) and salicylic acid (SA), together with specific processing conditions.5 The reactive additive, e.g. NH4OH, induced the formation of a similar HCP structure as was observed in the present study for the WG–urea films, while NaOH induced the formation of a tetragonal structure. The HCP structure was suggested to be related to improved film properties, including a high Young’s modulus and a low oxygen permeability.5 However, for the films used in the present study, previous studies have shown that the Young’s modulus was not as high18 as was reported for the WG–ammonium hydroxide films.14 In fact, the Young’s modulus of the WG–urea films was 10 times smaller18 compared to that of the WG–ammonium hydroxide films,5 even though both films exhibited similar HCP structures. A possible explanation is the plasticising effect of urea combined with its power to promote protein unfolding and stabilization of the denatured state.19 In the present films glycerol acts as an “external” plasticizer, while urea acts as an “internal” plasticizer and co-reacts with the protein matrix.
SAXS: X-ray beam parallel to the film plane and extrusion direction
WG–urea films containing 9, 10, 15 and 20% urea were further examined with the X-ray beam parallel to the film plane. SAXS patterns as a function of the scattering vector q showed three main Bragg peaks with a positional ratio (Fig. S1, ESI†).
(Fig. S1, ESI†).
In addition, the azimuthal intensity plot of I(φ) versus φ (φ = 0° when the scattering vector is at the equator and φ = 90° at the meridian) for these peaks was anisotropic and showed a local maximum distanced by 60°. Accordingly, the six-fold symmetry confirmed the bidimensional HCP structure, as was also observed with the X-ray beam perpendicular to the film plane in this study. For the WGG + 15U film the two-dimensional SAXS image and the corresponding azimuthal plots for the 1st and 3rd Bragg reflections are presented in Fig. 2 (positions of the rings are indicated by stars). In an ideal 2D HCP arrangement, the “spots” of the 1st and 3rd order reflections are typically placed at the angles φ = 30, 90, 150, 210, 270 and 310°.30 This was in agreement with the results observed in this study, although the higher intensity values were obtained at φ = 90 and 270° (Fig. 2b). A possible explanation for the different peak intensities is most likely due to the preferential orientation of the structure induced by the processing conditions.5
 | ||
| Fig. 2 SAXS patterns of the WGG + 15U film with the X-ray beam parallel to film plane and the extrusion direction: a) two dimensional (2D) image; the three main rings are indicated by *; b) azimuthal plot I(φ) versus φ for the 1st and 3rd Bragg reflections. Dotted lines indicate the expected position for a 6-fold symmetry in an ideal hexagonal lattice. | ||
For WGG + 9U, WGG + 10U and WGG + 20U samples the results of the azimuthal plots with the incoming X-ray beam parallel to the film plane and perpendicular to the extrusion direction showed two maxima for the Bragg peaks (Fig. S2, ESI†). This indicated that the elongated scattering objects of the HCP structure were oriented in the film plane along with the extrusion direction, as was for the WGG + 15U sample (Fig. 2 and S2†). The oriented HCP structure was observed previously for the NH4OH containing WG films,5 although higher intensity and better defined high order Bragg reflections were found for the urea films in comparison to NH4OH containing WG films. The development of the oriented HCP structure was most likely an effect of the similarities in the molecular/chemical structure of the additive and the protein, referring to e.g. the amino groups in both urea and ammonium hydroxide as well as in the protein.31 Also some differences should be noted, e.g. the extrusion temperature for the WG–urea films was higher (130 °C) compared to that of the WG–ammonium hydroxide films (120 °C), and this fact could have increased the urea reactivity.31 In addition, the contributing effect of glycerol on the HCP structure formation in both WG films should not be excluded, although, WGG itself did not form the HCP structure.
Urea film morphology revealed by WAXS
WAXS curves of the WGG control and the WG–urea films, displaying two main peaks and indicating an amorphous protein structure, are shown in Fig. 3. The peak at low q is related to the average distance of neighbouring helices (inter-helix distance), whereas the peak at the higher q-values is associated with the average backbone distance within the helix.10 Both these peaks have originated from the amorphous structure of WG proteins and are most likely due to the presence of α-helix structures observed previously in various systems.10,32,33 | ||
| Fig. 3 WAXS curves of WGG (control) and urea containing films. | ||
The scattering intensity of the two main peaks decreased with addition of urea in the samples (Fig. 3). The average position of the first peak displayed a d spacing of 9.7 Å, which was similar for all the studied films, including the WGG control and WGG + 20U films (Fig. 3). Although, some changes in the intensity and widening of the peak were observed, suggesting a more disordered inter-chain packing for WG–urea films with greater amounts of urea compared to those with lower amounts. For the peak at higher q-values the d spacing of ca. 4.5 Å was observed for the WGG control film and the lower urea concentration WG–urea films, i.e. WGG + 5U, WGG + 7U and WGG + 9U (Fig. 3). Interestingly, at a higher concentration of urea (i.e. WGG + 10U, WGG + 15U and WGG + 20U films), a clear shift in the maximum intensity of the same peak towards higher q values was observed. The peak observed had a characteristic d spacing of 4.2 Å (Fig. 3). A possible explanation for this is that a greater amount of urea leads to a greater extent of denaturation. This in turn favours unfolding of the protein, and the following large aggregation possibly leads to a “shrinking” more highly packed structure. At the same time some type of stabilisation of the WG–urea film structure by the protein–urea reactions occurs (urea reacts with transiently exposed, less-polar protein residues and the protein backbone).19,27,29 Also, the formation of β-sheets, reported previously,18 is highly significant.
Temperature effect on WG–urea film morphology
Polymer thermal treatment during processing results in internal molecular interactions (stresses) and a number of structural changes occur such as molecular weight, chain branching and extent of cross-linking through disulphide bond formation and other types of covalent bonding etc. Changes in protein structure and cross-linking of the gluten proteins after denaturation and at various applications have been described in a number of publications.34,35 In order to improve the understanding on the thermal stability of the HCP structure SAXS measurements were run at different temperatures and the results for the WGG + 15U film are presented in Fig. 4. At 35 °C or lower temperatures the WGG–urea films clearly showed the HCP structure (Fig. 4a). At higher than 25 °C temperature, the main Bragg peak moved towards greater q values, and at 45 °C, only the first Bragg peak of the HCP structure was observed and showed lower intensity (peak is smaller) compared to the lower temperatures (Fig. 4a). It is important to note that the SAXS pattern for the WGG + 15U film indicated a further change in the structure between 55 and 60 °C, where the HCP structure disappeared yielding only a broad peak with an average correlation distance of 67 Å. The same structural behaviour was observed for the WGG + 10U and WGG + 20U films (results not shown). For the WGG + 20U film no link between the small peak (Fig. 1 and S1†, indicated by star) and the HCP structure was observed. At 60 to 90 °C the peak shifted towards larger q values for the WGG + 15U film, indicating a reduction in the correlation distance till 61 Å at 90 °C (Fig. 4a). Also, the same shift of the broad correlation peak was observed for the WGG film at 60 to 90 °C temperature (Fig. 4b). A loss of the HCP organisation at 55 °C suggested that some of the bindings, i.e. hydrogen bonds (β-sheets), were disrupted at this temperature too. The shift of the broad peak with increasing temperature seemed to be due to the increase in mobility of the WG polymer chains and the formation of a more homogeneous protein organisation. Further study on the high temperature effect on the HCP structural phenomenon and its reversibility in the protein films is of interest to investigate.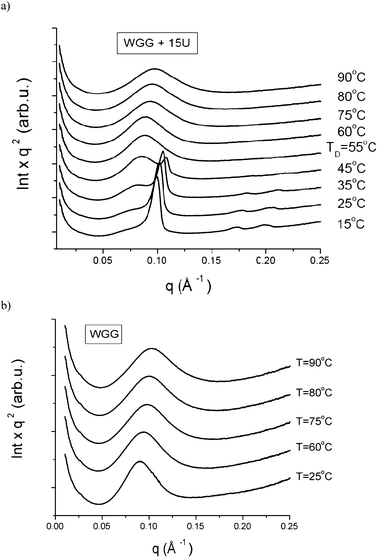 | ||
| Fig. 4 SAXS curves of WGG + 15U (a) and WGG (b) films: plots Iq2versus q as a function of temperature; TD, temperature where the HCP structure disappeared. | ||
Transmission electron microscopy (TEM)
The aim was to get a more detailed insight on the urea–WG film morphology using TEM. Due to the low contrast of biological samples in TEM, this task was however rather complex. In order to study the distribution of the hexagonal scattering objects we applied immunolabelling using two sizes of gold particles (2 and 10 nm). Gold particles were expected to be arranged in a manner related to the HCP structure in a given periodicity, or alternatively, the distribution of gold particles could possibly be related to inter-grain spaces. However, these type of patterns were not possible to reveal. Nevertheless, double immunolocalization of gliadin (2 nm gold) and HMW glutenins (10 nm gold) showed some difference between the WG films treated with or without urea (Fig. 5). Similarly, a difference between the WGG control and WG–urea films has been observed for the protein network structure by confocal laser scanning microscopy (CLSM).18 In this study, comparisons were made between a control sample WGG and WGG + 10U, since ultrathin sectioning of the WGG + 20U film was not possible due to the sample softness and stickiness. Fig. 5a–b shows that gliadins (small gold) were distributed bicontinuously in both samples treated with or without urea, following the more electron dense ridges in the samples. Control labelling without primary antibodies showed only occasional gold particles (Fig. 5c–d). Although immunolabelling was evident, HCP structures could not be visualized. This may be partially due to steric hindrance caused by the gold particles blocking the access of the secondary antibodies to the attached primary antibodies. | ||
| Fig. 5 TEM double immunolocalization of WG proteins. Large 10 nm gold particles show HMW glutenins and small 2 nm gold particles show gliadins. a) WGG (control) film treated with two primary antibodies; b) WGG + 10U film treated with two primary antibodies; c) WGG (control), control labelling without primary antibodies; d) WGG + 10U film, control labelling without primary antibodies; scale bar 100 nm. | ||
Conclusions
The molecular approach of protein structure is an attractive route towards understanding a structure-function relationship in bio-based protein materials. In this study, urea, a known denaturant of protein, has plasticised the WG films and showed an effect on WG film morphology. Independent on the urea concentration used in this study (5–20%), the HCP supramolecular structure was observed in all the studied WG–urea films. The WG proteins in the film were found to be in amorphous state. In the WG–urea films, the interdomain distance between the HCP scattering objects increased with increasing concentration of urea and varied between 68.2 and 74.8 Å (Table 1; schematically shown in Fig. 6). The HCP arrangement of the elongated scattering objects was also found to be dependent on the extrusion direction. The objects were oriented in the film plane, preferentially in the extrusion direction.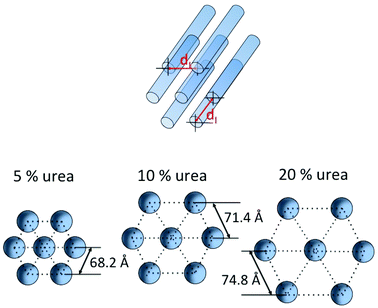 | ||
| Fig. 6 Schematic presentation of an idealized model of the supramolecular structure (HCP) for a WG protein polymer containing urea: the urea concentration effect on the HCP structure in WG–urea films; dI – distance between domains. | ||
The SAXS results on the temperature effect on WG structure showed that at a critical temperature, ≥55 °C, the HCP structure disappeared. The WAXS results indicated that the molecular packing of the WG protein polymer was distorted by the denaturation effect of urea. TEM images showed the denaturing effect of urea on the WG protein film structure. Further investigation on the temperature effect on the HCP structure is of interest in order to tailor bio-based polymers for innovative bio-based materials applications.
Acknowledgements
Governmental research program Trees and Crops for the Future (TC4F), VINNOVA and Bioraffinaderi Øresund are acknowledged for financial support. Professor Peter Shewry is thanked for the provided antibodies. MAX-lab is acknowledged for the beamtimes.References
- S.-W. Cho, H. Ullsten, M. Gällstedt and M. S. Hedenqvist, J. Biobased Mater. Bioenergy, 2007, 1, 56 CrossRef.
- M. A. Chan and J. M. Krochta, Tappi, 2001, 84, 1 Search PubMed.
- T. O. J. Blomfeldt, R. Kuktaite, E. Johansson and M. S. Hedenqvist, Biomacromolecules, 2011, 12, 1707 CrossRef CAS.
- S. W. Cranford, A. Tarakanova, N. M. Pugno and M. J. Buehler, Nature, 2012, 482, 72 CrossRef CAS.
- R. Kuktaite, T. S. Plivelic, Y. Cerenius, M. S. Hedenqvist, M. Gällstedt, S. Marttila, R. Ignell, Y. Popineau, O. Tranquet, P. R. Shewry and E. Johansson, Biomacromolecules, 2011, 12, 1438 CrossRef CAS.
- Z. Qin, L. Kreplak and M. J. Buehler, PLoS One, 2009, 4, 1 Search PubMed.
- I. Agnarsson, M. Kuntner and T. A. Blackledge, PLoS One, 2010, 5, e11234 Search PubMed.
- Z. Z. Shao and F. Vollrath, Nature, 2002, 418, 741 CrossRef CAS.
- N. Matsushima, G. Danno, N. Sasaki and Y. Izumi, Biochem. Biophys. Res. Commun., 1992, 2, 1057 CrossRef.
- N. H. Thomson, M. J. Miles, Y. Popineau, J. Harries, P. R. Shewry and A. S. Tatham, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1430, 359 CrossRef CAS.
- P. R. Shewry, M. J. Miles and A. S. Tatham, Prog Biophys. Mol. Biol., 1994, 61, 37 CAS.
- I. Olabarrieta, M. Gällstedt, I. Ispizua, J.-R. Sarasua and M. S. Hedenqvist, J. Agric. Food Chem., 2006, 54, 1283 CrossRef CAS.
-
M. Gällstedt, H. Ullsten, E. Johansson and M. S. Hedenqvist, Patent. PCT Int. Appl., 2010. CODEN: PIXXD2 WO 2010030234 A1 20100318 CAN 152:359236 AN 2010:330607, 1 Search PubMed.
- H. N. Ullsten, M. Gällstedt, G. M. Spencer, E. Johansson, S. Marttila, R. Ignell and M. S. Hedenqvist, Polym. Renew. Resour., 2010, 1, 173 Search PubMed.
- P. R. Shewry, Y. Popineau, D. Lafiandra and P. Belton, Trends Food Sci. Technol., 2001, 11, 433 CrossRef.
- H. Wieser, Food Microbiol., 2007, 24, 115 CrossRef CAS.
- F. Carrasco, P. Pagès, J. Gámez-Pérez, O. O. Santana and M. I. Maspoch, Polym. Degrad. Stab., 2010, 95, 116 CrossRef CAS.
- H. Türe, M. Gällstedt, R. Kuktaite, E. Johansson and M. S. Hedenqvist, Soft Matter, 2011, 7, 9416 RSC.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2003, 9, 5142 CrossRef.
- M. Gällstedt, A. Matozzi, E. Johansson and M. S. Hedenqvist, Biomacromolecules, 2004, 5, 2020 CrossRef.
- N. H. Ullsten, M. Gällstedt, E. Johansson, A. Gräslund and M. S. Hedenqvist, Biomacromolecules, 2006, 7, 771 CrossRef CAS.
- M. Knappila, C. Svensson, J. Barauskas, M. Zackrisson, S. S. Nielsen, K. N. Toft, D. Vestergaard, L. Arleth, U. Olsson, J. S. Pedersen and Y. J. Cerenius, J. Synchrotron Radiat., 2009, 16, 498 CrossRef.
- T. S. Plivelic, A. L. Labrador, K. Theodor, Y. Gapnonov, C. Svensson, J. Nygaard and Y. Cerenius, proceedings presented the 8th Nordic Workshop on Scattering from Soft Matter, Kjeller, Norway, ( 2011) Search PubMed.
- A. P. Hammersley, S. O. Svensson, A. Thompson, H. Graafsma, Å. Kvick and J. P. Moy, Rev. Sci. Instrum., 1995, 66, 2729 CrossRef CAS.
- C. B. Mammen, T. Ursby, Y. Cerenius, M. Thunnissen, J. Als-Nielsen, S. Larsen and A. Liljas, Acta Physica Polonica A, 2002, 101, 595 CAS.
- M. M. Thayer, R. C. Haillwanger, V. S. Allured, S. C. Gill and S. J. Gill, Biophys. Chem., 1993, 46165, 169 Search PubMed.
- L. Hua, R. Zhou, D. Thirumalai and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16928 CrossRef CAS.
- J. L. England and G. Haran, Annu. Rev. Phys. Chem., 2011, 62, 257 CrossRef CAS.
- A. Das and C. Mukhopadhyay, J. Phys. Chem. B, 2009, 113, 12816 CrossRef CAS.
- W. Ruland and B. Smarsly, J. Appl. Crystallogr., 2005, 38, 78 CrossRef.
- A. R. Goldfarb, Biochim. Biophys. Acta, Protein Struct., 1970, 1, 1 Search PubMed.
- W. Traub, J. B. Hutchinson and D. G. H. Daniels, Nature, 1957, 179, 769 CrossRef CAS.
- T. Blomfeldt, R. Kuktaite, T. S. Plivelic, F. Rasheed, E. Johansson and M. S. Hedenqvist, RSC Adv., 2012, 2, 6617 RSC.
- J. A. Delcour, I. J. Joye, B. Pareyt, E. Wilderjans, K. Brijs and B. Lagrain, in Annual Review of Food Science and Technology, Vol. 3 (Eds: M. P. Doyle, T. R. Klaenhammer), Annual Reviews, Palo Alto, CA, USA 2012, pages 469–492 Search PubMed.
- B. Lagrain, B. Goderis, K. Brijs and J. A. Delcour, Biomacromolecules, 2010, 11, 533 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c2ra21812g |
| This journal is © The Royal Society of Chemistry 2012 |