Intrinsic catalytic activity of Brønsted acid ionic liquids for the synthesis of triphenylmethane and phthalein under microwave irradiation†
Amol S.
Chaudhari
a,
Yogesh S.
Parab
b,
Vikas
Patil
a,
N.
Sekar
*a and
S. R.
Shukla
b
aTinctorial Chemistry Group, Department of Intermediate and Dyestuff Technology, Institute of Chemical Technology, N. P. Marg, Matunga, Mumbai 400 019, India
bDepartment of Textile and Fiber Processing Technology, Institute of Chemical Technology, N. P. Marg, Matunga, Mumbai 400 019. E-mail: nethi.sekar@gmail.com
First published on 18th September 2012
Abstract
A microwave-irradiated, ionic liquid-catalyzed, solvent-free method for the synthesis of triphenylmethane and a phthalein derivative has been developed from different aldehydes or anhydrides and substituted phenols or N,N-diaryl amines, respectively. Short reaction time, ambient reaction conditions, recyclability of catalyst, simple work up and high yields are some of the striking features of the present protocol. The immobilized catalyst could be easily recovered by simple filtration and recycled for up to four cycles without significant decrease in the catalytic activity.
Introduction
Phthalein, triphenylmethane and their analogues are vital heterocyclic motifs in dyes chemistry and have biological activities ranging from antibacterial, antiviral and anti-inflammatory, as well as being useful in photodynamic therapy.1–9 Other useful applications of these heterocycles are as fluorescent materials for visualization of bio-molecules and in laser technologies.10,11–13 Furthermore, these molecules find potential application in concurrent analytical assay expansion and measurement of intracellular pH while screening biologically active species for immunoassays, as biological probes, environmental sensors, molecular devices, ferric ion sensors, copper ion sensors and nerve gas sensors.14–16 Due to this wide range of applications, the synthesis of phthalein and triphenylmethane-type compounds has received considerable attention in recent years.15,16 A number of strategies have been reported for the preparation of triphenylmethane, fluorescein and rhodamine.17 Traditionally, Brønsted acids (H2SO4 or MeHSO3), niobium pentachloride, zinc(II) chloride, alkali disulphates, HClO4, hydrochloric acid, lead dioxide, manganese(IV) oxide, Lewis acids (BBr3, AlCl3 or ZnCl2), TiCl4, In(III) or mixtures thereof have been used in the synthesis of phthalein and triphenylmethane derivatives.18–26 These methodologies have drawbacks such as prolonged reaction time (4–5 h), tedious catalyst preparation and work up, formation of inevitable side products, and exhaustive usage of energy sources and solvents, which result in a lower yield of the desired product. Considering all these factors and with an emphasis on human health and environmental protection, the development of an eco-friendly methodology for the synthesis of phthalein and triphenylmethane derivatives is in great demand.Brønsted acid ionic liquids (BAILs) have been used in many areas due to their exclusive chemical and physical properties.27 They display the useful features of solid acids and mineral liquid acids, hence they are a good alternative to traditional mineral liquid acids in chemical procedures. Brønsted acid ILs have potential as dual solvents–catalysts in organic reactions.28 BAILs ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) have been successfully used in transesterification reactions,29 cyclocondensations,30 oxa-Michael addition reactions,31 and Prins reactions.32 Remarkably, the synthesis of triphenylmethane and phthalein derivatives using BAILs ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) as solvents–catalysts has not been reported yet.
In continuation of our previous research on fluorescent dyes and the development of green methodologies for the synthesis of heterocyclic compounds of biological importance,32–34 herein the microwave irradiated synthesis in the presence of ionic liquids ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) is reported. This is an efficient and reusable solvent–catalyst system for the synthesis of triphenylmethane derivatives (Scheme 1). In order to further extend the utility of ILs, the synthesis of phthalein derivatives is reported in Scheme 2. The reaction was completed smoothly and purification of the product was fairly simple. ILs ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) were conveniently separated from the products and easily recycled for another set of reactions.
 | ||
| Scheme 1 Schematic representation of the synthesis of triphenylmethane. | ||
 | ||
| Scheme 2 Schematic representation of the synthesis of phthalein. | ||
Results and discussion
Optimisation of the catalyst
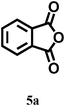
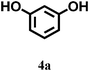
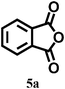
In our preliminary experiments, we carried out a reaction under solvent-free conditions as a model reaction (Scheme 3). The control reaction was carried out between phthalic anhydride 5a and resorcinol 4a under conventional as well as microwave heating in the presence of different solid acid catalysts, mineral liquid acid catalysts, ILs and in the absence of catalyst (Scheme 3, Table 1, entries1–8).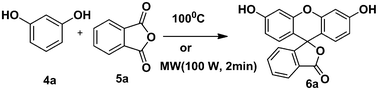 | ||
| Scheme 3 Reaction conditions: (2 mmol) resorcinol and (1 mmol) phthalic anhydride in 5 mmol (1 mL) of IL under microwave irradiation (100 W) for 2 min or reflux at 100 °C for 60 min. | ||
| Entry | Catalyst | Amount of catalyst (mmol) | Microwavea | Conventional b | ||
|---|---|---|---|---|---|---|
| Time (min) | Yield (%) | Time (h) | Yield (%) | |||
| a Reaction conditions: (2 mmol) resorcinol and (1 mmol) phthalic anhydride under microwave irradiation (100 W) in different catalyst. b Reaction conditions: (2 mmol) resorcinol and (1 mmol) phthalic anhydride reflux at 100 °C for 60 min in different catalyst. c Reaction carried out under solvent-free conditions (fused reaction). | ||||||
| 1 | Without catalyst | — | 2 | — | 5 | — |
| 2 | HCl | 5 | 2 | — | 5 | — |
| 3 | H2SO4 | 5 | 2 | 80 | 5 | 50 |
| 4 | AlCl3c | 2c | 2 | 75 | 4 | 35 |
| 5 | ZnCl2c | 2c | 2 | 75 | 4-5 | 45 |
| 6 | TiCl4 | 2 | 2 | — | 5 | — |
| 7 | ([HNMP]+ [CH3SO3]−) | 5 | 2 | 88 | 0.5 | 85 |
| 8 | ([HNMP]+ [HSO4]−) | 5 | 2 | 87 | 0.5 | 87 |
In this set we reacted the previously mentioned substrates in the presence of various catalysts viz. HCl, H2SO4, AlCl3, ZnCl2, TiCl4, ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) at the same temperature. This set was used for the purpose of screening for the most efficient catalyst by observing their effects through reaction monitoring. Effects of the catalysts and microwave irradiation (80–100 W) on the synthesis of triphenylmethane and phthalein analogues are summarized in Table 1, entries 1–8. It was interesting to note that the ionic liquids ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) offer the best results (Table 1, entries 7–8) owing to their ability to activate the carbonyl groups of anhydrides or aldehydes. This increases the rate of formation of C–C bonds via electrophilic substitution, followed by intramolecular cyclization rationalised by a proposed mechanism (Fig. 1).
![Plausible mechanism for the synthesis of phthalein derivatives promoted by [HNMP]+ MeSO3.](/image/article/2012/RA/c2ra21803h/c2ra21803h-f1.gif) | ||
| Fig. 1 Plausible mechanism for the synthesis of phthalein derivatives promoted by [HNMP]+ MeSO3. | ||
Optimisation of amount of ILs
We optimized the reaction conditions such as the amount of ionic liquid needed for the maximum yield of phthalein and triphenylmethane derivatives by using a model reaction (Table 1, Scheme 3). It was observed that the amount of ionic liquid plays a key role in catalyzing the formation of phthalein and triphenylmethane derivative (Scheme 3).It was found that the optimum reaction rate and yield could be achieved with 5 mmol (1 mL) of the ionic liquid (IL) with and without microwave irradiation under solvent-free conditions. We applied these conditions to the synthesis of various phthalein and triphenylmethane derivatives (Table 2, entries 1–15).
| Entry | Aldehyde | Substituted aniline or phenol | Product | Microwave | Conventional | |||
|---|---|---|---|---|---|---|---|---|
| Time (min)/MW (W) | Yield (%) | T/°C | Time (min) | Yield (%) | ||||
| 1 |

|

|

|
2/80 | 97 | 80 | 30 | 90 |
| 2 |

|

|

|
3/80 | 87 | 80 | 60 | 85 |
| 3 |
|

|

|
2/80 | 84 | 80 | 60 | 78 |
| 4 |

|

|

|
2/80 | 86 | 60 | 60 | 82 |
| 5 |

|

|

|
5/80 | 88 | 80 | 40 | 79 |
| 6 |

|

|

|
2/80 | 80 | 100 | 60 | 68 |
| 7 |
|

|

|
2/80 | 78 | 78 | 60 | 73 |
| 8 |
|

|

|
2/100 | 89 | 100 | 60 | 85 |
| 9 |

|

|
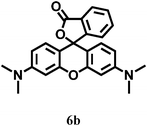
|
2/100 | 90 | 100 | 60 | 85 |
| 10 |
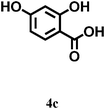
|
|

|
2/100 | 97 | 80 | 30 | 83 |
| 11 |

|

|

|
3/100 | 78 | 120 | 30 | 73 |
| 12 |
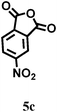
|
|

|
5/100 | 80 | 100 | 30 | 74 |
| 13 |
|

|
|
5/100 | 79 | 120 | 70 | 65 |
| 14 |

|

|

|
5/100 | 74 | 120 | 70 | 70 |
| 15 |

|

|

|
5/100 | 78 | 120 | 70 | 73 |
Optimisation of microwave and conventional heating
The synthesis of triphenylmethane and phthalein was optimised for catalysts ([HNMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−). The optimised microwave conditions were 80–100 W (9.6 × 103–1.2 × 104 J of heat) and conventional heating between 60–120 °C was investigated (Table 2, entries 1–15).With regard to the work up, the procedures were very simple and included the addition of sufficient amounts of ice cold water at the end of the reaction. The product was separated by filtration. Finally the crude product was recrystallised from ethanol. As is evident from the results, ([HNMP]+ [CH3SO3]−) and ([NMP]+ [CH3SO3]−) were found to be effective solvents–catalysts for the synthesis of the above mentioned reactions (Table 2, entries 1–15).
Reusability of ILs ([NMP]+ [CH3SO3]−) and ([HNMP]+ [HSO4]−) as catalysts
The recovery and reusability of catalysts (ILs) is a requirement for an eco-friendly catalytic process. In this protocol, we used ([HNMP]+ HSO4−) and ([HNMP]+ [CH3SO3]−) as solvents as well as catalysts under both microwave and conventional heating conditions (Scheme 3). After completion, the reaction mixture was cooled to room temperature then poured into a mixture of crushed ice and water. The crude product was separated by filtration. The ionic liquid was then recovered by evaporating the water under reduced pressure. The recovered ionic liquid was reused for the next four cycles of the same reaction without significant loss of activity. (Fig. 2)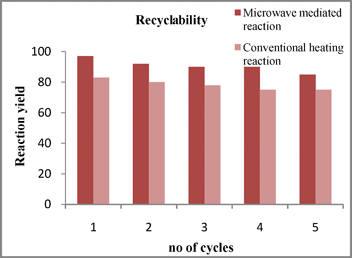 | ||
| Fig. 2 Recyclability study on the reaction depicted in Scheme 3. | ||
Experimental
All chemicals were commercially available and used without further purification. 1H NMR and 13C NMR spectra were obtained on a Varian Mercury plus 300 MHz NMR spectrometer. Chemical shift values are expressed in δ units relative to tetramethylsilane (TMS) signal as the internal reference in CDCl3. Data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, br = broad singlet, m = multiplet), coupling constant J (Hz).Preparation of ionic liquid N-methyl-2-pyrrolidonium methyl sulphate ([HNMP]+ [CH3SO3]−)35
Benzene (30 ml) was mixed with N-methyl-2-pyrrolidone (9.9 g, 0.1 mol) under vigorous stirring in a 100 mL flask. Then, methanesulphonic acid (9.6 g, 0.1 mol) was slowly added dropwise to the flask within 30 min in an ice bath. The reaction lasted for another 4 h at room temperature. Benzene was removed under reduced pressure and the product was further dried at 90 °C under 1–5 mmHg for 1 h. A light yellow viscous liquid [HNMP]+ [CH3SO3]− was obtained (yield: 97%) (Fig. 3a). | ||
| Fig. 3 a: Schematic presentation of synthesis of N-methyl-2-pyrrolidonium methyl sulphate, b: Schematic representation of the synthesis of N-methyl-2-pyrrolidonium hydrogen sulphate. | ||
Preparation of ionic liquid N-methyl-2-pyrrolidonium hydrogen sulphate ([HNMP]+ [HSO4]−)35
1-Methylpyrrolidone (0.2 mol) was placed in a 250 mL three necked flask with overhead stirrer. Concentrated sulphuric acid (0.2 mol) was slowly added dropwise to the flask within 40 min in an ice bath. The flask was then kept at 80 °C for 12 h to complete the reaction. The mixture was washed three times with ether to remove non-ionic residues and dried in vacuo by a rotary evaporator to obtain the viscous clear liquid ([NMP]+ HSO4−) (Fig. 3b). The preparation and purification of ionic liquids were discussed by Z. S. Qureshi and et al.28 and the products were used for further reactions as they were.General procedure for the preparation of triphenylmethane and phthalein derivatives‡
Microwave irradiation heating36
A 700 W Electrolux (17 L) domestic microwave oven was used. It was modified to allow fitting of a condenser as described in the previous communication.36 As mentioned in the above section, the same procedure was followed with a microwave oven being used as a heating source for various time periods between 2–5 min and 80–100 W for the synthesis of different derivatives.Conclusions
In summary, we have demonstrated a new, efficient and practical procedure for the synthesis of triphenylmethane and phthalein derivatives. Results showed that ([HNMP]+ HSO4−) and ([NMP]+ [CH3SO3]−) are efficient catalysts for the reaction of different aldehydes/anhydrides with different aryl amines/phenols. The advantages of our protocol are easy work up, fast reaction rates, mild reaction conditions, good yields, and reusability of ILs, which make the method an attractive and useful contribution to the existing methodologies.Acknowledgements
Authors are thankful to the Indian Institute of Technology, Mumbai for recording NMR and Mass Spectra. Amol Chaudhari is thankful to the Centre of Advanced Studies University Grant Commission for financial support by way of a SAP fellowship.References
- J. Culp and F. Beland, Int. J. Toxicol., 1996, 15, 219–238 CrossRef
.
- D. Alderman, J. Fish Dis., 1985, 8, 289–298 CrossRef CAS
- B. Cho, T. Yang, L. Blankenship, J. Moody, M. Churchwell, F. Beland and S. Culp, Chem. Res. Toxicol., 2003, 16, 285–294 CrossRef CAS
- A. Fryb, J. Gen. Microbiol., 1957, 16, 341–349 CrossRef
- R. Cooney, A. Pung, P. Harwood, A. Boynton, L. Zhang, M. Hossain and J. Bertram, Carcinogenesis, 1992, 13, 1107–1112 CrossRef CAS
- J. P. Goddard and J. L. Reymond, Curr. Opin. Biotechnol., 2004, 15, 314–322 CrossRef CAS
- L. D. Lavis, T. Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS
- J. Kao, A. Harootunian and R. Tsien, J. Biol. Chem, 1989, 264, 8179–8184 CAS
- J. Nicolas, V. Miguel, G. Mantovania and D. Haddleton, Chem. Commun., 2006, 45, 4697–4699 RSC
- J. R. Lakowicz, I. Gryczybki, V. Bogdano and J. Kdba, J. Phys. Chem., 1994, 98, 334–342 CrossRef CAS
- S. Rao Venugopal, A. A. Bettiol, K. C. Vishnubhatla, S. N. Bhaktha, D. Rao Narayana and F. Watt, Appl. Phys. Lett., 2007, 90, 101115–101117 CrossRef
- S. Rao, N. Srinivas and D. Rao, Chem. Phys. Lett., 2002, 361, 439–445 CrossRef
- S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831–7841 CrossRef CAS
- H. Kim, M. Lee, H. Kim and J. Kim, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC
- M. Beija, C. Afonso and Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC
- Y. Duan, M. Liu, W. Sun, M. Wang, S. Liu and Q. X. Li, Mini-Rev. Org. Chem., 2009, 6, 35–43 CrossRef CAS
- H. Jun-Tao, G. Jian- Wu and Z. Zhan-Hui, Monatsh. Chem., 2011, 142, 495–499 CrossRef
- P. Mariappan, J. Natarajan and B. Pandi, J. Org. Chem., 2000, 65, 3548–3550 CrossRef
- P. Mariappan, K. Neela and K. N. Jayakumar, Tetrahedron Lett., 2007, 48, 1955–1958 CrossRef
- B. M. Fox, J. D. Hepworth, D. Mason and G. Hallas, J. Chem. Soc., Perkin Trans. 2, 1982, 8, 987–991 RSC
- O. Fischer, Chem. Ber., 1881, 14, 2520–2529 CrossRef
- A. von Bayer, Chem. Ber., 1871, 5, 255–259 Search PubMed
- H. Mereyala and K. Sambaru, Ind. J. Chem., 2005, 44B, 615–617 CAS
- M. Clark, S. Hilderbrand and S. Lippard, Tetrahedron Lett., 2004, 45, 7129–7131 CrossRef CAS
- S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef CAS
- M. Picquet, I. Tkatchenko, I. Tommasi, P. Wassersscheid and J. Zimmermann, Adv. Synth. Catal., 2003, 345, 959–962 CrossRef CAS
- Z. S. Qureshi, K. M. Deshmukh, M. D. Bhor and B. M. Bhanage, Catal. Commun., 2009, 10, 833–837 CrossRef CAS
- P. P. Salvi, A. M. Mandhare, A. S. Sartape, D. K. Pawar, S. H. Han. and S. S. Kolekar, C. R. Chim., 2011, 14, 883–886 CrossRef CAS
- H. Guo, X. Li, J. Wang, X. Jin and X. Lin, Tetrahedron, 2010, 66, 8300–8303 CrossRef CAS
- W. Wang, L. Shao, W. Cheng, J. Yang and M. He, Catal. Commun., 2008, 9, 337–341 CrossRef CAS
- V. Patil, V. Padalkar, K. Phatangare, V. Gupta, P. Umape and N. Sekar, J. Phys. Chem. A, 2012, 116, 536–545 CrossRef CAS
- V. Padalkar, V. Patil, K. Phatangare, V. Gupta, P. Umape and N. Sekar, Green Chem. Lett. Rev., 2012, 5, 139–145 CrossRef CAS
- V. Padalkar, V. Patil, K. Phatangare, V. Gupta, P. Umape and N. Sekar, Synth. Commun., 2011, 41, 925–938 CrossRef CAS
- V. Patil, V. Padalkar and A. Chaudhari, Catal. Sci. Technol., 2012, 2, 1681–1684 CAS
- N. Pingale and S. Shukla, Eur. Polym. J., 2008, 44, 4151–4156 CrossRef CAS
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21803h |
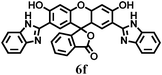
| ‡ 4,4′-(Cyclohexylmethylene) bis (N,N-dimethylaniline) 3c (Table 2, entry 2): Yield: 85%. M.p. 150–152 °C. FT-IR (KBr, cm−1): 3061, 2926, 1611, 1532, 1440, 1347. 1H NMR (CDCl3): δ 1.27–1.52 (m, 10H), 2.35 (m, 1H), 2.95 (s, 12H), 4.19 (d, 1H), 6.61 (d, 4H), 7.25 (d, 4H). 13C NMR (CDCl3): δ 14.83, 133.90, 128.66, 128.54, 113.13, 112.99, 54.03, 41.61, 41.05, 32.35, 29.79, 25.57. 4,4′-(3-Methylpentane-1,1-diyl) bis (N,N-dimethylaniline) 3d (Table 2, entry 3): Yield: 84%. M.p. > 250 °C. FT-IR (KBr, cm−1): 3070, 2915, 1465, 1375, 800. 1H NMR (CDCl3): δ 0.97 (t, 6H), 1.45 (m, 1H), 1.89 (t, 2H), 2.80 (s, 12H), 3.95 (d, 1H), 6.69 (d, 4H), 7.15 (d, 4H). 13C NMR (CDCl3)δ 149.05, 148.86, 134.76, 130.62, 129.50, 129.40, 128.40, 128.31, 113.26, 113.13, 46.85, 45.51, 43.30, 41.19, 41.06, 41.00, 40.93, 39.96, 29.77, 25.57, 22.81.4,4′-(3-Phenylprop-2-ene-1,1-diyl)bis (N,N-dimethylaniline) 3f (Table 2, entry 5): Yield: 88%. FT-IR (KBr, cm−1): 3065, 2924, 1611, 1470, 1532, 801. 1H NMR (CDCl3): δ 2.95 (s, 12H), 4.80 (d, 1H), 6.60 (d, 1H), 6.80 (d, 1H), 7.10 (m, 8H), 7.21–7.60 (m, 5H). 13C NMR (CDCl3): δ 144.09, 136.34, 134.74, 130.08, 129.92, 128.48, 127.08, 116.78, 113.09, 52.79, 40.78. 2′,7′-Di(1H-benzo[d]imidazol-2-yl)-3′,6′-dihydroxy-3H-spiro[isobenzofuran-1-9′-xanthen]-3-one) 6f (Table 2, entry 13): Yield: 79%. M. p. > 300 °C. FT-IR (KBr, cm−1): 3432, 3324, 1768 , 1740, 1689, 1642, 1578 ,1550,1515, 1441, 1402, 1339 (C–N), 1252, 1190, 1130. Mass: m/z 564 (M+). 1H NMR ((CD3)2SO, 300 MHz): δ 6.58 (s, 2H), 7.17 (d, 2H, J = 6.4 Hz), 7.34 (d, 2H, J = 6.4 Hz), 7.68 (d, 2H, J = 6.4 Hz), 7.72 (dd, 2H, J = 6.4 Hz; 14.9 Hz), 8.00 (s, 2H), 8.2–8.3 (d, 4H, J = 8.9 Hz), 8.4 (s, 2H), 11.3 (bs, 2H). 13CNMR ((CD3)2SO,75 MHz): 5.46, 166.00, 106.21, 11.92, 115.21, 115.39, 124.11, 25.83, 127.29, 130.74, 141.71, 151.73, 152.48, 152.93. |
| This journal is © The Royal Society of Chemistry 2012 |