Efficient removal of arsenate by versatile magnetic graphene oxide composites†
Guodong
Sheng
ab,
Yimin
Li
a,
Xin
Yang
b,
Xuemei
Ren
b,
Shitong
Yang
b,
Jun
Hu
b and
Xiangke
Wang
*b
aSchool of Chemistry and Chemical Engineering, Shaoxing University, Huancheng West Road 508, Shaoxing, Zhejiang 312000, P.R. China
bKey Laboratory of Novel Thin Film Solar Cells, Institute of Plasma Physics, Chinese Academy of Sciences, P.O. Box 1126, Hefei, 230031, P.R. China. E-mail: xkwang@ipp.ac.cn; Fax: +86 5515591310; Tel: +86 551 5592788
First published on 8th October 2012
Abstract
The magnetic graphene oxide (MGO) composites were prepared by coprecipitation of FeCl3·6H2O and FeCl2·4H2O on graphene oxide (GO) nanosheets and characterized in detail. The Fe3O4 was uniformly deposited on the surface of GO. The synthesized MGO composites were used as a versatile adsorbent for As(V) removal from aqueous solutions. The results showed that the adsorption of As(V) on MGO is an endothermic process and the adsorption kinetic fitted the pseudo-second-order model well. The MGO composites had a good adsorption capability for As(V) removal and the adsorption isotherms were described by the Langmuir model better than by the Freundlich model. The adsorption of As(V) on MGO decreased with ascending pH due to the electrostatic interaction. In addition, the adsorption of As(V) on MGO was greatly affected by the nature and concentration of coexisting cations and anions. The presence of coexisting anions showed an inhibiting effect on As(V) adsorption, which was more efficient at low pH, whereas the presence of coexisting cations showed an enhancing effect on As(V) adsorption, which was more efficient at high pH. The results of this work indicated that the combination of the excellent adsorption capacity of GO and the magnetic properties of Fe3O4 nanoparticles is very important in drinking water treatment due to the easy magnetic separation of MGO from aqueous solutions.
1. Introduction
The presence of hazardous contaminants in groundwater is an important issue in environmental pollution management due to their great consequences on human health.1,2 Arsenic, which is toxic to human and other living organisms, is commonly regarded as one of the most hazardous contaminants and can impose serious environmental problems throughout the world.2 Arsenic occurs naturally in soils, rocks, water and air, volcanic activity, the erosion of rocks, forest fires and anthropogenic activities are also responsible for arsenic release into the natural environment.2,3 In the natural soil and water environment, arsenic is mainly present in two species, i.e., arsenite (As(III)) and arsenate (As(V)), and arsenate (As(V)) is the main arsenic form under oxidizing conditions.4,5Thus far, a variety of remediation technologies, including nanofiltration, reverse osmosis, coagulation/coprecipitation, electrodialysis, ion exchange, adsorption, solvent extraction, foam flotation and bioremediation have been developed for As(V) removal.3 Based on our literature review, it was generally regarded that adsorption is a promising process among these remediation technologies because it is a simple, economic and suitable operation method. Therefore, a large number of adsorbents, such as iron(III) (hydr)oxides,5–9 natural and modified zeolite,10,11 titanium dioxide,12,13 pillared phyllosilicates14–17 and iron-based granular materials,3,11,18,19 have been studied for the efficient removal of As(V). However, there are still some issues that limit the practical application of these adsorbents, for example, the adsorption capacity of these materials is not high enough; and it is difficult to separate the adsorbents from aqueous solutions, which limits the application on a large scale in the practical applications of As(V) removal. In order to address these issues, it is of significant importance to explore new adsorbents with high adsorption capacity and the advantage of easy separation from solutions.
Graphene oxide (GO), which is considered as the oxidized graphene, is a lamellar flexible material with a wide range of oxygen-containing functional groups, such as epoxy (C–O–C), hydroxyl (OH) and carboxyl (COOH) on GO surfaces.20–22 Due to the oxygen-containing functional groups on the GO surface and its high surface area, GO has high adsorption capacity for the environmental contaminant cleanup in practical applications.20–22 Therefore, lots of graphene-based multifunctional hybrid materials have been synthesized using GO and applied for the removal of various environmental contaminants.20–29 More importantly, the introduction of magnetic materials into GO can combine the high adsorption capacity of GO and the separation convenience of magnetic materials. The preparation of graphene-based magnetic composites has been reported recently.22,27–29 However, to the best of our knowledge, the limited information about the adsorption performance and mechanism of graphene-based magnetic composites towards inorganic and organic contaminants severely limits the practical application of this versatile magnetic composite in environmental pollution management.
In this work, the magnetic graphene oxide (MGO) composites were synthesized and applied to the removal of As(V) from aqueous solutions by batch experiments. The objectives of this study were: (1) to investigate the effect of pH on As(V) adsorption; (2) to study the adsorption of As(V) onto MGO as affected by the nature and concentration of organic matters (oxalic acid (OA), succinic acid (SA), malic acid (MA), citric acid (CA), humic acid (HA) and fulvic acid (FA)), inorganic anions (fluoride (F−), chloride (Cl−), nitrate (NO3−), sulfate (SO42−), bicarbonate (HCO3−) and monohydrogen phosphate (HPO42−)) and coexisting cations (K+, Na+, Ca2+, Mg2+, Al3+ and Fe3+ ions); and (3) to discuss the interaction mechanism of As(V) with MGO under different experimental conditions. This study highlighted the broad applicability of this versatile material in environmental pollution cleanup.
2. Experimental section
2.1 Materials
Graphite powder (70 μm, Qingdao Graphite Company, China), ammonia solution, H2SO4 (98%), H2O2 (30%), KMnO4, FeCl3·6H2O, FeCl2·4H2O, glycol and all other chemicals in analytical purity were purchased from Sinopharm Chemical Reagent Co. Ltd. and used without further purification. Humic acid (HA) and fulvic acid (FA) were extracted from the soil of Hua-Jia county (Gansu province, China) and had been characterized in detail in our previous report.30 Milli-Q (Millipore, Billerica, MA, USA) water was used in all experiments.2.2 Preparation of GO and MGO composites
The preparation and characterization of GO and MGO composites had been reported in our previous study in detail.22 GO was prepared from natural graphite by the modified Hummers method.20,31,32 The MGO composites were synthesized by coprecipitation of FeCl3·6H2O and FeCl2·4H2O in the presence of GO (see Supplementary Information†).2.3 Adsorption experiments
MGO stock suspensions were prepared by ultrasonication of 0.1 g MGO in Milli-Q water to achieve the concentration of 4 g L−1. As(V) stock solution was prepared by dissolving Na2HAsO4·7H2O into Milli-Q water. For As(V) adsorption, at first, MGO aqueous solution was severely shaken to obtain a homogeneous suspension. Then, MGO suspension was transferred homogeneously to a 10 mL polyethylene (PE) tube using a syringe and finally, As(V) solution was added to the PE tube to achieve the desired concentrations of different components. The pH was adjusted by adding negligible volumes of 0.01 or 0.1 mol L−1 NaOH or HNO3. After the mixture was oscillated for 24 h, the solid and liquid were separated by the magnetic separation method. As(V) concentration was determined with atomic absorption spectrophotometry with vapour generation assembly (AAS-VGA). The removal percentage and adsorption capacity of As(V) were calculated from the difference between the initial concentration (C0, mg L−1) and the equilibrium one (Ce, mg L−1): Removal (%) = (C0 − Ce)/C0 × 100% and qe = (C0 − Ce) × V/m, where qe is the adsorbed amount of As(V) on MGO at equilibrium time (mg g−1), V is the solution volume (L) and m is the mass of MGO (g).3. Results and discussion
3.1 Characterization of MGO
Fig. 1 shows the typical scanning electron microscope (SEM) and transmission electron microscope (TEM) images of MGO. Compared with the SEM and TEM images (see Fig. S1†) of the GO sample, it can be clearly seen from Fig. 1A that GO and the matrix of Fe3O4 particles and some wrinkles can be observed on MGO surface, which may be attributed to the increased aggregation of GO. One can see from Fig. 1B that the Fe3O4 nanoparticles are well distributed on GO nanosheets.
The X-ray diffraction (XRD) pattern of the MGO composites is shown in Fig. S2†. The main peaks at 2θ = 30.21° (220), 35.71° (311), 43.31° (400), 53.7° (422), 57.35° (511) and 62.72° (440) indicate the characteristics of Fe3O4 with a face-centered cubic (fcc) structure on the MGO composites. The broad diffraction peaks are suggestive of nanoparticles with a very small size.33 The Fourier transform infrared spectroscopy (FTIR) spectrum of the MGO composites is shown in Fig. S3†. The peaks of epoxy C–O (∼1225 cm−1), aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (∼1578 cm−1) and O–H band (∼3389 cm−1) of H2O can be observed in the MGO composites (Fig. S3†). The peak around ∼584 cm−1 is attributed to Fe–O.33 The FTIR spectrum of the GO is also shown in Fig. S3† to make a comparison between the functional groups. It can be clearly seen that the peak around ∼584 cm−1, which is attributed to Fe–O, is not present in the FTIR spectrum of the GO. The magnetization property of MGO was investigated at room temperature by measuring the magnetization curve (Fig. S4†). The saturation magnetization (Ms) of MGO was 31 emu g−1 (magnetic field ±20 kOe), suggesting that MGO had a high magnetism.22 It can be obviously seen from Fig. S5† that MGO nanoparticles are attracted quickly toward the magnet and the clear solution can be easily removed by being decanted off. This simple magnetic separation experiment indicates that MGO can be used as a magnetic adsorbent to remove contaminants from aqueous solutions.
C (∼1578 cm−1) and O–H band (∼3389 cm−1) of H2O can be observed in the MGO composites (Fig. S3†). The peak around ∼584 cm−1 is attributed to Fe–O.33 The FTIR spectrum of the GO is also shown in Fig. S3† to make a comparison between the functional groups. It can be clearly seen that the peak around ∼584 cm−1, which is attributed to Fe–O, is not present in the FTIR spectrum of the GO. The magnetization property of MGO was investigated at room temperature by measuring the magnetization curve (Fig. S4†). The saturation magnetization (Ms) of MGO was 31 emu g−1 (magnetic field ±20 kOe), suggesting that MGO had a high magnetism.22 It can be obviously seen from Fig. S5† that MGO nanoparticles are attracted quickly toward the magnet and the clear solution can be easily removed by being decanted off. This simple magnetic separation experiment indicates that MGO can be used as a magnetic adsorbent to remove contaminants from aqueous solutions.
3.2 Adsorption kinetics
The kinetics of As(V) adsorption on MGO was determined to understand the adsorption property of As(V) on MGO. The effect of contact time on As(V) adsorption to MGO at pH = 5.0 and 6.5 is shown in Fig. 2. The adsorption can be divided into two stages: (1) a rapid uptake within the first 4 h of contact time, in which ∼88% of As(V) was removed at pH = 5.0 and ∼42% of As(V) was removed at pH = 6.5; and (2) a slow uptake thereafter until an equilibrium was reached. Based on our literature review, any adsorption process is normally governed by three steps: (i) transport of adsorbate from solution to the film surrounding adsorbent, (ii) from the film to the adsorbent surface, and (iii) from the surface to the active adsorbing sites.34 Herein, As(V) adsorption on MGO can be described as follows: first, As(V) transport from solution to the film surrounding MGO, then, from the film to MGO surface, and finally, from MGO surface to active adsorbing sites.![The adsorption kinetics of As(v) onto MGO, T = 298 K, C[As(v)] = 25 mg L−1, m/V = 0.4 g L−1, symbols represent experimental data, solid lines represent the kinetic model fitting. The inset in (A): the pseudo-second-order model fitting and (B): intra-particle diffusion model fitting.](/image/article/2012/RA/c2ra21623j/c2ra21623j-f2.gif) | ||
| Fig. 2 The adsorption kinetics of As(V) onto MGO, T = 298 K, C[As(V)] = 25 mg L−1, m/V = 0.4 g L−1, symbols represent experimental data, solid lines represent the kinetic model fitting. The inset in (A): the pseudo-second-order model fitting and (B): intra-particle diffusion model fitting. | ||
The pseudo-second-order kinetic model and intra-particle diffusion model have been widely used to fit the kinetic process of metal ion adsorption at the solid/water interfaces, therefore, these two models were used to fit the adsorption of As(V) on MGO in this work. The pseudo-second-order kinetic model fitted well with the experimental data (Fig. 2A). The kinetic equation was expressed as: t/qt = 1/(K2 × qe2) + t/qe, where qe (mg g−1) is the adsorption amount at equilibrium time and qt (mg g−1) is the amount of As(V) adsorbed at contact time t (h). K2 (g mg−1 h−1) is the pseudo-second-order rate constant.35–37 The values of qe calculated from the intercept and slope of the plot of t/qtvs. t are 61.4 mg g−1 at pH = 5.0 and 29.5 mg g−1 at pH = 6.5, respectively (Table S1†), which is consistent with the experimental data. The correlation coefficient (R2) is nearly close to 1.0. The high correlation coefficient and the good agreement of calculated and experimental qe values both suggest that the kinetic adsorption of As(V) on MGO follows the pseudo-second-order model very well. The intra-particle diffusion model (qt = kp × t0.5 + C, where kp (mg g−1 h−0.5) is indicative of the intra-particle diffusion rate constant, and C is indicative of the intercept of the line (mg g−1), which is proportional to the boundary layer thickness) was also applied to describe the kinetic adsorption of As(V) on MGO, from which the rate-determining step can be ascertained. The amount of adsorbed As(V) (qt) is proportional to the square root of contact time (t0.5).3,38 The plots of adsorbed As(V) per unit mass (qt) versus t0.5, at pH = 5.0 and 6.5 are presented in Fig. 2B, and the results indicate that the intra-particle diffusion model describes the kinetic adsorption process of As(V) on MGO well (Table S1†). According to this model, the plot linearity suggests that intra-particle diffusion also affects the reaction rate. The plots do not pass through the origin, suggesting that intra-particle diffusion is not the only rate-determining step. Intra-particle diffusion in liquid–porous solid systems may be controlled by pore volume diffusion or by surface diffusion or by all processes simultaneously.3,38 Two linear consecutive steps could be clearly observed in Fig. 2B, i.e., the first part with a much higher slope is a fast As(V) removal process, which is controlled by intra-particle diffusion and the second part is a slow As(V) removal process that is governed by complexation of adsorption reaction.3 These two distinct parts can be attributed to the difference in the rate of mass transfer at the initial and final adsorption stages. The first part is related to the instantaneous As(V) bonding at the most reactive adsorbing sites, such as COOH, and the second is related to the transport of As(V) to the less reactive adsorbing sites, such as CC.3
3.3 Adsorption isotherms
To evaluate the adsorption capacity of the as-prepared adsorbents, the adsorption of As(V) on MGO, GO and Fe3O4 was studied. It can be clearly seen from Fig. 3A that the adsorption isotherm keeps raising under our experimental conditions. For isotherm modeling, herein, the commonly used Langmuir and Freundlich models were applied to study the adsorption property of As(V) on MGO, GO and Fe3O4. The Langmuir model can be represented as: qe = bqmaxCe/(1 + bCe) and the Freundlich model can be described as: qe = kFCen, where qmax(mg g−1) and b(L g−1) are the Langmuir constants, which are related to adsorption capacity and adsorption energy, respectively, kF(mg1 − n Ln g−1) represents adsorption capacity and n represents the degree of dependence of adsorption at the equilibrium concentration.39–41 The adsorption isotherms were regressively simulated with the Langmuir and Freundlich models (Fig. 3A) and the values of the relative parameters calculated from the two isotherm models are listed in Table S2†. The R2 values of the Langmuir model are higher than those of the Freundlich model, indicating that the Langmuir model fits the adsorption isotherms better than the Freundlich model. The Langmuir model is valid for dynamic equilibrium adsorption on homogenous surfaces, whereas the Freundlich model is applicable to heterogeneous surfaces.39 Although the qmax value of As(V) on GO is calculated to be 80.1 mg g−1, the GO nanoparticles cannot be separated from solution easily. The magnetic separation experiment (Fig. S5†) obviously shows that the as-prepared MGO can be easily separated from solution in several minutes by inducing an external magnetic field. The qmax value of As(V) on MGO (59.6 mg g−1) is lower compared to that of As(V) on pure GO (80.1 mg g−1), which is caused by the reduction of the effective adsorbing sites after combination of Fe3O4 on GO.42 However, it is worth pointing out that the qmax value of As(V) on MGO is much higher than that of As(V) on other adsorbents (Table S3†), such as natural or modified zeolite,10,11 pillared montmorillonite,43 perlite incorporating α-MnO2 or γ-Fe2O3 nanocomposite44 and iron-modified activated carbon.45,46 With the costs of commercially available GO and MGO continuously decreasing, it is possible to use these novel nanomaterials for As(V) removal in wastewater treatment in the near future. More importantly, these findings demonstrate that the magnetic separable MGO is a promising material in the field of separation or purification-related technologies. In addition, the adsorption isotherm (Fig. 3B) is the highest at T = 338 K and the lowest at T = 298 K, suggesting that As(V) adsorption is favored at higher temperatures, which indicates that As(V) adsorption is an endothermic process. The increase in temperature may result in the increase in proportion and activity of As(V) in solution, the affinity of As(V) to an adsorbent surface, or the potential charge of an adsorbent.39,40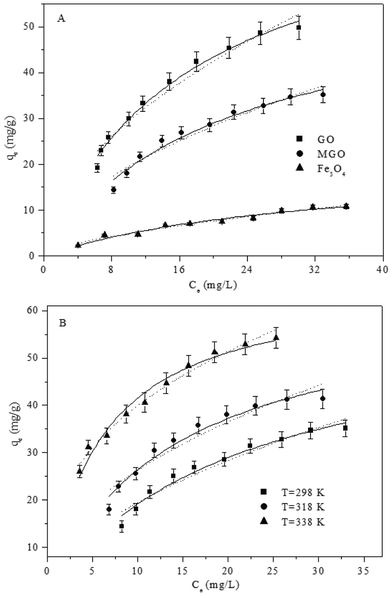 | ||
| Fig. 3 (A) The adsorption isotherms of As(V) onto GO, MGO and Fe3O4, T = 298 K, pH = 6.5, m/V = 0.4 g L−1; (B) the adsorption isotherms of As(V) onto MGO at three different temperatures, pH = 6.5, m/V = 0.4 g L−1. Symbols represent experimental data, solid lines represent the fitting of the Langmuir model and dot lines represent the fitting of the Freundlich model. | ||
The thermodynamic parameters (ΔH0, ΔS0 and ΔG0) can be calculated from the temperature-dependent adsorption isotherms. The ΔG0 value can be calculated from the equation: ΔG0 = −RTlnK0, where K0is adsorption equilibrium constant. The value of lnK0 can be obtained by plotting lnKdversus Ce and extrapolating Ce to zero, of which the intercept of the vertical axis gives the value of lnK0. The ΔS0 can be calculated using the equation: (QΔG0/QT)p = −ΔS0, and the ΔH0 is then calculated from ΔH0 = ΔG0 + TΔS0.30 The relative values are summarized in Tables S4 and S5†. The determination of thermodynamic parameters provides a mechanism insight into As(V) adsorption. The positive ΔH0 indicates that the adsorption is an endothermic process. The positive ΔS0 suggests the increase of randomness at the solid/liquid interface during the adsorption process, while the negative ΔG0 is indicative of a spontaneous adsorption process. Furthermore, the value of ΔG0 becomes more negative with increasing temperature, indicating more efficient As(V) adsorption at higher temperatures.30
3.4 pH impact on As(V) adsorption
The impact of solution pH on As(V) removal by MGO at pH 1.0–12.0 is shown in Fig. 4. As(V) removal on MGO maintains ∼95% at pH 1.0–4.0 and then decreases quickly at pH 4.0–8.0. At pH > 8.0, As(V) removal decreases slightly with increasing pH. The effect of pH on As(V) removal is the result of a variety of competing factors that are responsible for the adsorption reaction. Generally, the adsorption of As(V) on MGO is mainly governed by the electrostatic attraction or repulsion interaction of As(V) species with the MGO surface.47,48 Thereby, pH of zero point charge (pHZPC) of MGO and the species of As(V) are the main factors controlling the adsorption of As(V) on MGO. The degree of As(V) dissociation in aqueous solution is a function of pH. The major As(V) species in different pH ranges are also included in Fig. 4. When solution pH increases from an acidic region to an alkaline region, As(V) ions in solution exist mainly as H3AsO4 at pH < 2.2 (pKa1), H2AsO4− at pH 2.2–6.98 (pKa2), HAsO4−2 at pH 6.98–11.5 (pKa3) and AsO4−3 at pH > 11.5.47,48 It is well known that a solid surface is positively charged at pH < pHZPC and negatively charged at pH > pHZPC, leading to increased electrostatic attraction or repulsion with anionic As(V) species, thus resulting in more or less ready adsorption.49 The pHZPC of MGO was determined to be ∼4.3, below which the surface of MGO was positively charged and favorable for As(V) adsorption. As shown in Fig. 4, MGO surface is positively charged at pH < 4.3 and negatively charged at pH > 4.3. As pH increases, the surface of MGO becomes less positively charged and the interaction between MGO and As(V) becomes less and changes to a repulsive force at pH > 4.3, resulting in the significant decrease of As(V) adsorption. The change of the electrostatic force between As(V) species and MGO explains the pH impact on As(V) adsorption. This has been well documented in previous literatures of As(V) adsorption on various adsorbents.5,10,44,47,48 The ferric ion dissolution experiment of the MGO composites as a function of pH has been conducted and the results are shown in Fig. S6†. It can be seen from Fig. S6† that the ferric ion can hardly be dissolved from MGO into aqueous solution under alkaline conditions, therefore, the precipitates of Fe(OH)3 can not be formed from the dissolution of Fe3O4 particles, and thus can not affect the removal of arsenate. In addition, the change of pH value during the adsorption process has been determined and the results are shown in Table S6†. Under acidic conditions, the solution pH value increased a little due to the protonation reaction (i.e.,SOH + H+ = SOH2+) on the surfaces of MGO, while under alkaline conditions, the solution pH value decreased because of the surface deprotonation reaction (i.e., SOH = SO− + H+).
![The impact of pH on As(v) adsorption onto MGO, m/V = 0.4 g L−1, C[As(v)] = 25 mg L−1, T = 298 K.](/image/article/2012/RA/c2ra21623j/c2ra21623j-f4.gif) | ||
| Fig. 4 The impact of pH on As(V) adsorption onto MGO, m/V = 0.4 g L−1, C[As(V)] = 25 mg L−1, T = 298 K. | ||
3.5 Impact of competing anions on As(V) adsorption
The existence of various inorganic and organic competing anions in the water environment can present potential competition to As(V) adsorption, which may greatly affect As(V) removal.5,6 The most abundant inorganic competing anions present in the natural water environment are fluoride (F−), chloride (Cl−), nitrate (NO3−), sulfate (SO42−), bicarbonate (HCO3−) and monohydrogen phosphate (HPO42−).5,6 The impact of these inorganic competing anions (i.e., F−, Cl−, NO3−, SO42−, HCO3−, HPO42−) on As(V) adsorption at pH = 5.0 and 6.5 onto MGO was studied as a function of inorganic competing anion concentrations (Fig. 5). In addition, the most common organic competing anions in the natural water environment are low molecular mass organic acids, such as oxalic acid (OA), succinic acid (SA), malic acid (MA), citric acid (CA) and macromolecule organic acids, such as humic acid (HA) and fulvic acid (FA).5,50 The natural organic competing anions are not only ubiquitous in natural environments, but are also highly reactive toward metals and solid surfaces. Therefore, the impact of these organic competing anions (i.e., OA, SA, MA, CA, HA, FA) on As(V) adsorption onto MGO was also studied (Fig. 6).![Adsorption of As(v) onto MGO at pH 5.0 and 6.5 in the absence and presence of inorganic anions with various concentrations, T = 298 K, C[As(v)] = 25 mg L−1, m/V = 0.4 g L−1. (A) Cl−, (B) NO3−, (C) SO42−, (D) HCO3−, (E) F−, (F) HPO42−.](/image/article/2012/RA/c2ra21623j/c2ra21623j-f5.gif) | ||
| Fig. 5 Adsorption of As(V) onto MGO at pH 5.0 and 6.5 in the absence and presence of inorganic anions with various concentrations, T = 298 K, C[As(V)] = 25 mg L−1, m/V = 0.4 g L−1. (A) Cl−, (B) NO3−, (C) SO42−, (D) HCO3−, (E) F−, (F) HPO42−. | ||
![Adsorption of As(v) onto MGO at pH 5.0 and 6.5 in the absence and presence of organic anions with various concentrations, T = 298 K, C[As(v)] = 25 mg L−1, m/V = 0.4 g L−1. (A) OA, (B) SA, (C) MA, (D) CA, (E) HA, (F) FA.](/image/article/2012/RA/c2ra21623j/c2ra21623j-f6.gif) | ||
| Fig. 6 Adsorption of As(V) onto MGO at pH 5.0 and 6.5 in the absence and presence of organic anions with various concentrations, T = 298 K, C[As(V)] = 25 mg L−1, m/V = 0.4 g L−1. (A) OA, (B) SA, (C) MA, (D) CA, (E) HA, (F) FA. | ||
From Fig. 5, one can see that the presence of Cl− and NO3−, even at very high concentrations (0.1 mol L−1), shows little inhibition effect on As(V) adsorption, while the presence of SO42− and HCO3− shows moderate inhibition effect on As(V) adsorption. However, F− and HPO42− could significantly decrease As(V) adsorption onto MGO, which is consistent with the results of Tang et al.6 and Jeong et al.51 From Fig. 6 it is clear that the presence of OA, SA, MA, CA, HA and FA shows a significant inhibition effect on As(V) adsorption onto MGO. These findings suggest that, when competing anions coexist with As(V) in solutions, the adsorption of As(V) onto MGO is primarily affected by the type and concentration of competing anions. Additionally, increasing the concentration of a competing anion leads to the decrease of As(V) adsorption onto MGO. This is due to the competition between the competing anions and As(V) on the adsorbing sites of MGO, and/or the reduction of the surface charge of MGO adsorbent.5,6,50 The inhibition efficiency (IE) of each competing anion on As(V) adsorption can be calculated using the expression IE(%) = (1 − As adsorbed in the presence of an anion/As adsorbed in the absence of an anion) × 100%,5 and the data are summarized in Tables S7 and S8†.
For inorganic competing anions at pH 5.0, when the concentration of each competing anion increases from 0.001 to 0.1 mol L−1, the IE values of inhibiting As(V) adsorption increase from 2.7% to 5.9% for Cl−, from 1.6% to 7.5% for NO3−, from 9.1% to 27.4% for SO42−, from 11.8% to 29.5% for HCO3−, from 21.9% to 52.0% for F− and from 20.4% to 54.7% for HPO42−. At pH 6.5, when the concentration of each competing anion increases from 0.001 to 0.1 mol L−1, the IE values inhibiting As(V) adsorption increase from 2.2% to 5.4% for Cl−, from 1.1% to 4.3% for NO3−, from 6.5% to 25.9% for SO42−, from 8.7% to 24.9% for HCO3−, from 20.6% to 49.7% for F− and from 15.2% to 54.1% for HPO42−. The ability of inorganic competing anions to inhibit As(V) adsorption onto MGO is as follows: Cl− ≈ NO3− < SO42− ≈ HCO3− < F− ≈ HPO42− (Table S7†). The stronger the interactions of these competing anions with the active adsorbing sites on the adsorbent surface, the more pronounced the inhibition of As(V) adsorption is.3 For organic competing anions at pH 5.0, when the concentration of each competing anion increases from 1 to 20 mg L−1, the IE values inhibiting As(V) adsorption increase from 12.9% to 32.7% for OA, from 10.7% to 35.9% for SA, from 13.9% to 31.7% for MA, from 12.9% to 32.2% for CA, from 23.6% to 52.0% for HA and from 24.7% to 51.5% for FA. At pH 6.5, when the concentration of each competing anion increases from 1 to 20 mg L−1, the IE values inhibiting As(V) adsorption increase from 11.9% to 32.5% for OA, from 8.7% to 28.1% for SA, from 7.6% to 20.2% for MA, from 10.8% to 27.1% for CA, from 21.6% to 46.5% for HA and from 18.4% to 49.8% for FA. The ability of organic competing anions to inhibit As(V) adsorption onto MGO is as follows: OA ≈ SA ≈ MA ≈ CA < HA ≈ FA (Table S8†). In comparison with low molecular mass organic ligands, macromolecule organic ligands have more oxygen-containing functional groups and can impose stronger interactions with MGO, leading to an inhibition effect on As(V) adsorption which is more obvious. The efficiency of competing anions in inhibiting As(V) adsorption on MGO at pH 5.0 was higher than that at pH 6.5, which could be attributed to the stronger interaction of the competing anions with the active adsorbing sites on MGO surface at lower pH values. Besides, one can see that the inorganic and organic competing anions showed a tremendous difference in inhibiting As(V) adsorption, i.e., organic competing anions were more effective in inhibiting As(V) adsorption on MGO than inorganic competing anions. This might be attributed to the stronger interaction of organic anions with MGO by hydrogen bonding, the hydrophobic effect, electrostatic interaction and/or π–π electron-donor-acceptor (EDA) interaction between the aromatic functional group of organic anions and the polarizable graphene nanosheets of MGO.52 Therefore, more adsorbing sites on MGO could be occupied by organic competing anions, and thus reduced As(V) adsorption more greatly.
3.6 Impact of coexisting cations on As(V) adsorption
The impact of coexisting cations (i.e., K+, Na+, Ca2+, Mg2+, Al3+and Fe3+ ions) on As(V) adsorption at pH = 5.0 and 6.5 onto MGO were also studied (Fig. S7†). The presence of coexisting cations enhances As(V) adsorption, especially at pH 6.5. Besides, the increase in the concentration of coexisting cations can further enhance As(V) adsorption. The enhancement efficiency (EE) of each coexisting cation can be calculated using the following expression: EE(%) = (As adsorbed in the presence of an anion/As adsorbed in the absence of an anion − 1) × 100% and the data are summarized in Table S9†. The EE values of competing cations on As(V) adsorption at pH 6.5 are higher than those at pH 5.0. The EE values in As(V) adsorption are affected by solution pH and the nature and concentration of coexisting cations. Similar results of coexisting cation-induced enhancement in As(V) adsorption were also reported by Zhang et al.52 It is believed that the enhancement of As(V) uptake is most likely attributed to the participation of coexisting cations in the adsorption process. The adsorption of cations (K+, Na+, Ca2+, Mg2+, Al3+, Fe3+) on MGO has been carried out and the results are shown in Fig. S8†. It can be clearly seen from Fig. S8† that a significant number of cations can be adsorbed on MGO. It is the adsorption of coexisting cations that results in a positively charged surface of MGO, and thus obviously facilitates the adsorption of anionic As(V) species.534. Conclusion and implication
In conclusion, the MGO-bound As(V) can be quickly separated and recovered from aqueous solution by an easy magnetic separation method. The adsorption of As(V) by MGO is greatly affected by solution chemistry. The magnetic MGO composites are very suitable materials for the removal of As(V) from aqueous solution in natural environmental pollution cleanup. With the development of GO preparation technology, the cost of GO preparation will be reduced and GO or GO-based composites will be used in real work in the near future. The finding in this paper highlights the practical application of a magnetic separation method and graphene oxide-based magnetic composites in environmental pollution management.Acknowledgements
Financial supports from 973 projects (2011CB933700), NSFC (21207092, 21071107, 20971126, 21107115), Doctoral Fund of Ministry of Education of China (20110111110002) and Natural Science Foundation of Anhui Province (Grant No:1208085QB32) are acknowledged.References
- J. O. Nriagu and J. M. Pacyna, Nature, 1988, 333, 134–139 CrossRef CAS
.
- A. K. Mishra and S. Ramaprabhu, J. Phys. Chem. C, 2010, 114, 2583–2590 CAS
- Z. Velivckovic, G. D. Vukovic, A. D. Marinkovic, M. S. Moldovan, A. A. Peric-Grujic, P. S. Uskokovic and M. D. Ristic, Chem. Eng. J., 2012, 181, 174–181 CrossRef
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS
- J. Zhu, M. Pigna, V. Cozzolino, A. G. Caporale and A. Violante, J. Hazard. Mater., 2011, 189, 564–571 CrossRef CAS
- W. Tang, Q. Li, S. Gao and J. Shang, J. Hazard. Mater., 2011, 192, 131–138 CAS
- S. Dixit and J. Hering, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS
- K. P. Raven, A. Jain and R. H. Loeppert, Environ. Sci. Technol., 1998, 32, 344–349 CrossRef CAS
- S. Fendorf, M. J. Eick, P. Grossl and D. L. Sparks, Environ. Sci. Technol., 1997, 9, 315–320 CrossRef
- L. M. Camacho, R. R. Parra and S. Deng, J. Hazard. Mater., 2011, 189, 286–293 CrossRef CAS
- K. Payne and T. Abdel-Fattah, J. Environ. Sci. Health A, 2005, 40, 723–749 CAS
- M. Pirilä, M. Martikainen, K. Ainassaari, T. Kuokkanen and R. L. Keiski, J. Colloid Interface Sci., 2011, 353, 257–262 CrossRef
- S. Bang, M. Patel, L. Lippincott and X. Meng, Chemosphere, 2005, 60, 389–397 CrossRef CAS
- C. Luengo, V. Puccia and M. Avena, J. Hazard. Mater., 2011, 186, 1713–1719 CrossRef CAS
- B. A. Manning and S. Goldberg, Clays Clay Miner., 1996, 44, 609–623 CAS
- Z. Li, R. Beachner, Z. McManama and H. Hanlie, Microporous Mesoporous Mater., 2007, 105, 291–297 CrossRef CAS
- A. Saada, D. Breeze, C. Crouzet, S. Cornu and P. Baranger, Chemosphere, 2003, 51, 757–763 CrossRef CAS
- S. Santra, R. Tapec, N. Theodoropoulou, J. Dobson, A. Hebard and W. Tan, Langmuir, 2001, 17, 2900–2906 CrossRef CAS
- S. Zhang, X. Li and J. P. Chen, Carbon, 2010, 48, 60–67 CrossRef CAS
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS
- G. Zhao, X. Ren, X. Gao, X. Tan, J. Li, C. Chen, Y. Huang and X. Wang, Dalton Trans., 2011, 40, 10945–40952 RSC
- M. Liu, C. Chen, J. Hu, X. Wu and X. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CAS
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS
- G. Zhao, J. Li and X. Wang, Chem. Eng. J., 2011, 173, 185–190 CrossRef CAS
- L. Hao, H. Song, L. Zhang, X. Wan, Y. Tang and Y. Lv, J. Colloid Interface Sci., 2012, 369, 381–387 CrossRef CAS
- A. Kumar and M. S. Ramaprabhu, Desalination, 2011, 282, 39–45 CrossRef
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS
- C. Wang, C. Feng, Y. Gao, X. Ma, Q. Wu and Z. Wang, Chem. Eng. J., 2011, 173, 92–97 CrossRef CAS
- Y. Yao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS
- S. T. Yang, G. D. Sheng, X. L. Tan, J. Hu, J. Z. Du, G. Montavon and X. K. Wang, Geochim. Cosmochim. Acta, 2011, 75, 6520–6534 CrossRef CAS
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1341 CrossRef CAS
- M. Jahan, Q. L. Bao, J. X. Yang and K. P. Loh, J. Am. Chem. Soc., 2010, 132, 14487–14495 CrossRef CAS
- X. Yang, C. Chen, J. Li, G. Zhao, X. Ren and X. Wang, RSC Adv., 2012, 2, 8821–8826 RSC
- Y. C. Sharma, V. Srivastava and A. K. Mukherjee, J. Chem. Eng. Data, 2010, 55, 2390–2398 CrossRef CAS
-
Y. S. Ho, Ph.D. thesis. UK: University of Birmingham; 1995 Search PubMed
- Y. S. Ho and G. McKay, Water Res., 2000, 34, 735–742 CrossRef CAS
- Y. S. Ho, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS
- J. C. Igwe, A. A. Abia and C. A. Ibeh, Int. J. Environ. Sci. Tech., 2008, 5, 83–92 CAS
- G. Sheng, D. Shao, X. Ren, X. Wang, J. Li, Y. Chen and X. Wang, J. Hazard. Mater., 2010, 178, 505–516 CrossRef CAS
- S. Yang, Z. Guo, G. Sheng and X. Wang, Sci. Total Environ., 2012, 420, 214–221 CrossRef CAS
- Z. Q. Guo, Y. Li, S. W. Zhang, H. H. Niu, Z. S. Chen and J. Z. Xu, J. Hazard. Mater., 2011, 192, 168–175 CrossRef CAS
- N. Li, M. Zheng, X. Chan, G. Ji, H. Lu, L. Xue, L. Pan and J. P. Cao, J. Solid State Chem., 2011, 184, 953–958 CrossRef CAS
- S. Zhao, C. Feng, X. Huang, B. Li, J. Niu and Z. Shen, J. Hazard. Mater., 2012, 203-204, 317–325 CrossRef CAS
- D. N. Thanh, M. Singh, P. Ulbrich, N. Strnadova and F. Štepánek, Sep. Purif. Technol., 2011, 82, 93–101 CrossRef
- Z. Gu, J. Fang and B. Deng, Environ. Sci. Technol., 2005, 39, 3833–3843 CrossRef CAS
- W. Chen, R. Parette, J. Zou, F. S. Cannon and B. A. Dempsey, Water Res., 2007, 41, 1851–1858 CrossRef CAS
- Q. Chang, W. Lin and W. Ying, J. Hazard. Mater., 2010, 184, 515–522 CrossRef CAS
- H. Zhu, Y. Jia, X. Wu and H. Wang, J. Hazard. Mater., 2009, 172, 1591–1596 CrossRef CAS
- G. Sheng, J. Hu, H. Jin, S. Yang, X. Ren, J. Li, Y. Chen and X. Wang, Radiochim. Acta, 2010, 98, 291–299 CrossRef CAS
- L. Weng, W. H. Van Riemsdijk and T. Hiemstra, Environ. Sci. Technol., 2009, 43, 7198–7204 CrossRef CAS
- Y. Jeong, M. H. Fan, J. V. Leeuwen and J. F. Belczyk, J. Environ. Sci., 2007, 19, 910–919 CrossRef CAS
- G. Zhao, T. Wen, C. Chen and X. Wang, RSC Adv., 2012, 2, 9286–9303 RSC
- G. Zhang, H. Liu, J. Qu and W. Jefferson, J. Colloid Interface Sci., 2012, 366, 141–146 CrossRef CAS
Footnote |
| † Electronic Supplementary Information (ESI) available: Preparation of GO and MGO, SEM and TEM of GO, XRD, IR, magnetization curve of MGO, the kinetic and thermodynamic parameters, effect of coexisting cations on As(V) removal and impact of adsorbent content. See DOI: 10.1039/c2ra21623j |
| This journal is © The Royal Society of Chemistry 2012 |