Cetyltrimethylammonium bromide intercalated graphene/polypyrrole nanowire composites for high performance supercapacitor electrode†
Lu
Mao
a,
Hardy Sze On
Chan
*a and
Jishan
Wu
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmcsoh@nus.edu.sg (H.S.O. Chan); chmwuj@nus.edu.sg (J. Wu); Fax: (65) 6779 1691
bInstitute of Materials Research and Engineering, A*Star, 3 Research Link, 117602, Singapore. E-mail: wuj@.imre.a-star.edu.sg
First published on 6th September 2012
Abstract
Polypyrrole (PPy) nanowires were prepared by in situ polymerization of pyrrole in the presence of cetyltrimethylammonium bromide stabilized graphene (GCR). PPy nanowire/GCR composites with different loading ratios were tested as supercapacitor electrode materials in both 1 M H2SO4 (aq) and 1 M KCl (aq) electrolytes, and different electrochemical behaviors were observed. The incorporation of GCR nanosheets into the PPy nanowire matrix obviously improved the electrochemical performance of PPy and a high specific capacitance of 492 F g−1 can be obtained for PPyGCR91 in 1 M H2SO4 at a current density of 0.2 A g−1 with good cycling stability.
1. Introduction
Electrically conducting polymers (ECPs) have attracted considerable attention as promising electrode materials for high performance supercapacitors because of their unusual electronic properties. In addition to a relatively high conductivity in oxidized and doped states, simple ECPs, such as polypyrrole, polyaniline, and polythiophene are characterized by high specific capacitances via a redox pseudocapacitive charge storage mechanism originating from the reversible oxidation and reduction of the conjugated double bonds in polymer networks, as well as relatively fast charge–discharge processes.1–6 However, all known simple ECPs are mechanically weak and have to be oxidized and doped by counter anions to achieve significant conductivities.6,7 Of all known ECPs, polypyrrole (PPy) is particularly appropriate for this application because of the water solubility of the pyrrole monomer as well as it being more environmentally ‘‘friendly’’ with regard to the carcinogenic risks associated with the degradation products of polyaniline.8Alternatively, supercapacitors made from high surface area carbons are attractive for their excellent rates of charge/discharge and cyclability, properties stemming from their high surface area and double-layer charge storage mechanism.1 Double-layer supercapacitors store charge in an electrochemical double-layer formed at their interface with the electrolyte, which can increase the rate of response and cycling stability while limit the total amount of charge that can be stored relative to redox pseudocapacitive materials. Graphene, a two-dimensional carbon material, has received significant research attention in energy storage devices due to its extraordinarily high electrical and thermal conductivities, great mechanical strength, large specific surface area, and potentially low manufacturing cost.9–11
Recently, much effort was devoted to produce composites of redox pseudocapacitive PPy and double-layer capacitive carbon material graphene to create a synergetic effect resulting in significantly enhanced performances and stabilities. Zhang et al. demonstrated the preparation of layered graphene oxide structures with sandwiched PPy of different morphologies in which a high specific capacitance of over 500 F g−1 at a current density of 0.3 A g−1 was realized.12 Biswas et al. combined graphene nanosheets with polypyrrole nanowires in a multilayered configuration to create a binder free multilayered composite structure electrode, exhibiting a high specific capacitance of ∼165 F g−1 at 1 A g−1 discharge current density.13 Many groups focus on combining graphene with polypyrrole using an electropolymerization method to achieve higher electrochemical performances.14–16 Xu, Zhang and Bose et al. fabricated graphene/polypyrrole composites via in situ polymerization, and specific capacitances of 318.6 F g−1 at a scan rate of 2 mV s−1, 482 F g−1 at a current density of 0.5 A g−1, and 267 F g−1 at a scan rate of 100 mV s−1 were obtained. 17–19
One-dimensional (1D) nanostructures exhibit superior electrochemical properties compared to three-dimensional structures due to their smaller dimensions and high aspect ratios.20 Zhang et al. reported a simple template strategy for the synthesis of 1D wire-like polypyrrole nanostructures in the presence of cetyltrimethylammonium bromide (CTAB).21 In the present work, based on the CTAB-stablized graphene (GCR) material developed by our group recently,22 which has good dispersibility in aqueous solvents and high specific capacitances, we have, for the first time, prepared PPy nanowire-graphene composites using a simple one-step in situ polymerization. The electrochemical performances of the as-prepared PPy nanowire/GCR composites with different loading ratios were investigated in both 1 M H2SO4 (aq) and 1 M KCl (aq), and different capacitive behaviors were observed. Upon the combination of PPy nanowire and GCR sheet, a synergetic effect was achieved through the complementary advantages of an electrical double layer capacitor (EDLC) and a faradaic pseudocapacitor. Meanwhile, the unique nanowire–sheet morphology is beneficial for ionic accessibility and electron transportation, which can be ascribed to the better electrochemical performance obtained.
2. Experimental
2.1. Material synthesis
Graphene oxide (GO) was synthesized from natural graphite by a modified Hummers method.23 CTAB stabilized graphene material, termed as GCR, was prepared by the chemical reduction of GO in the presence of a surfactant in water followed by filtration and washing.22 PPy nanowires were prepared through a simple strategy using lamellar inorganic/organic mesostructures as templates.21Typically, 0.3 mmol CTAB was dissolved in 30 mL of distilled water to form a homogeneous solution. Then, 60 mL of pyrrole monomer was added and the mixture was stirred vigorously for 10 min and then cooled to 0–5 °C. Pre-cooled ammonium persulfate (APS) aqueous solution (0.90 mmol, 6.5 mL) was added dropwise into the mixture with simultaneous vigorous stirring. The solution was allowed to stand at 0–5 °C for a further 24 h. The solid PPy was collected by vacuum filtration, washed with D.I. water and ethanol, and dried in a vacuum oven at 80 °C for 12 h. For PPy/GCR composites, a similar procedure was followed except different amounts of GCR were added to the surfactant aqueous solution and sonicated for 15 min before adding the pyrrole monomer, as illustrated in Fig. 1. Hydrophobic pyrrole molecules will predominately locate themselves in the interior of the micelles in aqueous solutions of the cationic surfactant CTAB.12,21 In the process, the pyrrole monomer has been adsorbed onto the surface of the graphene via π–π stacking as well as by electrostatic interactions.17–19,24 Lamellar mesostructural (CTA)2S2O8 formed in situ during polymerization between surfactant cations and oxidising anions acted as a template for the formation of 1D PPy nanowires.21 The weight feed ratio of pyrrole to GCR was varied (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, 1:1 and 1:3), and the resulting composites were named as PPyGCR91, PPyGCR51, PPyGCR11, and PPyGCR13, respectively. The real mass ratios of polypyrrole and GCR in the composites are estimated as 9.46:1, 5.12:1, 1.07:1 and 1:3.48, respectively, by weighing the powder before and after polymerization.
:1, 5:1, 1:1 and 1:3), and the resulting composites were named as PPyGCR91, PPyGCR51, PPyGCR11, and PPyGCR13, respectively. The real mass ratios of polypyrrole and GCR in the composites are estimated as 9.46:1, 5.12:1, 1.07:1 and 1:3.48, respectively, by weighing the powder before and after polymerization.
 | ||
| Fig. 1 Illustration of the process for the preparation of PPy/GCR composites. | ||
2.2. Material characterization
The structure and morphology of the products were characterized by transmission electron microscopy (TEM; JEOL 2010 FEG, 200 keV), scanning electron microscopy (SEM; JEOL-6300F, 5 kV), and X-ray diffraction (XRD; Bruker-AXS D8 DISCOVER, GADDS powder X-ray diffractometer, Copper KR line, λ = 1.5406 Å). The thermal data of the products were determined by thermogravimetric analysis (TGA; TA Instruments 2960) at a heating rate of 10 °C min−1 under nitrogen flow. Raman spectra were measured on a Renishaw inVia Raman microscope using a 514 nm laser under ambient conditions. The N2 adsorption-desorption isotherms of the samples were measured at 77 K using NOVA1200 (Quantachrome, USA). Prior to measurements, the samples were vacuum-degassed at 90 °C for 12 h. The specific surface areas (SBET) were determined according to the Brunauer–Emmett–Teller (BET) method in the relative pressure range of 0.05–0.3. The pore size distribution (PSD) curves were derived from the Barrett–Joyner–Halenda (BJH) method using the desorption branches. IR spectra were obtained on a Varian 3100 FT-IR instrument using pressed KBr pellets. A three-electrode cell system was used to evaluate the electrochemical performance by electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV), and galvanostatic charge–discharge techniques on an Autolab PGSTAT302N at room temperature. The working electrode was prepared by casting a nafion-impregnated sample onto a glassy carbon electrode with a diameter of 5 mm. Typically, 5 mg of a composite was dispersed in 1 mL of ethanol solution containing 5 μL of nafion solution (5 wt% in water) by sonication for 30 min. Then, 20 μL of this sample was dropped onto the glassy carbon electrode and dried in an oven before the electrochemical test. Both 1 M H2SO4 and 1 M KCl aqueous solutions were added as the electrolytes. A platinum sheet and a AgCl/Ag electrode were used as the counter and the reference electrodes, respectively.3. Results and discussion
3.1. Microstructure characterizations
The XRD patterns of pure PPy, pure GCR, and the PPy/GCR composites are shown in Fig. 2a. Pure PPy nanowires exhibit two broad bands at about 2θ = 26 (i.e., d = 0.34 nm) and 42.5°, indicating their amorphous nature. For GCR, the diffraction peaks at 2θ = 21.4° (i.e., d = 0.41 nm) and 42.6° can be attributed to the graphite-like structure (002) and (100), respectively. The weak diffraction peak around 9.4° is caused by the residual surfactant CTAB intercalated.22,25 It can be seen that the two weak broad peaks of PPy somewhat overlap with peaks from GCR at 21.4 and 42.6°. However, the character of the diffraction peaks from GCR becomes more obvious with an increasing loading ratio of GCR in the composites. As for PPyGCR13, reflection peaks with higher intensity can be seen. From the XRD patterns, it can be concluded that the amorphous nature of PPy was preserved and no additional crystalline phase changes occurred in the composites. | ||
| Fig. 2 XRD patterns (a) and FT-IR spectra (b) of PPy, GCR and representative PPy/GCR composites. | ||
The FT-IR absorption spectra of PPy, PPyGCR91 and GCR are shown in Fig. 2b. The characteristic polypyrrole peaks located at 1550 and 1464 cm−1 are due to the symmetric and antisymmetric ring-stretching modes, respectively. A broad band at 3000–3500 cm−1 describes the N–H stretching vibrations.18,26 The bands at 1046 and 1310 cm−1 are attributed to C–H deformation vibrations and C–N stretching vibrations, respectively.19 Meanwhile, peaks near 1193 and 915 cm−1 indicate the doping state of polypyrrole and the bands at 792 and 968 cm−1 verify the presence of polymerized pyrrole.17,25 The broad peak at 3422 cm−1 of GCR could be assigned to the O–H stretching vibration, suggesting residual oxide groups exist. The peaks at 2916 and 2845 cm−1 are designated as the asymmetric stretching and symmetric vibrations of CH2, respectively, indicating the existence of surfactant CTAB in GCR.12 All of the peaks can be seen in the PPy/GCR composites, showing the presence of both PPy and GCR in these composites.
The Raman spectrum of GCR (Fig. 3a) shows characteristic D and G bands at 1348 and 1580 cm−1, respectively. The G band represents the first-order scattering of the E2g vibrational mode, and the D band is assigned to the dispersive, defect-induced vibrations.27 The Raman spectrum of PPy shows a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C band at around 1589 cm−1, corresponding to the CC backbone stretching of PPy. The band at 1360 cm−1 is attributed to the ring stretching mode of PPy. The peak at 1052 cm−1 is attributed to the C–H in-plane deformation. The bands located at about 978 and 938 cm−1 correspond to ring deformations associated with the dication (bipolaron) and the radical cation (polaron), respectively.28–32 The Raman spectrum of the PPy/GCR51 composite shows the bands for PPy and an enhanced intensity of the band around 1350 cm−1, which indicates the interaction between pyrrole and graphene sheets.27
C band at around 1589 cm−1, corresponding to the CC backbone stretching of PPy. The band at 1360 cm−1 is attributed to the ring stretching mode of PPy. The peak at 1052 cm−1 is attributed to the C–H in-plane deformation. The bands located at about 978 and 938 cm−1 correspond to ring deformations associated with the dication (bipolaron) and the radical cation (polaron), respectively.28–32 The Raman spectrum of the PPy/GCR51 composite shows the bands for PPy and an enhanced intensity of the band around 1350 cm−1, which indicates the interaction between pyrrole and graphene sheets.27
 | ||
| Fig. 3 Raman spectra (a) and TGA curves (b) of PPy, GCR and representative PPy/GCR composites. | ||
The thermal stabilities of PPy, GCR and the PPy/GCR composites were studied by TGA (Fig. S3, ESI†), and representative curves are shown in Fig. 3b. It can be seen from this figure that all the materials show a little mass loss around 100 °C due to the deintercalation of H2O. GCR shows a steep weight loss at around 200 °C, ascribed to the decomposition of CTAB in the interlayer of graphene, and a total weight loss of 48% at 900 °C. Meanwhile, PPy displays a 58% mass loss from 100 to 900 °C. PPy/GCR composites show similar TGA features to GCR and PPy with 55, 51, 50, and 46% mass loss for PPy/GCR91, PPy/GCR51, PPy/GCR11 and PPy/GCR13, respectively, indicating a better thermal stability with increasing GCR loading.
PPy/GCR composites exhibit a type IV nitrogen adsorption isotherm (Fig. S4, ESI†), indicating samples having relatively large pores.33Fig. 4 shows the N2 adsorption–desorption isotherm and BJH pore-size distribution curve (inset) of sample PPyGCR91. Data in Table 1 show that the PPy/GCR composites have much higher surface areas than pure GCR due to the incorporation of PPy nanowires. Furthermore, PPyGCR91 and PPyGCR11 exhibit higher specific surface areas than other composites, which may result from an optimum combination of GCR sheet and PPy nanowire morphology due to the specific mass ratios and thus can contribute to a higher electrochemical performance. The morphology and structure of PPy, GCR and the PPy/GCR composites were characterized using SEM and TEM (Fig. 5 and 6). The pure polypyrrole prepared by the described procedure shows uniform nanowire morphology with diameters around 25 nm and lengths up to several micrometers. GCR prepared here exhibits a curved, thin, flaky appearance. In all PPy/GCR composites, PPy retains its wire-like morphology with similar size to the pure PPy nanowires, and GCR retains its layer-like structure. For PPyGCR91, where PPy nanowires are in the majority, GCR sheets are embedded in the PPy nanowires with rather uniform distribution. For the PPyGCR51 composite, PPy nanowires homogeneously coat the surface of GCR sheets, and are distributed both on the surface and between the GCR sheets. For other PPy/GCR composites with a reduced amount of PPy, wire-like PPy mainly adsorbs on the surface or intercalates between GCR sheets, and the wrinkled, folded, layer-like morphology of GCR is well reserved.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms of PPyGCR91. The inset is the corresponding pore-size distributions. | ||
 | ||
| Fig. 5 SEM images of PPy nanowire (a), GCR (b), PPyGCR91 (c), PPyGCR51 (d), PPyGCR11 (e), and PPyGCR13 (f). | ||
 | ||
| Fig. 6 TEM images of PPyGCR91 (a), PPyGCR51 (b), PPyGCR11 (c) and PPyGCR13 (d). Insets are the corresponding SAED patterns. | ||
| Samples | PPy:GCR mass ratio |
1 M KCl | 1 M H2SO4 | BET surface area (m2 g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| 1 A g−1 | 0.5 A g−1 | 0.2 A g−1 | 1 A g−1 | 0.5 A g−1 | 0.2 A g−1 | |||
| PPy | — | 120 | 128 | 141 | 122 | 194 | 301 | 73.45 |
| GCR | — | 42 | 47 | 55 | 126 | 179 | 220 | 27.34 |
| PPyGCR91 | 9.46:1 |
182 | 195 | 214 | 177 | 256 | 492 | 105.7 |
| PPyGCR51 | 5.12:1 |
162 | 178 | 204 | 170 | 248 | 361 | 49.87 |
| PPyGCR11 | 1.07:1 |
136 | 143 | 158 | 156 | 246 | 324 | 102.7 |
| PPyGCR13 | 1:3.48 |
78 | 83 | 94 | 130 | 163 | 200 | 57.99 |
Since GCR has good crystalline character and PPy nanowires have amorphous nature, it can be seen from the selected-area electron diffraction (SAED) patterns of PPyGCR91, PPyGCR51, PPyGCR11 and PPyGCR13 that with an increase of graphene ratio, the composites show an increase of crystalline character (Fig. 6, insets), which is consistent with the XRD results discussed above.
3.2 Electrochemical behavior
The performance of the as-prepared PPy/GCR composites as electrode materials for supercapacitors was tested by standard CV, galvanostatic charge–discharge technique and EIS. All electrochemistry measurements were conducted in a three-electrode cell. The average specific capacitance was estimated from the discharge slope according to the following equation:| Cavg = I × Δt/(ΔV × m) | (1) |
The electrochemical tests were performed in both 1 M H2SO4 and 1 M KCl aqueous solutions, within potential windows from −0.2 to 0.8 V and −0.5 to 0.5 V vs. AgCl/Ag, respectively. Fig. 7 compares the cyclic voltammetry characteristics of different composite electrodes in different electrolytes at a scan rate of 100 mV s−1.
 | ||
| Fig. 7 The cyclic voltammograms of different composite electrodes at a scan rate of 100 mV s−1 in 1 M H2SO4 (a) and 1 M KCl (b). | ||
It can be seen that the CV curve of GCR in 1 M H2SO4 (aq) has an almost rectangular shape, indicating a good charge propagation and ion response within the electrodes.22 The CV curve of PPy nanowire shows a broad oxidation peak around 0.4 V. Both oxidation and reduction currents decrease toward the negative potential end, which is an indication of the polymer gradually becoming inactive and resistive.8 In the same potential range, the CV curves of PPyGCR composites show more rectangular shapes compared with that of PPy nanowires, indicating faster charging and discharging behavior of the composites. Meanwhile, PPyGCR91 shows a much higher output current than the other composites, suggesting a larger electrochemical capacity.
The CV curve of GCR in 1 M KCl exhibits an almost rectangular shape but quite a low current output, indicating a relatively low specific capacitance under this neutral condition. The CV curve of PPy in 1 M KCl shows a more distorted feature, and the current significantly decreases in the negative potential region, which shows that the conductivity of the polymer drastically decreases as it becomes neutral (undoped).34 In contrast, the CV curves of PPyGCR composites in 1 M KCl electrolyte display voltage reversal at each end potential with respect to the zero-current line and rapid current responses. The rectangular-like and almost symmetric I–E responses of ideal capacitive behavior match the requirement of the application as supercapacitors, which also suggests a better cyclability.8 It can be clearly seen that with the increasing GCR loading in the hybrid composites, the redox current decreases which indicates a lower specific capacitance. Meanwhile, it can be deduced that PPyGCR91 has the highest electrochemical performance.
The specific capacitance was calculated from the galvanostatic discharge branch. Fig. 8 presents the charge/discharge curves of PPy, GCR and various PPyGCR composites in 1 M H2SO4 (Fig. 8a) and 1 M KCl (Fig. 8b) aqueous solutions at a current density of 1 A g−1. More detailed results are showed in Table 1.
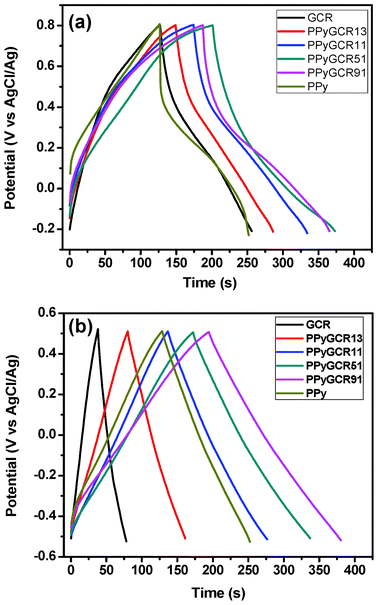 | ||
| Fig. 8 Galvanostatic charge–discharge curves of different composites electrode at a current density of 1 A g−1 in 1 M H2SO4 (a) and 1 M KCl (b). | ||
The galvanostatic charge–discharge curve of GCR in 1 M H2SO4 is almost linear, indicating its good double-layer capacitive behavior. The small deviation from an ideal triangle shape may due to the residual oxygen functional groups on the edge. The curve of PPy is distorted from a symmetrical triangle shape due to the existence of the faradaic reactions. In comparison, the charge–discharge curves of the PPy/GCR composites show less distorted and more symmetric features, indicating a combined effect of double-layer and redox capacitive behavior. The PPy/GCR composite electrodes have much longer charge–discharge durations, which means enhanced charge storage capacities. PPyGCR91 exhibits the highest specific capacitance of 177 F g−1 under such conditions, much higher than either PPy or GCR alone (120 and 126 F g−1, respectively). A specific capacitance of as high as 492 F g−1 was obtained at a current density of 0.2 A g−1, due to a more complete faradaic reaction at a lower current density. With the decreasing PPy loading in the composites, the specific capacitances of the PPyGCR composites decrease accordingly, which may result from a lower contribution to pseudocapcitance from PPy.
Highly linear and triangle-shaped charge–discharge curves of the as-prepared composites in 1 M KCl electrolyte can be seen from Fig. 8a, suggesting the increased EDL capacitive behavior of these composites in such neutral conditions. Upon the incorporation of GCR, the specific capacitances of PPyGCR91 and PPyGCR51 are obviously improved from 120 to 182 and 162 F g−1, respectively, which is due to a synergetic effect derived from PPy and GCR. Since the specific capacitance of GCR under such conditions is quite low (42 F g−1), the increasing loading of GCR in these composites inhibits higher specific capacitances from composites PPyGCR11 and PPyGCR13. However, the incorporation of GCR into PPy nanowire matrix can obviously enhance the utilization of the pseudocapacitance from PPy in the composites. The specific capacitance of PPy in the composite is quite good if extracting the capacitance contribution from GCR (Cppy = 203 F g−1 for PPyGCR13).
The significantly improved ion transport behavior of the PPyGCR composites was further characterized using EIS technique. EIS is a powerful technique, complementary to galvanostatic cycling, that provides more information on the electrochemical frequency behavior of the system. The EIS data were analyzed using Nyquist plots and are presented in Fig. 9.
 | ||
| Fig. 9 Nyquist plots of different composite electrodes in 1 M H2SO4 (a) and 1 M KCl (b). | ||
Fig. 9a shows the impedance spectra of PPy, GCR and the PPyGCR composites in 1 M H2SO4 electrolyte. The equivalent series resistance (ESR) of the electrode can be obtained from the intercept of the Nyquist plots with the real impedance (Z′), which includes the resistance of the electrolyte, the intrinsic resistance of the active material, and the contact resistance at the interface of the active material/current collector.35 All the materials show comparable ESR at about 5 Ω. For PPy, PPyGCR91, PPyGCR51 and PPyGCR11, the curves are similar in form, composed of a semicircle at high-frequency and a straight line in the low-frequency region. The curved region of the pure PPy nanowire is much larger than the other three PPyGCR composites, indicating a high interfacial charge-transfer resistance, which can be attributed to its poor electrical conductivity. The 45° sloped portion of the Nyquist plots, the so called Warburg resistance, is a result of the frequency dependence of ion transport in the electrolyte. The smaller Warburg region of PPyGCR11 among these four composites indicates fewer variations in ion diffusion path lengths and less obstruction of ion movement. GCR and PPyGCR13 show nearly vertical Nyquist plots at the low frequency part, indicating an almost ideal capacitor response.
The Nyquist plots of PPy, GCR and the PPyGCR composites in 1 M KCl aqueous solution are presented in Fig. 9b. The equivalent series resistances (ESR) of all the materials are about 15 Ω. The negligible high frequency curves for all samples, including pure PPy nanowires, indicate that interfacial charge-transfer resistance is significantly low or body impedance is too large.18 GCR exhibits a larger Warburg region in the 1 M KCl electrolyte compared to the 1 M H2SO4, revealing a relatively weaker charge propagation and ion response in this neutral condition. PPy nanowires show a larger Warburg resistance compared with other composites, indicating a higher ion diffusion resistance and more obstruction of the ion movement. As for PPyGCR composites, the slopes of the impedance increase obviously at low frequency, demonstrating good capacitive behavior without diffusion limitation.
The cycling electrochemical stability of the PPyGCR91 based electrode was tested by galvanostatic charge–discharge at a current density of 2 A g−1 (Fig. S5, ESI†). It was found that 84% of the original capacitance was retained after 1000 cycles in 1 M KCl aqueous solution, higher than the 70% retention in 1 M H2SO4, due to the more dominant EDL capacitive behavior of these composites in this neutral condition, which is consistent with the CV and EIS results. For pure PPy, only 43 and 32% of the original capacitance was retained in 1 M KCl and 1 M H2SO4 aqueous solutions (not shown), respectively. The decrease in capacity could be mainly ascribed to the degradation of the PPy chains during doping/de-doping processes, which would break the polymer chains and generate soluble oligomers, resulting in a significant loss of polymer mass.36 Due to the presence of graphene sheets as substrate, the mechanical strengths of the composite materials can be efficiently enhanced, which is highly favorable for prolonged charge–discharge cycle ability.
The above results demonstrate that the electrochemical performances of the as-prepared PPyGCR composites were greatly improved upon the incorporation of graphene sheets into the PPy nanowire matrix. On the one hand, the morphology of the conducting polymer played a very important role in the electrochemical performance. PPy with 1D nanowire-like morphology in the composite material possesses superior electrochemical properties due to the combination of the properties of low-dimensional organic conductors with high surface area materials.37 On the other hand, the presence of graphene sheets as a conductive material provides a large surface area, which enhances the utilization of PPy. The electrochemical accessibility of the electrolyte through PPy was accelerated in the presence of the graphene sheets. The conducting network formed by GCR can accelerate charge-transfer within these composites. Meanwhile, the unique nanowire-sheet morphology can favor ionic accessibility and transportation during charging–discharging processes. Above all, the unique structure and design not only reduce the diffusion resistance of the electrolytes in the electrode material but also enhance the electrochemical performance due to the synergistic effect between the GCR sheets and the PPy nanowires. PPyGCR91 exhibits the best capacitive performance in both 1 M H2SO4 and 1 M KCl with a specific capacitance of 177 and 182 F g−1, respectively, at a current density of 1 A g−1, and a specific capacitance of 492 and 214 F g−1 at a lower current density of 0.2 A g−1, respectively, exhibiting different capacitive behaviors and rather good electrochemical performance, which is comparable with the results reported recently by other groups.12,13,17–19
Conclusions
PPyGCR composites with different mass ratios were prepared through a simple one step in situ polymerization, with PPy nanowire–GCR nanosheet morphology well preserved and homogeneously mixed. The as-prepared composites exhibit different electrochemical behaviors in 1 M H2SO4 and 1 M KCl electrolytes. Higher specific capacitances and more obvious pseudocapacitive behaviors can be found in the acidic electrolyte, while higher cycling stability and more dominant EDL capacitive behavior were observed in the 1 M KCl electrolyte. It can also be seen that PPyGCR91 exhibits the highest specific capacitance compared to other composites, due to possessing a larger amount of PPy nanowires, which can result in an effective pseudocapacitive contribution to the specific capacitance of the electrodes. A high specific capacitance of 492 F g−1 can be obtained for PPyGCR91 in 1 M H2SO4 at a current density of 0.2 A g−1 with good cycling stability.Acknowledgements
This work was financially supported by the Singapore A*STAR SERC Thematic Strategic Research Program—Sustainable Materials: Composites & Lightweights (No. 092 137 0011) and A*STAR IMRE core funding IMRE/12-1P0902.References
- M. Hughes, G. Z. Chen, M. S. P. Shaffer, D. J. Fray and A. H. Windle, Chem. Mater., 2002, 14, 1610 CrossRef CAS.
- X. Zhao, B. M. Sanchez, P. J. Dobson and P. S. Grant, Nanoscale, 2011, 3, 839 RSC.
- K. S. Ryu, S. K. Jeong, J. Joo and K. M. Kim, J. Phys. Chem. B, 2007, 111, 731 CrossRef CAS.
- B.C. Kim, J. M. Ko and G. G. Wallace, J. Power Sources, 2008, 177, 665 CrossRef CAS.
- R. K. Sharma, A. C. Rastogi and S. B. Desu, Electrochem. Commun., 2008, 10, 268 CrossRef CAS.
- M. Mastragostino, C. Arbizzani and F. Soavi, Solid State Ionics, 2002, 148, 493 CrossRef CAS.
- G. Z. Chen, M. S. P. Shaffer, D. Coleby, G. Dixon, W. Z. Zhou, D. J. Fray and A. H. Windle, Adv. Mater., 2000, 12, 522 CrossRef CAS.
- J. Zhang, L. B. Kong, H. Li, Y. C. Luo and L. Kang, J. Mater. Sci., 2010, 45, 1947 CrossRef CAS , and references therein.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS.
- L. L. Zhang, S. Zhao, X. N. Tian and X. S. Zhao, Langmuir, 2010, 26, 17624 CrossRef CAS.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667 CrossRef CAS.
- P. A. Mini, A. Balakrishnan, S. V. Nair and K. R. V. Subramanian, Chem. Commun., 2011, 47, 5753 RSC.
- A. Liu, C. Li, H. Bai and G. Shi, J. Phys. Chem. C, 2010, 114, 22783 CAS.
- A. Davies, P. Audette, B. Farrow, F. Hassan, Z. Chen, J.-Y. Choi and A. Yu, J. Phys. Chem. C, 2011, 115, 17612 CAS.
- C. Xu, J. Sun and L. Gao, J. Mater. Chem., 2011, 21, 11253 RSC.
- D. Zhang, X. Zhang, Y. Chen, P. Yu, C. Wang and Y. Ma, J. Power Sources, 2011, 196, 5990 CrossRef CAS.
- S. Bose, N. H. Kim, T. Kuila, K. T. Lau and J. H. Lee, Nanotechnology, 2011, 22 Search PubMed.
- X. Lu, W. Zhang, C. Wang, T.-C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671 CrossRef CAS.
- X. T. Zhang, J. Zhang, Z. F. Liu and C. Robinson, Chem. Commun., 2004, 1852 RSC.
- K. Zhang, L. Mao, L. L. Zhang, H. S. On Chan, X. S. Zhao and J. Wu, J. Mater. Chem., 2011, 21, 7302 RSC.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392 CrossRef CAS.
- L. Mao, K. Zhang, H. S. On Chan and J. Wu, J. Mater. Chem., 2012, 22, 80 RSC.
- Y. Q. Han, X. T. Qing, S. J. Ye and Y. Lu, Synth. Met., 2010, 160, 1159 CrossRef CAS.
- S. Bose, T. Kuila, M. E. Uddin, N. H. Kim, A. K. T. Lau and J. H. Lee, Polymer, 2010, 51, 5921 CrossRef CAS.
- V. Chandra and K. S. Kim, Chem. Commun., 2011, 47, 3942 RSC.
- Y.-C. Liu and B.-J. Hwang, Synth. Met., 2000, 113, 203 CrossRef CAS.
- Y.-C. Liu, B.-J. Hwang, W.-J. Jian and R. Santhanam, Thin Solid Films, 2000, 374, 85 CrossRef CAS.
- J. Duchet, R. Legras and S. Demoustier-Champagne, Synth. Met., 1998, 98, 113 CrossRef CAS.
- A. B. Gonçalves, A. S. Mangrich and A. J. G. Zarbin, Synth. Met, 2000, 114, 119 CrossRef.
- G. Han, J. Yuan, G. Shi and F. Wei, Thin Solid Films, 2005, 474, 64 CrossRef CAS.
- C. Zhou, S. Kumar, C. D. Doyle and J. M. Tour, Chem. Mater., 2005, 17, 1997 CrossRef CAS.
- S. Y. Liew, W. Thielemans and D. A. Walsh, J. Phys. Chem. C, 2010, 114, 17926 CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825 CrossRef CAS.
- Q. Fu, B. Gao, H. Dou, L. Hao, X. Lu, K. Sun, J. Jiang and X. Zhang, Synth. Met, 2011, 161, 373 CrossRef CAS.
- D. Li, J. X. Huang and R. B. Kaner, Acc. Chem. Res., 2009, 42, 135 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional FT-IR spectra, Raman spectra, TGA curves, capacity retention figure, N2 adsorption–desorption isotherms and the corresponding pore-size distributions. See DOI: 10.1039/c2ra21617e |
| This journal is © The Royal Society of Chemistry 2012 |