In(OTf)3-catalyzed tandem aza-Piancatelli rearrangement/Michael reaction for the synthesis of 3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives†
B. V. Subba
Reddy
*a,
Y. Vikram
Reddy
a,
P. Subba
Lakshumma
a,
G.
Narasimhulu
a,
J. S.
Yadav
a,
B.
Sridhar
b,
P. Purushotham
Reddy
c and
A. C.
Kunwar
c
aNatural Product Chemistry, Indian Institute of Chemical Technology, Hyderabad 500007, India
bCentre for Nuclear Magnetic Resonance, Indian Institute of Chemical Technology, Hyderabad 500007, India
cLaboratory of X-ray Crystallography, Indian Institute of Chemical Technology, Hyderabad 500007, India
First published on 6th September 2012
Abstract
Furan-2-yl(phenyl)methanol derivatives undergo smooth aza-Piancatelli rearrangement with 2-aminothiophenol and 2-aminophenol in the presence of 10 mol% In(OTf)3 in acetonitrile at room temperature to afford the corresponding 3,4-dihydro-2H-benzo[b][1,4]thiazine or oxazine derivatives respectively in good yields with high selectivity in short reaction times. The salient features of this methodology are good yields, high selectivity, low catalyst loading and faster reaction times. The structure of the products was established by nOe studies and X-ray crystallography.
Introduction
The benzo[b][1,4]oxazine and thiazine structural units are frequently found in various biologically active molecules (Fig. 1).1 They are known to display aldose reductase inhibitory activity2 and potential therapeutic properties.3 In addition to this, benzoxazinones exhibit a diverse range of pharmacological properties such as anti-candida-albicans,4 antifungal,5 kinase inhibitory,6 antagonism to progesterone receptor, antitumor, antiviral, antithrombotic, antimycobacterial, anti-inflammatory, antidiabetic and hypolipidaemic effects.7 Furthermore, 2-arylidene-4-aminoalkyl-2H-1,4-benoxazin-(4H)-ones and related compounds were found to exhibit significant CNS (central nervous system) depression.8 Thus, benzo[b][1,4]oxazines have been recognized as privileged structures for the generation of drug like libraries in drug-discovery. Consequently, various methods have been devised for the synthesis of 3,4-dihydro-2H-benzo[b][1,4]oxazine or thiazine derivatives.9 Despite the fact that numerous methods are reported for the synthesis of benzo[b][1,4]oxazine derivatives, the development of new methods for generating annulated benzo[b][1,4]oxazine and thiazine derivatives would certainly expand the synthetic utility of the Piancatelli rearrangement in medicinal chemistry.10–12![Examples of biologically active benzo[b][1,4]oxazine and thiazine derivatives.](/image/article/2012/RA/c2ra21591h/c2ra21591h-f1.gif) | ||
| Fig. 1 Examples of biologically active benzo[b][1,4]oxazine and thiazine derivatives. | ||
Results and discussion
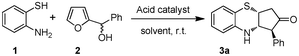
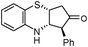
In continuation of our interest on catalytic application of In(III) salts,13 we herein report a novel strategy for the synthesis of cis-3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives via a tandem aza-Piancatelli rearrangement/Michael reaction. As a preliminary experiment, we first attempted the reaction of 2-aminothiophenol (1) with furan-2-yl(phenyl)methanol (2) in the presence of 10 mol% In(OTf)3 in acetonitrile. Interestingly, the reaction proceeded smoothly at room temperature to furnish the corresponding 3,4-dihydro-2H-benzo[b][1,4]thiazine 3a in 86% yield (Scheme 1, Table 1).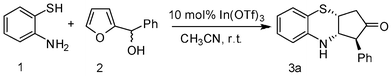 | ||
| Scheme 1 Reaction of 2-aminothiophenol with furan-2-yl(phenyl)methanol. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid catalyst | Amount of catalyst | Solvent | Time (h) | Yield (%)b |
| a The reaction was performed at 0.5 mmol scale. b Isolated yield. c 20% of cyclopentenone without Michael addition. d With respect to the total weight of the reactants. | |||||
| a | Montmorillonite K10 | 67%d | CH3CN | 12.0 | 45c |
| b | Amberlyst-15 | 67%d | CH3CN | 12.0 | 70 |
| c | Phosphomolybdic acid | 10 mol (%) | CH3CN | 2.5 | 75 |
| d | In(OTf)3 | 10 mol (%) | THF | 8.0 | 60 |
| e | In(OTf)3 | 10 mol (%) | DCE | 8.0 | 65 |
| f | In(OTf)3 | 5 mol (%) | CH3CN | 6.0 | 62 |
| g | In(OTf)3 | 10 mol (%) | CH3CN | 2.0 | 86 |
| h | InCl3 | 10 mol (%) | CH3CN | 4.0 | 65 |
| i | InBr3 | 10 mol (%) | CH3CN | 4.0 | 70 |
| j | CeCl3·7H2O | 10 mol (%) | CH3CN | 12.0 | 40 |
| k | Al(OTf)3 | 10 mol (%) | CH3CN | 4.0 | 72 |
In order to optimize the reaction conditions, we performed the above reaction with various acid catalysts such as InCl3, InBr3, In(OTf)3, CeCl3·7H2O and Al(OTf)3. Among these Lewis acids, In(OTf)3 was found to be more effective than others in terms of reaction time and yields (Table 1). Of the various Brønsted acids like heteropoly acids, K10 clay and ion-exchange resin, i.e. Amberlyst-15®, used for this reaction, phosphomolybdic acid was found to be superior to other solid acid catalysts (Table 1). Next we examined the effect of various solvents such as 1,2-dichloroethane, tetrahydrofuran and acetonitrile. Of these, acetonitrile appeared to give the best results (Table 1). As seen in Table 1, 10 mol% In(OTf)3 in acetonitrile is crucial for this conversion.
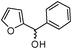
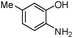
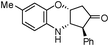
Inspired by the above results, we extended this process to other substrates. Interestingly, various furan-2-yl(aryl)methanols reacted well with 2-aminothiophenols under the optimized conditions to afford the corresponding cis-fused 3,4-dihydro-2H-benzo[b][1,4]thiazine derivatives in excellent yields (entries f and kTable 2). On the other hand, 4-chloro-2-aminothiophenol also participated in this reaction (entries d and i, Table 2). Next we attempted the coupling of 2-aminophenols with furan-2-yl(aryl)methanol derivatives. Though the condensation of furan-2-yl(phenyl)methanol with 5-methyl-2-aminophenol proceeds at room temperature, the reaction requires a relatively long reaction time. The reason may be attributed to the lower reactivity of 2-aminophenol than 2-aminothiophenol (Scheme 2).
| Entry | Substrate (1) | Arylamine (2) | Product (3)b | Time (h) | Yield (%)c |
|---|---|---|---|---|---|
| a The reactions were performed at 1 mmol scale. b All the products were characterized by 1H and 13C NMR, IR and mass spectroscopy. c Yield refers to pure products after column chromatography. | |||||
| a |
|
|
|
2.0 | 86 |
| b |
|
|
|
3.5 | 73 |
| c |
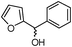
|
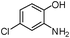
|
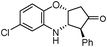
|
3.5 | 78 |
| d |
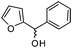
|
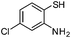
|
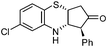
|
2.5 | 85 |
| e |
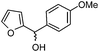
|
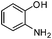
|
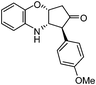
|
3.0 | 75 |
| f |
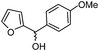
|
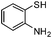
|
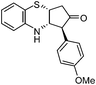
|
2.0 | 87 |
| g |
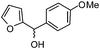
|
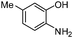
|
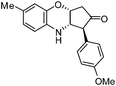
|
2.5 | 80 |
| h |
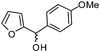
|
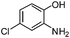
|
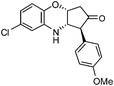
|
2.5 | 82 |
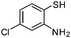
| i |
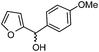
|
|
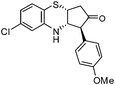
|
2.5 | 88 |
| j |
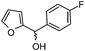
|
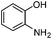
|
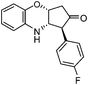
|
3.5 | 81 |
| k |
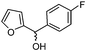
|
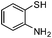
|
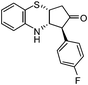
|
2.5 | 85 |
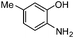
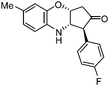
| l |
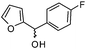
|
|
|
3.5 | 78 |
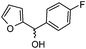
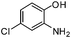
| m |
|
|
|
3.5 | 80 |
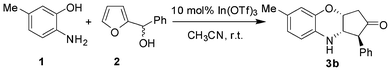 | ||
| Scheme 2 Reaction of 5-methyl-2-aminophenol with furan-2-yl(phenyl)methanol. | ||
Next we studied the reactivity of various 2-aminophenols with furan-2-yl(aryl)methanol derivatives. The scope of the reaction is illustrated with respect to various substrates and the results are summarized in Table 2. As seen from Table 2, halogenated 2-aminophenols or 2-aminothiophenols gave comparatively higher yields than methyl-substituted derivatives.
The structure of the compound 3g was characterized by detailed NMR studies including 2-D double quantum filtered correlation spectroscopy (DQFCOSY) and nuclear Overhauser effect spectroscopy (NOESY). The 1H NMR experiments provided coupling constants of 3JH1–H2 (pro-R) = 4.4, 3JH1–H4 = 3.1 and 3JH3–H4 = 10.7 and 3JH1–H2 (pro-S)∼0 Hz. From these coupling values, along with the presence of a nOe correlations, H2(pro-R)/H4 (medium intensity) and H2(pro-S)/H3 (weak intensity), the 5-membered cis-fused ring was found to take a twist conformation. In addition a ω-coupling, 4JH3–H2(pro-S) = 1.6 Hz, and nOe correlation between H5/H6 further supports the structure. An energy minimized structure14 is consistent with the NMR findings which are given in Fig. 2.
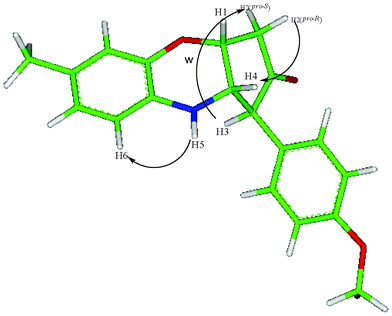 | ||
| Fig. 2 Characteristic nOe's and energy minimized structure of 3g. | ||
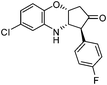
Furthermore, the structure of 3e was confirmed by X-ray crystallography† (Fig. 3).15
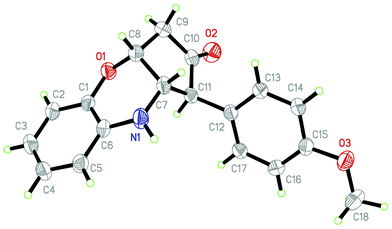 | ||
| Fig. 3 ORTEP diagram of 3e. | ||
Mechanistically, the reaction is expected to proceed via the formation of an oxocarbenium ion from furan-2-yl(phenyl)methanol, likely after activation through In(III). This is followed by attack of 2-aminothiophenol or 2-aminophenol resulting in the formation of an aminal. An acid catalyzed rearrangement of the aminal followed by thia-/oxa-Michael reaction respectively would give the desired product as shown in Scheme 3.
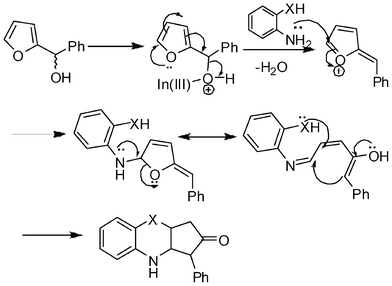 | ||
| Scheme 3 A plausible reaction pathway. | ||
In the absence of In(OTf)3, no aza-Piancatelli rearrangement was observed even in refluxing acetonitrile. The effect of various oxidants such as ceric ammonium nitrate, molecular iodine and ferric chloride was studied. The use of 10 mol% of molecular iodine in acetonitrile facilitates the SN2 substitution of OH rather than aza-Piancatelli rearrangement. Instead of product 3a, the cyclopentenone derivative was formed without the Michael addition when the reaction was performed either by 10 mol% ceric ammonium nitrate or by 10 mol% FeCl3. Thus the reaction was quite successful only with 10 mol% In(OTf)3. This method is quite simple and convenient to synthesise a wide range of 3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives in a single step process.
Conclusions
In summary, we have demonstrated a novel strategy for the synthesis of 3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives via a tandem aza-Piancatelli/Michael reaction between furan-2-yl(phenyl)methanols and 2-aminothiophenols and 2-aminophenols respectively. This is the first report on the synthesis of annulated 3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives by means of aza-Piancatelli rearrangement.Experimental
General
The solvents were dried according to a standard literature procedure. The reactions were performed in oven-dried round bottom flasks under a nitrogen atmosphere. Glass syringes were used to transfer the solvent. The products were purified by column chromatography on silica gel of 60–120 mesh. Thin layer chromatography plates were visualized by the ultraviolet light and/or by exposure to iodine vapours and/or by exposure to acidic methanolic solution of p-anisaldehyde followed by heating (<1 min) on a hot plate (∼250 °C). Organic solutions were concentrated on rotary evaporator at 35–40 °C. IR spectra were recorded on FT-IR spectrometer. 1H and 13C NMR spectra were recorded in CDCl3 using 300 or 500 MHz NMR spectrometers. The chemical shifts (δ) were reported in parts per million (ppm) with respect to TMS as an internal standard. The coupling constants (J) are quoted in Hertz (Hz). Mass spectra were recorded on mass spectrometer by Electrospray ionization (ESI) technique.
Typical procedure: A mixture of furan-2-yl(phenyl)methanol (1, 1.2 mmol), 2-aminophenol or 2-aminothiophenol (2, 1 mmol) and In(OTf)3 (56 mg, 10 mol%) in acetonitrile (4 mL) was stirred at room temperature for a specified time as required to complete the reaction (see Table 1). After complete conversion, as indicated by TLC, the reaction mixture was diluted with water and extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, concentrated in vacuo and purified by column chromatography on silica gel (Merck, 60–120 mesh, ethyl acetate–hexane, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) to afford the pure 3,4-dihydro-2H-benzo[b][1,4]oxazine or thiazine derivative.
:7) to afford the pure 3,4-dihydro-2H-benzo[b][1,4]oxazine or thiazine derivative.
3a: IR (neat): νmax 3880, 3020, 2923, 2853, 1743, 1591, 1485, 1260, 1154, 1074, 749 cm−1; 1H NMR (500 MHz, CDCl3): δ 2.61 (d, 1H, J = 19.4 Hz), 2.87 (dd, 1H, J = 7.0, 19.0 Hz), 3.72 (d, 1H, J = 10 Hz), 3.81 (t, 1H, J = 5.0 Hz), 4.17–4.26 (m, 1H), 4.31–4.40 (brs, 1H), 6.53 (d, 1H, J = 8.0 Hz), 6.70 (t, 1H, J = 8.0 Hz), 6.97 (t, 1H, J = 7.0 Hz), 7.09 (d, 3H, J = 7.0 Hz), 7.30 (t, 1H, J = 7.0 Hz), 7.36 (t, 2H, J = 8.0 Hz); 13C NMR (75 MHz, CDCl3): δ 35.3, 44.8, 59.8, 60.1, 115.3, 115.5, 118.5, 126.4, 127.5, 127.6, 128.9, 129.0, 135.8, 139, 212.9; ESI-MS: m/z 282 (M+H); HRMS calcd for C17H16NOS: 282.0947, found: 282.0944.
3b: Solid, m.p. 163–165 °C; IR (neat): νmax 3390, 3053, 2915, 2857, 1753, 1642, 1527, 1463, 1295, 1241, 1121, 1038, 759 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.24 (s, 3H), 2.65 (dd, 1H, J = 4.5, 18.8 Hz), 2.76–2.86 (m, 1H), 3.35 (d, 1H, J = 10.5 Hz), 3.75 (dd, 1H, J = 3.0, 11.3 Hz), 4.34 (t, 1H, J = 3.0 Hz), 4.64–4.71 (m, 1H), 6.67–6.80 (m, 2H), 7.04 (dd, 1H, J = 1.5, 7.5 Hz), 7.24–7.31 (m, 2H), 7.33–7.45 (m, 2H), 7.56 (dd, 1H, J = 2.3, 6.0 Hz); 13C NMR (75 MHz, CDCl3): δ 20.3, 46.1, 56.6, 56.9, 69.7, 116.5, 117.9, 122.9, 127.7, 128.8, 128.9, 129.5, 135.6, 136.3, 137.8, 142.9, 161.7, 212.3; ESI-MS: m/z 280 (M+H).
3c: Solid, m.p. 136–138 °C; IR (neat): νmax 3396, 3027, 2924, 2856, 1742, 1602, 1498, 1287, 1121, 1016, 755 cm−1; 1H NMR (500 MHz, CDCl3): δ 2.70 (dd, 1H, J = 4.7, 19.1 Hz), 2.90 (d, 1H, J = 19.1 Hz), 3.47 (d, 1H, J = 10.3 Hz), 4.00–4.08 (m, 1H), 4.19–4.28 (brs, 1H), 4.62 (t, 1H, J = 3.9 Hz), 6.58 (d, 1H, J = 2.4 Hz), 6.65 (dd, 1H, J = 2.4, 7.9 Hz), 6.80 (d, 1H, J = 8.7 Hz), 7.09 (d, 2H, J = 7.2 Hz), 7.31 (t, 1H, J = 7.2 Hz), 7.37 (t, 2H, J = 7.9 Hz); 13C NMR (125 MHz, CDCl3): δ 45.4, 58.2, 58.5, 70.5, 114.5, 118.2, 118.4, 127.1, 127.7, 128.8, 129.1, 131.1, 135.4, 140.6, 212.1; ESI-MS: m/z 300 (M+H); HRMS calcd for C17H15NO2Cl: 300.07858, found: 300.07855.
3d: IR (neat): νmax 3402, 2951, 2894, 1736, 1582, 1526, 1322, 1239, 1165, 1073, 895, 754 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.61 (d, 1H, J = 18.8 Hz), 2.90 (dd, 1H, J = 6.8, 18.8 Hz), 3.69 (d, 1H, J = 9.8 Hz), 3.77 (t, 1H, J = 4.5 Hz), 4.18–4.30 (m, 1H), 4.46 (d, 1H, J = 4.5 Hz), 6.53 (d, 1H, J = 2.3 Hz), 6.67 (dd, 1H, J = 2.3, 8.3 Hz), 7.01 (d, 1H, J = 8.3 Hz), 7.05–7.13 (m, 2H), 7.28–7.43 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 34.9, 44.6, 59.6, 60.3, 113.4, 114.8, 118.4, 127.6, 128.5, 128.9, 129.1, 131.7, 135.5, 140, 212.4; ESI-MS: m/z 316 (M+H).
3e: Solid, m.p. 119–121 °C; IR (KBr): νmax 3388, 2922, 2841, 1889, 1739, 1608, 1249, 1026, 826, 747 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.68 (dd, 1H, J = 4.5, 18.8 Hz), 2.89 (d, 1H, J = 18.8 Hz), 3.47 (d, 1H, J = 10.5 Hz), 3.80 (s, 3H), 4.01 (dd, 1H, J = 2.3, 10.5 Hz), 4.65 (t, 1H, J = 3.7 Hz), 6.61 (dd, 1H, J = 1.5, 7.5 Hz), 6.72 (dd, 1H, J = 1.5, 7.5 Hz), 6.72 (dd, 1H, J = 1.5, 7.5 Hz), 6.81–6.95 (m, 4H), 7.03 (d, 2H, J = 9.1 Hz); 13C NMR (75 MHz, CDCl3): δ 45.3, 55.2, 57.3, 58.6, 70.3, 114.4, 115.1, 117.1, 118.6, 122.4, 127.5, 129.8, 130.1, 142.1, 158.9, 213.1; ESI-MS: m/z 297 (M+H); HRMS calcd for C18H18NO3: 296.1281, found: 296.1276.
3f: IR (KBr): νmax 3373, 2923, 2853, 1735, 1587, 1463, 1248, 1178, 1026, 753 cm−1; 1H NMR (500 MHz, CDCl3): δ 2.61 (d, 1H, J = 18.6 Hz), 2.87 (dd, 1H, J = 6.5, 18.6 Hz), 3.68 (d, 1H, J = 10.3 Hz), 3.75–3.84 (m, 1H), 3.80 (s, 3H), 4.16–4.23 (m, 1H), 4.37 (brs, 1H), 6.55 (d, 1H, J = 8.4 Hz), 6.71 (t, 1H, J = 7.5 Hz), 6.91 (d, 1H, J = 8.4 Hz), 6.98 (t, 1H, J = 8.4 Hz), 7.03 (d, 2H, J = 8.4 Hz), 7.10 (d, 1H, J = 7.5 Hz); 13C NMR (75 MHz, CDCl3): δ 45.3, 55.2, 57.3, 58.5, 70.2, 114.4, 115.1, 117.1, 118.6, 122.4, 127.5, 129.8, 130.1, 142.0, 158.9, 213.2; ESI-MS: m/z 310 (M+H).
3g: Solid, m.p. 127–129 °C; IR (neat): νmax 3396, 3013, 2925, 1743, 1612, 1514, 1460, 1296, 1251, 1123, 1028, 757 cm−1; 1H NMR (500 MHz, CDCl3): δ 2.23 (s, 3H), 2.62 (dd, 1H, J = 4.4, 18.7 Hz), 2.83 (d, 1H, J = 18.7 Hz), 3.42 (d, 1H, J = 10.7 Hz), 3.77 (s, 3H), 3.93 (dd, 1H, J = 2.2, 11.0 Hz), 4.59 (t, 1H, J = 3.1 Hz), 6.49 (d, 1H, J = 8.8 Hz), 6.64 (d, 1H, J = 7.7 Hz), 6.69 (s, 1H), 6.88 (d, 2H, J = 7.7 Hz), 6.98 (d, 2H, J = 8.8 Hz); 13C NMR (75 MHz, CDCl3): δ 20.5, 45.5, 55.3, 56.9, 58.7, 70.5, 114.5, 115.3, m117.6, 122.9, 127.3, 127.6, 128.6, 129.8, 124.1, 142.1, 159, 213; ESI-MS: m/z 310 (M+H); HRMS calcd for C19H20NO3: 310.1437, found: 310.1436.
3h: IR (neat): νmax 3385, 2925, 2853, 1744, 1606, 1250, 1126, 1029, 757 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.68 (dd, 1H, J = 4.5, 19.1 Hz), 2.87 (d, 1H, J = 19.1 Hz), 3.41 (d, 1H, J = 10.5 Hz), 3.79 (s, 3H), 3.99 (dd, 1H, J = 3.0, 10.7 Hz), 4.13–4.42 (brs, 1H), 4.60 (t, 1H, J = 3.7 Hz), 6.58 (d, 1H, J = 2.3 Hz), 6.60–6.70 (m, 1H), 6.78 (d, 1H, J = 8.5 Hz), 6.90 (d, 2H, J = 8.7 Hz), 7.01 (d, 2H, J = 8.7 Hz); ESI-MS: m/z 330 (M+H); HRMS calcd for C18H17NO3Cl: 330.0891, found: 330.0893.
3i: IR (neat): νmax 3406, 2957, 2875, 1754, 1593, 1536, 1373, 1257, 1215, 1064, 849, 764 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.31 (t, 1H, J = 7.5 Hz), 2.68 (dd, 1H, J = 3.7, 18.8 Hz), 2.88 (d, 1H, J = 18.8 Hz), 3.42 (d, 1H, J = 10.6 Hz), 3.80 (s, 3H), 3.99 (d, 1H, J = 10.6 Hz), 4.60 (brs, 1H), 6.58 (s, 1H), 6.64 (d, 1H, J = 8.3 Hz), 6.79 (d, 1H, J = 9.0 Hz), 6.90 (d, 2H, J = 8.3 Hz), 7.01 (d, 2H, J = 8.3 Hz); ESI-MS: m/z 346 (M+H).
3j: Solid, m.p. 138–140 °C; IR (KBr): νmax 3433, 3026, 2918, 1739, 1603, 1305, 1216, 1119, 838, 732 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.70 (dd, 1H, J = 4.5, 18.8 Hz), 2.92 (d, 1H, J = 18.1 Hz), 3.53 (d, 1H, J = 10.5 Hz), 4.03 (d, 1H, J = 10.6 Hz), 4.09–4.21 (brs, 1H), 4.65 (t, 1H, J = 3.8 Hz), 6.64 (dd, 1H, J = 1.5, 7.5 Hz), 6.74 (dd, 1H, J = 1.5, 7.5 Hz), 6.82–6.93 (m, 2H), 7.08 (d, 4H, J = 6.8 Hz); 13C NMR (75 MHz, CDCl3): δ 45.4, 57.3, 58.7, 70.2, 115.2, 115.8, 116, 117.2, 118.8, 122.5, 129.9, 130.3, 130.4, 131.3, 142, 160.5, 163.8, 212.5; ESI-MS: m/z 284 (M+H); HRMS calcd for C17H15NO2F: 284.1081, found: 284.1079.
3k: Solid, m.p. 140–142 °C; IR (KBr): νmax 3432, 2924, 2854, 1738, 1590, 1506, 1310, 1219, 1156, 1095, 840, 737 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.64 (d, 1H, J = 19.3 Hz), 2.40 (dd, 1H, J = 6.6, 19.2 Hz), 3.75 (d, 1H, J = 10.4 Hz), 3.82 (t, 1H, J = 5.3 Hz), 4.15–4.26 (m, 1H), 4.35 (brs, 1H), 6.57 (d, 1H, J = 7.9 Hz), 6.73 (t, 1H, J = 7.5 Hz), 7.00 (t, 1H, J = 7.3 Hz), 7.08 (d, 4H, J = 6.9 Hz), 7.12 (d, 1H, J = 1.1 Hz); 13C NMR (75 MHz, CDCl3): δ 35.2, 44.7, 59.4, 59.9, 115.4, 115.6, 115.8, 116.1, 118.6, 126.5, 127.6, 130.5, 130.6, 131.4, 138.9, 160.6, 163.8, 212.5; ESI-MS: m/z 300 (M+H); HRMS calcd for C17H15NOSF: 300.0852, found: 300.0851.
3l: IR (neat): νmax 3396, 2923, 2855, 1741, 1597, 1509, 1293, 1229, 1155, 1020, 808, 758 cm−1; 1H NMR (500 MHz, CDCl3): δ 2.25 (s, 3H), 2.68 (dd, 1H, J = 4.9, 19.8 Hz), 2.90 (d, 1H, J = 19.8 Hz), 3.51 (d, 1H, J = 10.8 Hz), 3.99 (d, 1H, J = 7.9 Hz), 4.65 (t, 1H, J = 2.9 Hz), 6.55 (d, 1H, J = 7.9 Hz), 6.67 (d, 1H, J = 7.9 Hz), 6.72 (s, 1H), 7.07 (d, 4H, J = 6.9 Hz); 13C NMR (75 MHz, CDCl3): δ 20.5, 45.5, 56.9, 58.8, 70.4, 115.3, 115.8, 116.1, 117.7, 123, 128.8, 130.3, 130.4, 131.3, 163.9, 212.6; ESI-MS: m/z 298 (M+H).
3m: IR (KBr): νmax 3405, 3076, 2974, 1739, 1582, 1563, 1540. 1387, 1274, 1106, 1172, 1037, 964, 860, 752 cm−1; 1H NMR (300 MHz, CDCl3): δ 2.67 (dd, 1H, J = 4.5, 18.8 Hz), 2.87 (d, 1H, J = 19.6 Hz), 3.44 (d, 1H, J = 10.5 Hz), 3.98 (dd, 1H, J = 3.0, 10.5 Hz), 4.22–4.50 (brs, 1H), 4.56 (t, 1H, , J = 3.0 Hz), 6.56 (d, 1H, J = 2.3 Hz), 6.63 (dd, 1H, J = 2.3, 8.3 Hz), 6.78 (d, 1H, J = 9.1 Hz), 7.04 (d, 4H, J = 6.8 Hz); 13C NMR (75 MHz, CDCl3): δ 45.2, 57.5, 58.3, 70.3, 114.5, 115.8, 116.1, 118.2, 118.3, 127.1, 130.3, 130.4, 131, 140.5, 160.5, 163.8, 212.2; ESI-MS: m/z 318 (M+H). HRMS calcd for C17H13FClNO2: (M+H): 318.1008; found: 318.1014.
Acknowledgements
GNL thanks CSIR, New Delhi for the award of a fellowship.References
- (a) S. Tanimori, Y. Kato and M. Kirihata, Synthesis, 2004, 2103 Search PubMed; (b) M. Ohno, Y. Tanaka, M. Miyamoto, T. Takeda, K. Hoshi, N. Yamada and A. Ohtake, Bioorg. Med. Chem., 2006, 14, 2005 Search PubMed; (c) J. F. Bower, P. Szeto and T. Gallagher, Org. Lett., 2007, 9, 3283 CrossRef CAS.
- (a) T. Aotsuka, H. Hosono, T. Kurihara, Y. Nakamura, T. Matsui and F. Kobayashi, Chem. Pharm. Bull., 1994, 42, 1264 CAS; (b) A. Ota, H. Suhara and Y. Kawashima, Chem. Pharm. Bull., 1992, 40, 833 CAS.
- (a) D. W. Combs, M. S. Rampulla, S. C. Bell, D. H. Klaubert, A. J. Tobia, R. Falotico, R. B. Hertlein, C. Lakas-Weiss and C. J. B. Morre, J. Med. Chem., 1990, 33, 380 CrossRef CAS; (b) A.-S. Bourlot, I. Sanchez, G. Dureng, G. Guillaumet, R. Massingham, A. Monteil, E. Winslow, M. D. Pujol and J.-Y. Merour, J. Med. Chem., 1998, 41, 3142 CrossRef CAS; (c) M. Largeron, H. Dupuy and M. B. Fleury, Tetrahedron, 1995, 51, 4953 CrossRef CAS; (d) A. K. Willard, R. L. Smith and E. J. Cragoe, J. Org. Chem., 1981, 46, 3846 CrossRef CAS.
- R. Fringuelli, D. Pietrella, F. Schiaffella, I. A. Guarrac, S. Perito, F. Bistoni and A. Vecchiarelli, Bioorg. Med. Chem., 2002, 10, 1681 CrossRef CAS.
- A. Macchiarulo, G. Costantino, F. Fringuelli, A. Vecchiarelli, F. Schiaffella and R. Fringuelli, Bioorg. Med. Chem., 2002, 10, 3415 CrossRef CAS.
- I. Sheikhshoaie, F. Belaj and A. Kamali, Bull. Chem. Soc. Ethiop., 2010, 24, 283 Search PubMed.
- A. D. Meijere, I. D. Kuchuk, V. V. Sokolov, T. Labahn, K. Rauch, M. Es-Sayed and T. Krämer, Eur. J. Org. Chem., 2003, 985 CrossRef CAS.
- (a) M.-Z. Huang, F.-X. Luo, H.-B. Mo, Y.-G. Ren, X.-G. Wang, X.-M. Ou, M.-X. Lei, A.-P. Liu, L. Huang and M.-C. Xu, J. Agric. Food Chem., 2009, 57, 9585 CrossRef CAS; (b) C. F. Turk, J. Krapcho, I. M. Michel and I. Weinryb, J. Med. Chem., 1977, 20, 729 CrossRef CAS.
- (a) K. C. Nicolaou, P. S. Baran, Y.-L. Zhong and K. Sugita, J. Am. Chem. Soc., 2002, 124, 2212 CrossRef CAS; (b) S. Bhadra, L. Adak, S. Samanta, A. K. M. M. Islam, M. Mukherjee and B. C. Ranu, J. Org. Chem., 2010, 75, 8533 CrossRef CAS; (c) R. Koteshwar Rao and G. Sekar, Tetrahedron: Asymmetry, 2011, 22, 948 Search PubMed; (d) M. A. Abdel-Rahman, A. Khodairy, A.-B. A. G. Ghattas and S. Younes, J. Chin. Chem. Soc., 2004, 51, 103 Search PubMed.
- (a) G. Piancatelli, A. Scettri and S. Barbadoro, Tetrahedron Lett., 1976, 17, 3555 CrossRef; (b) A. Scettri, G. Piancatelli, M. D'Auria and G. David, Tetrahedron, 1979, 35, 135 CrossRef CAS; (c) A. N. Faza, C. S. Lopez, R. Alvarez and I. R. Lera, Chem.–Eur. J., 2004, 10, 4324 CrossRef CAS.
- (a) J. Dauvergne, A. M. Happe, V. Jadhav, D. Justice, M. C. Matos, P. J. McCormack, M. R. Pitts, S. M. Roberts, S. K. Sing, T. J. Snape and J. Whitall, Tetrahedron, 2004, 60, 2559 Search PubMed; (b) F. A. Davis and Y. Z. Wu, Org. Lett., 2004, 6, 1269 Search PubMed; (c) J. Dauvergne, A. M. Happe and S. M. Roberts, Tetrahedron, 2004, 60, 2551 CrossRef CAS; (d) M. Zaja and S. Blechert, Tetrahedron, 2004, 60, 9629 CrossRef CAS.
- (a) G. K. Veits, D. R. Wenz and J. R. Alaniz, Angew. Chem., Int. Ed., 2010, 49, 9484 Search PubMed; (b) L. I. Palmer and J. R. Alaniz, Angew. Chem., Int. Ed., 2011, 50, 7167 Search PubMed; (c) B.V. S. Reddy, G. Narasimhulu, P. S. Lakshumma, Y. V. Reddy and J. S. Yadav, Tetrahedron Lett., 2012, 53, 1776 Search PubMed.
- (a) J. S. Yadav, B. V. S. Reddy, K. V. Rao, K. Saritha Raj, A. R. Prasad, S. K. Kumar, A. C. Kunwar, P. Jayaprakash and B. Jagannadh, Angew. Chem., Int. Ed., 2003, 42, 5198 Search PubMed; (b) J. S. Yadav, B. V. S. Reddy, T. Maity and G. G. K. S. N. Kumar, Tetrahedron Lett., 2007, 48, 8874 CrossRef CAS; (c) B. V. S. Reddy, P. Borkar, J. S. Yadav, P. P. Chakravarti, A. C. Kunwar, B. Sridhar and G. Rene, Org. Biomol. Chem., 2012, 10, 1349 RSC; (d) J. S. Yadav, B. V. S. Reddy, A. V. Hara Gopal and K. S. Patil, Tetrahedron Lett., 2009, 50, 3493 CrossRef CAS; (e) J. S. Yadav, B. V. S. Reddy, S. Aravind, G. G. K. S. N. Kumar and A. S. Reddy, Tetrahedron Lett., 2007, 48, 6117 CrossRef CAS; (f) B. V. S. Reddy, P. S. Reddy, Y. J. Reddy and J. S. Yadav, Tetrahedron Lett., 2011, 52, 5789 Search PubMed; (g) B. V. S. Reddy, M. Sreelatha, Ch. Kishore, P. Borkar and J. S. Yadav, Tetrahedron Lett., 2012, 53, 2748 Search PubMed; (h) J. S. Yadav, V. Sunitha, P. P. Das and E. Gyanchander, Tetrahedron Lett., 2008, 49, 855 Search PubMed.
- Energy-minimized structure was deduced using the INSIGHT-II, Discover model on SGI workstation, Version 2.98; Biosym molecular simulation, San Diego, CA, 1995 Search PubMed.
- The crystal (C18H17NO3, M = 295.33, colorless block, 0.21 × 0.18 × 0.08 mm3) belongs to the orthorhombic crystal system, space group is Pbcn (No. 60), a = 18.360(3), b = 10.7617(18), c = 15.277(3) Å, V = 3018.5(9) Å3, Z = 8, Dc = 1.300 g cm−3, F000 = 1248, CCD Area Detector, Mo-Kα radiation, λ = 0.71073 Å, T = 294(2)K, 2θmax = 50.0°, 27253 reflections collected, 2661 unique (Rint = 0.0281). Final GooF = 1.053, R1 = 0.0341, wR2 = 0.0898, R indices based on 2219 reflections with I >2σ(I) (refinement on F2), 204 parameters, 0 restraints. μ = 0.089 mm−1. CCDC 890425 contains supplementary Crystallographic data for the structure†.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21591h |
| This journal is © The Royal Society of Chemistry 2012 |