Silver–gold nanotubes containing hot spots on their surface: facile synthesis and surface-enhanced Raman scattering investigations†
Jean C. S.
Costa
,
Paola
Corio
* and
Pedro H. C.
Camargo
*
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Lineu Prestes, 748 05508-000, São Paulo-SP, Brazil. E-mail: paola@iq.usp.br; camargo@iq.usp.br; Tel: 55 1130919148
First published on 4th September 2012
Abstract
We report the synthesis of silver–gold nanotubes containing hot spots along their surface. The Ag–Au nanotubes exhibited exceptional SERS properties compared to silver nanowires, enabling the detection of crystal violet in the 10−10 M regime, as well as 9-nitroanthracene and benzo[a]pyrene at 3.3 × 10−7 M.
Bimetallic nanostructures containing hollow interiors comprise an interesting class of materials, as they enable not only the combination of properties from the metal components, but also provide higher surface to volume ratios than their solid counterparts.1–4 Among the various approaches for their synthesis, the galvanic replacement reaction represents a simple strategy to obtain, in one step and in aqueous phase, bimetallic nanostructures with hollow interiors and thin walls.5–9 With this method, the shape, composition and structure of the resulting materials can be tailored by employing sacrificial templates with different morphologies and/or by controlling the molar ratio between the reactants and the extent of the reaction. For instance, the galvanic reaction between silver (Ag) nanocrystals as sacrificial templates and HAuCl4 to produce silver–gold (Ag–Au) nanostructures with well-defined shapes, composition and structures has been comprehensively investigated.5–10
Among bimetallic and hollow systems, Ag–Au nanomaterials are of particular interest for the development of substrates for the identification of trace chemicals by the surface-enhanced Raman scattering effect (SERS).11–15 Firstly, increased surface areas offer a higher number of sites for analyte adsorption relative to the solid equivalents. In this case, it is important to mention that molecules must be located close to the surface of the Ag or Au nanostructures for the SERS effect to take place in the visible range, according to the electromagnetic mechanism of enhancement.11–13 Also, bimetallic Ag–Au nanostructures enable the fine-tuning of surface plasmon resonance (SPR) frequencies.10,16,17 This is important as it has been reported that matching of the SPR frequency and the SERS excitation wavelength gives rise to improved SERS intensities.11,18
In this work, we describe the use of the galvanic reaction between Ag nanowires (Ag NWs) and HAuCl4 as a facile strategy to produce Ag–Au nanotubes (Ag–Au NTs) with porous walls, followed by their application as SERS substrates for the detection of trace chemicals. More specifically, we started by investigating the morphology of the Ag–Au NTs produced by this approach. Then, we studied the optical and SERS properties of the Ag–Au NTs compared to the Ag NWs. Finally, we demonstrate the application of the Ag–Au NTs as SERS substrates for the detection of crystal violet in the 10−10 M regime, as well as the detection of polycyclic aromatic hydrocarbons at 3.3 × 10−7 M concentrations.
We started our study with the synthesis of pentagonal Ag NWs with well-defined size and shape via the polyol process.6,19 In a typical synthesis, 5 mL of ethylene glycol (EG) was transferred to a round bottom flask and heated to 160 °C under stirring for 1 h. Then, 3 mL of a 0.1 M AgNO3 solution and 3 mL of a 0.6 M PVP solution (both in EG) were added simultaneously, using a peristaltic pump at the rate of 0.4 mL min−1, to the pre-heated EG. The reaction was allowed to proceed at this temperature for 4 h before the product was collected by centrifugation, washed three times with water and re-suspended in water. A SEM image of the produced Ag NWs is shown in Fig. S1.†6,19 The Ag NWs were 93.5 ± 5.0 nm in width and >3 μm in length. After their synthesis, the Ag NWs were employed as sacrificial templates in a galvanic replacement reaction with HAuCl4 to yield Ag–Au NTs in a single step according to the following equation:7,8
| 3Ag(s) + [AuCl4]−(aq) → Au(s) + 3Ag+(aq) + 4Cl−(aq) |
For the synthesis of the Au–Ag NTs, 100 μL of the suspension containing the Ag NWs was added to 5 mL of an aqueous PVP solution (1 mg mL−1) under magnetic stirring. This system was heated to 100 °C for 10 min. Then, 2 mL of a 0.1 M HAuCl4·3H2O aqueous solution was added dropwise, causing a change in color from light brown to violet. The solution was kept at 100 °C for another 10 min and the product was collected by centrifugation, washed with saturated NaCl aqueous solution and re-suspended in water. Fig. 1A and B show SEM and TEM (inset) images of the Ag–Au NTs produced by this approach. The images confirm that the 1D morphology was maintained during the reaction, in which Au was deposited on the surface of the sacrificial templates. The Ag–Au NTs were uniform, being 144.8 ± 6.9 nm in width. This result indicates a wall thickness of ∼25 nm, which is in agreement with the TEM (inset) image. Fig. 1A and B also reveal that the surface of the nanotubes was porous and comprised of Ag–Au islands, ∼10–25 nm in size, that are closely joined together.
 | ||
| Fig. 1 SEM and TEM (inset) images of Ag–Au NTs obtained by the galvanic reaction between Ag NWs and HAuCl4. The scale bar in the inset corresponds to 50 nm. | ||
It has been established that the galvanic replacement reaction between Ag and HAuCl4 consists of two major stages. In the first, Ag is oxidized to Ag+ and continuously dissolved, while Au is epitaxially deposited over the surface of the template. In this case, an Ag–Au alloy is formed due to the interdiffusion between Ag and Au, leading to hollow nanostructures with smooth surfaces.5–9 In the second stage, as the galvanic replacement continues, the Ag–Au walls are dealloyed. During this process, Ostwald ripening can induce morphological reconstruction and the formation of pinholes on the walls. Eventually, complete dealloying could lead to the rupture of the material into Au nanoparticles.7 Therefore, according to our results, is plausible to assume that the amount of HAuCl4 employed allowed the galvanic replacement process to reach its second stage, in which Ag–Au dealloying and Ostwald ripening processes enabled the formation of porous walls. Information on the composition of the Ag–Au NTs was obtained by energy dispersive spectroscopy (EDS). Our EDS analysis showed that the atomic percentages of Au and Ag in the nanotubes were 47.2 ± 5.7% and 52.8 ± 5.8%, respectively. This indicates a Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au molar ratio of 1.1, and is in agreement with previously reported results involving Ag nanocrystals as templates in which dealloying and reconstruction processes took place at, to give Au molar percentages higher than 30%.7,14
:Au molar ratio of 1.1, and is in agreement with previously reported results involving Ag nanocrystals as templates in which dealloying and reconstruction processes took place at, to give Au molar percentages higher than 30%.7,14
The extinction spectra recorded from the initial Ag NWs and the produced Ag–Au NTs aqueous suspensions are shown in Fig. 2. The spectrum for the Ag NWs shows a well-defined localized SPR peak centered at 396 nm, which is assigned to transversal plasmon excitation.20,21 No bands due to longitudinal plasmon modes were detected, as a result of the large aspect ratio of the nanowires (Fig. S1†). For the Ag–Au NTs, the SPR peak shifted to 530 nm. The red shift of the SPR peak after the galvanic replacement reaction is in agreement with the conversion of the nanowires into nanotubes, with the incorporation of Au on the nanotubes’ surface and the formation of hollow interiors.7,17
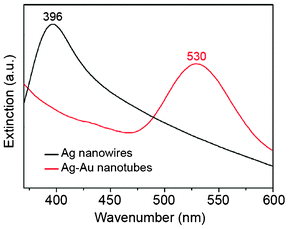 | ||
| Fig. 2 Extinction spectra recorded from aqueous suspensions containing Ag NWs (black line) and Ag–Au NTs (red line). | ||
The morphological features of the Ag–Au NTs make them attractive for the development of highly sensitive SERS substrates,13,22 as the porous walls comprised of Ag–Au islands closely joined together may afford a large number of electromagnetic hot spots on the surface of the NTs.14,23 In SERS, electromagnetic hot spots can be defined as junctions or gaps between two or more closely spaced particles in which colossal electromagnetic enhancements may arise, as opposed to individual nanoparticles.23–25 The general method for obtaining substrates containing hot spots relies on the uncontrolled aggregation of silver or gold nanoparticles.26 While these aggregates can provide strong SERS signals, the poor reproducibility of their fabrication may hinder their practical application in analytical sensors. In this work, rather than relying on the uncontrolled aggregation of colloids, hot spots were generated in a controlled manner and over well-defined sites in the substrate, corresponding to the Ag–Au NTs walls. Thus, after their synthesis, we turned our attention to the investigation of the SERS properties of the Ag–Au NTs. First, we compared the SERS spectra recorded from the aqueous suspension containing the Ag NWs with that obtained from the aqueous suspension containing the Ag–Au NTs. In these experiments, we aimed to compare the performances of Ag NWs and Ag–Au NTs as SERS substrates by employing the same concentrations of crystal violet (CV) as the probe molecule, and the same concentration of nanostructures (NWs or NTs) in the suspensions to acquire the spectra. Fig. 3 compares the SERS spectra for CV obtained using Ag–Au NTs (trace A) and Ag NWs (trace B) as substrates. The results shown in Fig. 3 indicate that the Ag–Au NTs yielded much higher CV SERS intensities than the Ag NWs. More specifically, the CV band at 1172 cm−1 displayed a 124-fold increase in terms of area for the Ag–Au NTs relative to the initial Ag NWs. Based on the morphology of the Ag–Au NTs (Fig. 1) such an effect can be attributed to both the increase in surface area, as well as the porous surface comprised of Ag–Au islands of 10–25 nm in size that provide a large number of electromagnetic hot spots along the nanotube walls (that are not present in Ag NWs). The increase in resonance between the excitation wavelength employed in the SERS experiments (632.8 nm) and the SPR peak may also contribute to the increased SERS intensities of the nanotubes relative to the nanowires.18 The 124-fold increase in the area of the CV band at 1172 cm−1 relative to Ag NWs was estimated assuming that each Ag NW was converted to a Ag–Au NT during the synthesis, and there were no losses during the isolation and purification stages. It is important to note that it is plausible that some particles were lost during these steps. Hence, the SERS enhancement reported here for the Ag–Au NTs relative to the Ag NWs will likely be an underestimate rather than an overestimate of the actual values. A precise estimation of the enhancement factor of Ag–Au NTs was not included here, as their complex morphology hampers the precise estimation of their surface area and, thus, the number of probe molecules contributing to the SERS intensities. It also limits the quantification of the SERS improvement in terms of surface area.
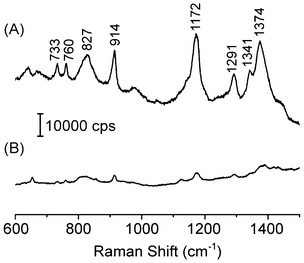 | ||
| Fig. 3 SERS spectra for crystal violet (10−7 M) recorded from aqueous suspensions containing (A) Ag–Au NTs and (B) Ag NWs as substrates. The spectra were recorded by employing approximately the same concentration of nanostructures in each suspension. | ||
The exceptional SERS properties of the Ag–Au NTs were also investigated by recording the spectrum of CV in the single molecule regime (10−10 M range). In this study, we deposited a suspension of the Ag–Au NTs over a glass slide. After drying under ambient conditions, this system was immersed in a 10−10 M CV aqueous solution for 1 h. The glass slide containing the Ag–Au nanotubes were then washed with water, dried under ambient conditions, and the SERS spectra were recorded. Fig. 4A shows an optical microscopy image of the 100 × 140 μm Ag–Au NTs film substrate that was obtained by this procedure.
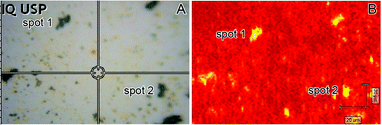 | ||
| Fig. 4 (A) Optical microscopy image of a 100 × 140 μm film substrate containing the Ag–Au NTs. (B) SERS image of the film shown in (A) acquired after its immersion in a 1.0 × 10−10 M CV aqueous solution and employing the 1170 cm−1 signal to build the intensity map (high and low intensity regions are marked as yellow and red, respectively). | ||
Although the concentration of Ag–Au NTs seemed to be low, they could be easily detected in some regions of the film (such as spots 1 and 2 marked in Fig. 4A). Fig. 4B shows a SERS intensity map of the region shown in Fig. 4A employing the CV Raman band at 1170 cm−1 registered under the aforementioned conditions. It can be observed that only the locations where the Ag–Au NTs are present (as evidenced by dark spots in the optical image, such as spots 1 and 2 in Fig. 4A) produced strong SERS signals. This becomes clear in Fig. S2,† which shows two typical SERS spectra for CV obtained from spots 1 and 2 (marked in Fig. 4A and B). Therefore, our SERS mapping results for CV in the 10−10 M regime are in agreement with the presence of highly localized electromagnetic hot spots on the surface of the Ag–Au NTs due to their unique morphology. It is important to note that these results do not represent a straightforward proof of detection at the single molecule regime, because some preferential adsorption of the probe molecules on the surface during the evaporation process might occur.
The Ag–Au NTs were also employed as a SERS substrate for the detection of polycyclic aromatic hydrocarbons, which represent an important class of atmospheric pollutants. The SERS spectra recorded employing Ag–Au NTs as substrates (Fig. 5) show that the SERS signal of 9-nitroanthracene and benzo[a]pyrene in aqueous solution at 3.33 × 10−7 M concentrations could be readily identified. Thus, the Ag–Au NTs represent a promising alternative for the development of reproducible SERS analytical sensors operating at low analyte concentration levels.
![SERS (top trace) and ordinary Raman (bottom trace) spectra recorded for (A) 9-nitroanthracene and (B) benzo[a]pyrene employing the Ag–Au NTs as substrates. While the ordinary Raman spectra were recorded from the solid samples, the SERS spectra were recorded from aqueous suspensions containing the Ag–Au NTs and 3.33 × 10−7 M 9-nitroanthracene or benzo[a]pyrene, respectively.](/image/article/2012/ra/c2ra21562d/c2ra21562d-f5.gif) | ||
| Fig. 5 SERS (top trace) and ordinary Raman (bottom trace) spectra recorded for (A) 9-nitroanthracene and (B) benzo[a]pyrene employing the Ag–Au NTs as substrates. While the ordinary Raman spectra were recorded from the solid samples, the SERS spectra were recorded from aqueous suspensions containing the Ag–Au NTs and 3.33 × 10−7 M 9-nitroanthracene or benzo[a]pyrene, respectively. | ||
In summary, we have described a facile and versatile strategy based on the galvanic replacement reaction between Ag NWs and HAuCl4 for the one step synthesis of porous Ag–Au NTs whose surface is comprised of Ag–Au islands 10–25 nm in size. This unique morphology enabled the concentration of a large number of electromagnetic hot spots along the nanotubes surface. The investigation of the SERS properties of the Ag–Au NTs showed that they displayed superior performance relative to Ag NWs. Also, our results showed that the Ag–Au NTs were effective SERS substrates for the detection of crystal violet (CV) in the 10−10 M regime. Finally, application of the Ag–Au NTs for the SERS detection of polycyclic aromatic hydrocarbons at 10−7 M concentration was also demonstrated. The results presented herein indicate that the Ag–Au NTs are promising candidates as substrates for the development and application of SERS sensors in the detection of environmentally relevant species at low concentration levels.
This work was supported by FAPESP (grant numbers 2011/06847-0, 2006/58748-7 and 2006/06629-4), CNPq (grant number 309297/2009-5) and start-up funds from USP (grant numbers 11.1.25042.1.0 and 2012-145).
References
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- K. An and T. Hyeon, Nano Today, 2009, 4, 359–373 CrossRef CAS.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS.
- M. B. Cortie and A. M. McDonagh, Chem. Rev., 2011, 111, 3713–3735 CrossRef CAS.
- S. E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS.
- J. Chen, B. J. Wiley and Y. Xia, Langmuir, 2007, 23, 4120–4129 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, Adv. Mater., 2004, 16, 264–268 CrossRef CAS.
- X. Lu, H.-Y. Tuan, J. Chen, Z.-Y. Li, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2007, 129, 1733–1742 CrossRef CAS.
- M. V. Petri, R. A. Ando and P. H. C. Camargo, Chem. Phys. Lett., 2012, 531, 188–192 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. R. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS.
- N. P. W. Pieczonka and R. F. Aroca, Chem. Soc. Rev., 2008, 37, 946–954 RSC.
- N. L. Netzer, C. Qiu, Y. Y. Zhang, C. K. Lin, L. F. Zhang, H. Fong and C. Y. Jiang, Chem. Commun., 2011, 47, 9606–9608 RSC.
- S. E. Hunyadi and C. J. Murphy, J. Mater. Chem., 2006, 16, 3929–3935 RSC.
- L. Au, Y. C. Chen, F. Zhou, P. H. C. Camargo, B. Lim, Z. Y. Li, D. S. Ginger and Y. N. Xia, Nano Res., 2008, 1, 441–449 CrossRef CAS.
- M. Rycenga, K. K. Hou, C. M. Cobley, A. G. Schwartz, P. H. C. Camargo and Y. N. Xia, Phys. Chem. Chem. Phys., 2009, 11, 5903–5908 RSC.
- W. A. T. V. Hermoso, F. R. Ornellas and P. H. C. Camargo, Eur. Phys. J. D, 2012, 66, 135 CrossRef.
- P. H. C. Camargo, C. M. Cobley, M. Rycenga and Y. N. Xia, Nanotechnology, 2009, 20, 434020 CrossRef.
- J. C. S. Costa, R. A. Ando, P. H. C. Camargo and P. Corio, J. Phys. Chem. C, 2011, 115, 4184–4190 CAS.
- A. Tao, F. Kim, C. Hess, J. Goldberger, R. R. He, Y. G. Sun, Y. N. Xia and P. D. Yang, Nano Lett., 2003, 3, 1229–1233 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, Annu. Rev. Phys. Chem., 2012, 63, 65–87 CrossRef CAS.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS.
- E. C. Le Ru, M. Meyer, E. Blackie and P. G. Etchegoin, J. Raman Spectrosc., 2008, 39, 1127–1134 CrossRef CAS.
- Y. Li, N. Koshizaki, H. Wang and Y. Shimizu, ACS Nano, 2011, 5, 9403–9412 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental details and more characterization results. See DOI: 10.1039/c2ra21562d |
| This journal is © The Royal Society of Chemistry 2012 |