Unprecedented spiro-annelated sugar isoureas, guanidines and amidines as new families of glycosidase inhibitors†‡
Penélope
Merino-Montiel
,
Óscar
López
* and
José G.
Fernández-Bolaños
Departamento de Química Orgánica, Facultad de Química, Universidad de Sevilla, c/Profesor García González 1, E-41012, Seville, Spain. E-mail: osc-lopez@us.es; Fax: +34 954624960; Tel: +34 954559997
First published on 25th September 2012
Abstract
We report the synthesis and evaluation of the hitherto unknown spiranic isoureas, guanidines and amidines derived from carbohydrates as novel glycosidase inhibitors. The effect of the heterocyclic moiety and its substituents on the inhibition was studied, and as a result, a potent α-galactosidase inhibitor (Ki 35 μM) bearing an isoureido motif was achieved.
Introduction
Carbohydrate-processing enzymes, like glycosidases, glycosyl transferases, and glycogen phosphorylase, are ubiquitous biomolecules involved in crucial biological processes, like glycoconjugate catabolism, cell wall biosynthesis, or molecular recognition, among others.1–3Such enzymes are currently considered to be attractive targets in Medicinal Chemistry, as their anomalous functioning favours the development of numerous diseases, like cancer, diabetes or lysosomal storage disorders. Moreover, carbohydrate-mediated recognition processes are also associated with microbial infections.2
In this context, strong and selective inhibitors of glycosidases are promising candidates for developing pharmaceutical drugs.4 Most of such derivatives belong to the iminosugar or azasugar families; that is, carbohydrate mimics where the endocyclic oxygen, or a ring carbon atom, respectively, have been replaced by a nitrogen atom.5 A great number of iminosugars or azasugars are capable of mimicking either the charge or the shape developed in the transition state of glycosidase-mediated hydrolysis, a fact that has been used in the rational design of new iminosugar derivatives.6
In vitro and in vivo tests conducted on a plethora of glycosidase inhibitors revealed that many of them have good activities as antidiabetic,7 antimicrobial8 and antitumor agents9 or that they might be suitable for combating lysosomal storage disorders10 and neurodegenerative diseases.11
Incorporation of spiranic heterocyclic motifs, mainly on the C-1 position of carbohydrate residues has also allowed the development of very potent inhibitors, especially against glycogen phosphorylase.12,13 Nevertheless, reports concerning the use of such templates in any other position rather than the anomeric carbon are relatively scarce.14–16
Although the presence of free hydroxyl groups on C-3 positions of sugar moieties is generally accepted for strong inhibitor–enzyme interactions, some carbohydrate derivatives bearing heterocyclic motifs on C-3 have been reported to possess relevant glycosidase inhibition activity.17
In previous work, we reported18 the preparation of spiranic N-substituted thiohydantoins at C-3 (carbohydrate nomenclature) using O-protected α-aminoester 2 as the key intermediate. Coupling of 2 with different isothiocyanates afforded, after acidic deprotection, thiohydantoins 4 (Scheme 1). Although quite weak, compounds 4 were, to the best of our knowledge, the first spiranic thiohydantoins at a position different from the anomeric one, exhibiting inhibition against glycosidases.
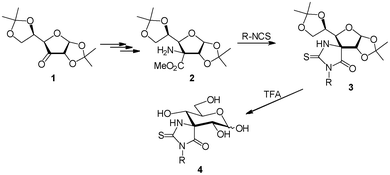 | ||
| Scheme 1 Preparation of sugar-derived spiranic thiohydantoins at C-3. | ||
Some authors claim that thiohydantoins behave as promiscuous scaffolds, exhibiting interactions with a large number of biological targets, making it difficult sometimes to be used as pharmacophores.19
Stimulated by the reports in the literature concerning glycosidase inhibition exerted by C-3-substituted carbohydrates,17 together with the strong enzyme affinity exhibited by many carbohydrates incorporating basic heterocycles,20,21 we explored the possibility of introducing different heterocyclic motifs on C-3 in order to analyse in a qualitative fashion their structural influence on the inhibitory activity.
Results and discussion
Herein we have extended our studies on spiranic N-substituted heterocycles at C-3 searching for more potent glycosidase inhibitors than 4. For that purpose, we envisioned the possibility of preparing a small library of compounds by changing both the basicity and geometry of the heterocycle. We postulated that the insertion of a more basic heterocyclic motif, ionized at physiological pH, might contribute to stronger ionic interactions within the enzyme active site, and therefore might lead to improved glycosidase inhibitors.Taking this consideration in mind, we selected cyclic isoureas, guanidines and amidines as interesting heterocyclic motifs, instead of the thiohydantoin fragment of compounds 4. Such heterocycles, which form part of numerous pharmacophores, are basic appendages, and thus the existence of the ionized spiro-compound in aqueous solution is guaranteed. It is remarkable that no reports on the preparation of spiranic carbohydrates bearing these heterocycles can be found in the literature.
Considering the retrosynthetic analysis depicted in Scheme 2, the three families of 5,6-membered spiro-compounds 5–7 can be accessed using D-arabino-configured ulose 1 as a common starting material. In particular, we propose the synthesis of oxazoline derivative 5 from amino alcohol 11, and both guanidines 6 and amidines 7 from diamino derivative 12.
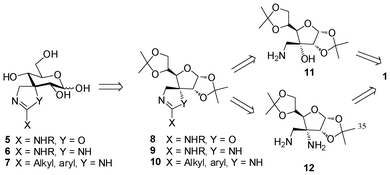 | ||
| Scheme 2 Retrosynthetic analysis for the synthesis of spiro-annelated 2-amino-oxazolines, guanidines and amidines 5–7. | ||
Ulose 1 was accessed starting from commercially-available di-O-isopropylidene glucofuranose 13 by oxidation with PCC.22 Subsequent treatment with KCN furnished cyanohydrin 1423 in a totally stereoselective fashion. Reduction of the cyano moiety with LiAlH4 led to the formation of D-allo-configured β-amino alcohol 11, recently prepared in our group,24 and herein our starting material in the preparation of spiro-annelated cyclic isoureas (Scheme 3).
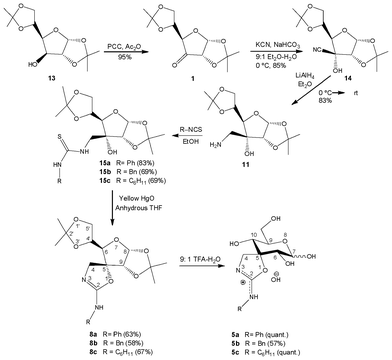 | ||
| Scheme 3 Synthesis of 2-amino-oxazolines 5a–c. | ||
Amino alcohol 11 was treated with a series of alkyl and aryl isothiocyanates (phenyl, benzyl and cyclohexyl) to give hydroxy-thioureas 15a–c in good to excellent yields.
Conversion of thioureas into spiranic 2-amino-2-oxazolines 8a–c was achieved by a cyclodesulfurization reaction mediated by yellow mercury(II) oxide (Scheme 3) in 58–67% yields.
Formation of spiranic isoureas must proceed via transient carbodiimides 16a–c; undetected by TLC, such intermediates undergo a spontaneous nucleophilic attack involving the free hydroxyl group (Scheme 4, pathway A), affording the spirocyclization reaction.
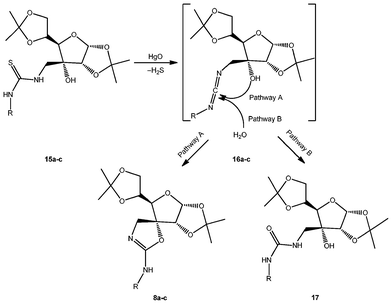 | ||
| Scheme 4 Mechanism of the formation of protected isoureas 8a–c. | ||
In order to avoid side-formation of ureas 17 (Scheme 4, pathway B) by competing H2O attack on the carbodiimides, and to ensure the spirocyclization reaction is the only process, treatment with HgO was accomplished in anhydrous THF. We have previously reported25 the reaction of transient carbodiimides generated from per-O-acetylated glycopyranosyl thioureas with H2O to afford the corresponding ureas.
NMR spectra of the final products confirmed the complete absence of the potential ureas 17, and the formation of the spiro-compounds as the sole products; on the one hand, 13C-NMR indicated the disappearance of the thioureido group at roughly 182 ppm, together with the appearance of a new signal in the range 155.6–160.1 ppm, assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N framework (C-2 in the spiro nomenclature). Furthermore, carbons located on the oxazoline ring were deshielded in comparison with the parent non-cyclized structures, especially C-4 (roughly 11 ppm) and C-5 (8.2–9.2 ppm).
N framework (C-2 in the spiro nomenclature). Furthermore, carbons located on the oxazoline ring were deshielded in comparison with the parent non-cyclized structures, especially C-4 (roughly 11 ppm) and C-5 (8.2–9.2 ppm).
Deprotection of compounds 8a–c using aqueous TFA furnished unprotected derivatives 5a–c. Such compounds were found to exist in their pyranose form, evidenced by the deshielding of C-9 in comparison with C-10; furthermore, vicinal coupling constants suggest a non-distorted 4C1 conformation.
Derivatives 5a–c were found to exist as an anomeric mixture in solution, the β-anomer being the major one (J6,7 = 7.5–8.1 Hz). Moreover, the very strong deshielding found for C-5 (95.4–97.4 ppm) must be indicative of the final compounds existing as protonated species. The absence of TfO− signals in 13C-NMR spectra induced us to postulate that spiranic isoureas 5a–c were isolated as their hydrate form.
We have also succeeded in the preparation of sugar-derived spiroguanidines 9. For that purpose we envisioned the possibility of using spiranic imidazolidine-2-thione 19 (Scheme 5), recently prepared in our group.24 Such a compound was accessed starting from ulose 1 by treatment with NH3 followed by stereoselective cyanide addition to the transient imine.26 Reduction with LiAlH4 gave β-diamino 18, which in turn was treated with 1,1′-thiocarbonyldiimidazole to furnish compound 19.24 We have attempted two different synthetic approaches for the synthesis of the sugar-derived spiroguanidines 9.
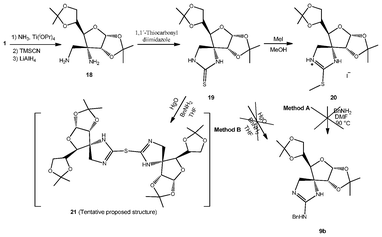 | ||
| Scheme 5 Attempted synthesis of spiroguanidines. | ||
On the one hand, reaction of 19 with MeI was attempted with the aim of obtaining the corresponding isothiouronium salt 20 by methylation of the sulfur atom. The reaction was followed by TLC, and the formation of a more polar compound was observed. The crude reaction was then heated in the presence of BnNH2 in order to obtain cyclic guanidine 9b (Scheme 5, Method A). Nevertheless, a complex mixture of compounds was obtained instead.
On the other hand, imidazolidine-2-thione 19 was treated with yellow HgO and BnNH2 (Scheme 5, Method B). After two days stirring at rt, the reaction mixture was filtered off and the filtrate was concentrated to dryness to give a yellowish solid. This compound, which could not be properly purified and characterized, was tentatively assigned only by HR-MS to dimeric sulfide 21. Formation of 21 could be explained considering that yellow HgO complexes with imidazolidine-2-thione 19, and the sulfur atom of a second molecule could act as a nucleophile, displacing the S–Hg–O fragment. Analogous compounds can be found in the literature upon reductive desulfurization of thiohydantoins with RANEY® Ni,27 and in the oxidation28 of imidazoline-2-thiones with IrCl63−, or chlorination29 of the same heterocycle.
Given that the synthesis of guanidines from 19 did not provide the expected results, we changed the methodology, and started instead from diamino compound 18. We accomplished the regioselective synthesis of aminothioureas 22a–c by coupling an excess of diamino 18 with commercially-available isothiocyanates (Scheme 6) in order to favour reaction through the less sterically-hindered aminomethyl moiety; thioureas were obtained in good to quantitative yields.
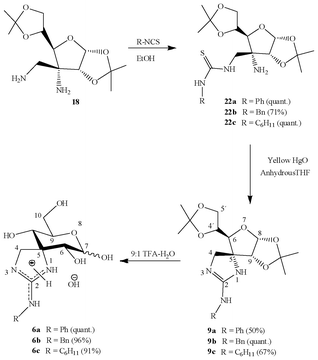 | ||
| Scheme 6 Synthesis of spiro-annelated guanidines. | ||
In a similar way as described for isoureas 8a–c, we conducted a cyclodesulfurization reaction involving aminothioureas 22a–c and yellow HgO, where a transient carbodiimide must be trapped by the free amino group, furnishing spiroguanidines 9a–c. Deprotection with aqueous TFA (Scheme 6) gave access to derivatives 6a–c as anomeric mixtures of the pyranosic forms. Surprisingly, and unlike analogous isoureas, the major isomer found for guanidines was the α-anomer; furthermore, this result also strongly contrasts with spiranic thiohydantoins, where the β-isomer is present almost exclusively.18,24 A plausible explanation for this fact could be the existence of stabilizing opposite dipoles on C-5 and C-7 of the α-configured guanidine derivatives, together with a strong intramolecular hydrogen bond involving the charged heterocyclic motif and the anomeric hydroxyl group (Fig. 1).
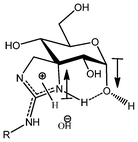 | ||
| Fig. 1 Dipoles and intramolecular hydrogen bond postulated for unprotected guanidines 6a–c. | ||
The third family of spiranic carbohydrates that we have accessed herein comprises imidazolines, or cyclic amidines. For that purpose we have studied the condensation of diamino compound 18 with commercially-available amidine hydrochlorides (Scheme 7). 1H-NMR spectra of the crude reactions using benzamidine and acetamidine hydrochlorides showed a mixture of the expected amidines 10a,b, together with partially deprotected derivatives 23a,b in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
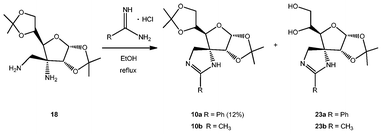 | ||
| Scheme 7 Synthesis of spiranic imidazolines. | ||
Amidines 10a and 23a were separated by liquid–liquid extraction; the former, located in the organic phase, was purified by column chromatography and was obtained in a modest yield (12%). The latter compound, soluble in the aqueous phase, could not be separated from the excess of benzamidine hydrochloride.
Regarding reaction with acetamidine, none of the compounds, 10b nor 23b, could be purified properly by column chromatography, so they were totally deprotected in order to attempt a better separation from the reaction side-products.
Spiranic amidine 10a and the mixture of compounds 10b and 23b were subjected to acidic treatment (Scheme 8) to remove the isopropylidene groups, giving compounds 7a (quantitative yield) and 7b (14% yield for the two steps), respectively. The modest yield for compounds 7a and 7b was due to partial decomposition in the condensation step.
 | ||
| Scheme 8 Deprotection of amidines. | ||
Fully unprotected amidine 7b was found to be in an α/β anomeric mixture of the pyranose and furanose forms in a 7.1:5.9:1.1:1 (αp/βp/αf/βf) ratio.
Enzymatic inhibition studies
We have evaluated the inhibitory properties of unprotected derivatives 5–7 against five commercially-available glycosidases (α-glucosidase from baker's yeast, β-glucosidase from almonds, α-galactosidase from green coffee beans, β-galactosidase from E. coli and from Aspergillus oryzae) and against glycogen phosphorylase type b from rabbit muscle. Ki values are depicted in Table 1 and compared with structurally related N-phenyl and N-cyclohexyl thiohydantoins 4a and 4b.| Compound | α-Glucosidase (baker’s yeast) | β-Glucosidase (almonds) | α-Galactosidase (green coffee beans) | β-Galactosidase (Asp. oryzae) | β-Galactosidase (E. coli) | Glycogen phosphorylase b (rabbit muscle) |
|---|---|---|---|---|---|---|
| a K i in μM. b No inhibition. c Not tested. | ||||||
| 4a (R = Ph) | N.I.b | 4070 | N.I. | 8% | N.I. | N.I. |
| 4b (R = C6H11) | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. |
| 5a (R = Ph) | N.I. | N.I. | 35 | N.I. | 8% | N.I. |
| 5b (R = Bn) | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. |
| 5c (R = C6H11) | N.I. | N.I. | N.I. | N.I. | N.I. | 13% |
| 6a (R = Ph) | 210 | N.I. | 103 | N.I. | N.I. | N.I. |
| 6b (R = Bn) | 230 | N.I. | 560 | N.I. | N.I. | 6% |
| 6c (R = C6H11) | N.I. | N.I. | 8% | N.I. | N.I. | 3% |
| 7a (R = Ph) | N.I. | 7% | N.I. | 2000 | N.I. | 6% |
| 7b (R = Me) | N.I. | 9% | —c | — | — | 14% |
Replacement of the thiohydantoin scaffold in 418 by a cyclic isoureido moiety bearing an N-aryl substituent (compound 5a) afforded a spectacular shift from no inhibition to strong and selective inhibition of α-galactosidase (Ki 35 μM). This compound combines both a basic fragment to induce stabilizing ionic interactions within the active site of the enzyme, together with a lipophilic residue probably involved in van der Waals interactions with hydrophobic subsites.
The mode of inhibition of compound 5a was confirmed by using the Lineweaver–Burk graphical analysis by plotting 1/v vs. 1/[S] at different inhibitor concentrations (Fig. 2). Considering the different plots depicted in Fig. 2 it is clear that compound 5a behaves as a competitive inhibitor of α-galactosidase.
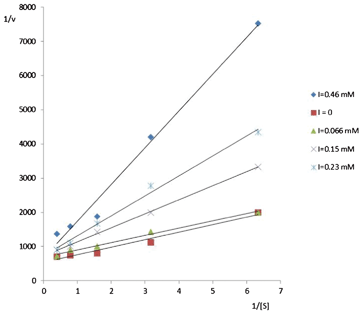 | ||
| Fig. 2 Mode of α-galactosidase inhibition by 5a. | ||
Guanidines 6a and 6b, bearing an N-aryl and aryl-alkyl substituent, respectively, were found to be moderate inhibitors of both α-glucosidase and α-galactosidase, with Ki values in the range 103–560 μM. Although somehow less active and selective than their analogue 5a, the title guanidines are still much more potent than their thiohydantoin counterparts; imidazolines 7a and 7b were quite poor inhibitors. None of the tested compounds showed remarkable activity against glycogen phosphorylase b.
Conclusions
We have carried out the expeditious synthesis of the first examples of spiranic sugar-derived isoureas, guanidines and amidines, starting from a common protected hexosulose as the main intermediate. The first two families were accessed by transforming the ulose into a β-amino alcohol or a β-diamino compound, respectively, conversion into the corresponding hydroxy(amino) thioureas, and a final cyclodesulfurization reaction promoted by yellow mercury(II) oxide. Cyclic amidines were obtained by direct condensation of the protected β-diamino compound with commercially-available amidines. Inhibitory activities conducted on the unprotected 5,6-membered spiro-derivatives revealed a spectacular increase in potency of isoureas and guanidines bearing aromatic substituents, especially against α-glucosidase and α-galactosidase, when compared with previously reported spiranic thiohydantoin analogues. In particular, the N-phenyl isourea was found to be the hitherto most potent spiranic inhibitor against α-galactosidase. These results clearly demonstrate how the insertion of a basic heterocyclic motif on C-3, together with an aromatic residue can provide moderate to good glycosidase inhibitors.Experimental
General procedures
Optical rotations were measured with a Jasco P-2000 polarimeter. 1H (300 MHz) and 13C (75.5 MHz) NMR spectra were recorded on Bruker Avance-300 spectrometer at rt. The assignments of 1H and 13C signals were confirmed by homonuclear COSY and heteronuclear 2D correlated spectra, respectively. Mass spectra (CI and LSI) were recorded on Micromass AutoSpec-Q mass spectrometer with a resolution of 1000 or 10000 (10% valley definition). For LSI spectra, ions were produced by a beam of xenon atoms and Cs+ ions, respectively, using thioglycerol as matrix and NaI as additive. TLC was performed on aluminium pre-coated sheets (E. Merck Silica Gel 60 F254); spots were visualized by UV light, by charring with 10% H2SO4 in EtOH or with 3% ninhydrin in EtOH. Column chromatography was performed using E. Merck Silica Gel 60 (40–63 μm). Microanalyses were performed at the Instituto de Investigaciones Químicas CSIC-US, using an elemental analyzer LECO Truspec CHN/CHNS.
Enzymatic assays were carried out in a Hitachi U-2900 spectrophotometer. Commercially-available α-glucosidase (baker's yeast), β-glucosidase (almonds), α-galactosidase (green coffee beans), β-galactosidase (E. coli), β-galactosidase (Aspergillus oryzae), and glycogen phosphorylase type b (rabbit muscle) were used as received, without further purification. Assays were accomplished as described previously.30 Each glycosidase assay was performed by preparing ten 2 mL samples in PS cuvettes containing 0.1 M phosphate buffer (pH 6.8) and substrate solution (p-nitrophenyl-α-D-glucopyranoside, p-nitrophenyl-β-D-glucopyranoside, p-nitrophenyl-α-D-galactopyranoside, or o-nitrophenyl-β-D-galactopyranoside). The concentration of the substrates ranged from 0.25 to 4.0 Km. Water or inhibitor solution plus water were also made up to a constant volume of 1.9 mL for the Km or the Ki measurement, respectively. Reactions were started by adding 0.1 mL of dilute enzyme solution at 25 °C and the formation of the p-nitrophenolate was monitored for 2 min by measuring the increase in absorbance at 400 nm.
Initial rates were calculated from the slopes of each reaction and were used to obtain two Hanes plots ([S]/V vs. [S]), one with and another without inhibitor. Inhibition constants (Ki) were obtained from the formula Ki = [I]/(Km′/Km − 1), where [I] is the inhibitor concentration in the cuvette and Km and Km′ are the enzymatic Michaelis–Menten constants in the absence and in the presence of the inhibitor, respectively.
Glycogen phosphorylase activities were assayed in the direction of glycogen synthesis31 by using commercial glycogen phosphorylase b (rabbit muscle, 10 μg ml−1) at 30 °C in the presence of 1% oyster glycogen, α-D-glucose-1-phosphate, 1 mM AMP, with or without inhibitors at pH 6.8 (50 mM triethanolamine/HCl, 1 mM dithiothreitol and 1 mM EDTA buffer). Pi concentration was determined using the method developed by Taussky and Shorr.32 Hanes plots were built using the initial velocity from each reaction, as indicated above for glycosidases.
General procedure for the preparation of thioureas 15a–c
To a solution of amino alcohol 11 (100 mg, 0.35 mmol) in EtOH (4 mL) was added the corresponding commercially-available isothiocyanate, and the solution was kept stirring during the time and at the temperature indicated in each case. The solvent was then removed under reduced pressure, and the residue was purified by column chromatography using the eluant indicated in each case.1,2:5,6-Di-O-isopropylidene-3-C-(3′-phenylthioureido)methyl-α-D-allofuranose (15a)
Phenyl isothiocyanate (62 μL, 0.52 mmol) was used. The reaction was kept stirring for 1 h at 25 °C. Column chromatography (9:1 → 7:3 hexane–EtOAc) gave 15a (122 mg, 83%); [α]24D +18 (c 1.23, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 8.26 (m, 1H, Ph-NH), 7.45–7.40 (m, 2H, Ar–H), 7.31–7.23 (m, 3H, Ar–H), 6.81 (m, 1H, NH-CH2), 5.88 (d, 1H, J1,2 = 3.9 Hz, H-1), 4.54 (d, 1H, H-2), 4.40 (dd, 1H, JCHa,CHb = 14.1 Hz, JCHa,NH = 8.4 Hz, N–CHa), 4.15–4.08 (m, 2H, H-5, H-6a), 3.90 (m, 1H, H-6b) 3.74 (d, 1H, J4,5 = 8.4 Hz, H-4), 3.69 (dd, 1H, JCHb,NH = 3.3 Hz, N–CHb), 2.95 (brs, 1H, OH), 1.57 (s, 3H, CH3), 1.37 (s, 6H, 2CH3), 1.27 (s, 3H, CH3); 13C-NMR (75.5 MHz, CDCl3): δ 181.0 (CS), 135.9, 130.2, 127.3, 125.0 (Ar–C), 112.8, 109.9 (2CMe2), 103.8 (C-1), 81.4 (C-4), 80.5 (C-2), 79.9 (C-3), 73.0 (C-5), 68.2 (C-6), 46.1 (CH2), 26.6 (×3), 25.0 (4CH3); CIMS m/z 425 ([M+H]+, 6%); HRCI-MS calcd for C20H29N2O6S ([M+H]+): 425.1746, found: 425.1743; Anal. calcd for C20H28N2O6S: C, 56.59; H, 6.65; N, 6.60; S, 7.55, found: C, 56.86; H, 6.83; N, 6.29; S, 7.15.
3-C-(3′-Benzylthioureido)methyl-1,2:5,6-di-O-isopropylidene-α-D-allofuranose (15b)
Benzyl isothiocyanate (68 μL, 0.51 mmol) was used. The reaction was kept stirring for 5 h at 25 °C. Column chromatography (7:3 → 1:1 hexane–EtOAc) gave 15b (107 mg, 69%); [α]23D +30 (c 1.26, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.36–7.25 (m, 5H, Ar–H), 7.06 (brs, 1H, NH-Bn), 6.81 (brs, 1H, NH-CH2), 5.80 (d, 1H, J1,2 = 3.9 Hz, H-1), 4.60 (brs, 2H, CH2Ph), 4.37 (brs, 1H, H-2), 4.13–4.10 (m, 1H, N-CHa), 4.13–4.06 (m, 2H, H-5, H-6a), 3.89 (dd, 1H, J5,6b = 8.1 Hz, J6a,6b = 11.7 Hz, H-6b), 3.73 (d, 1H, J4,5 = 7.8 Hz, H-4), 3.64 (dd, 1H, JCHb,NH = 2.7 Hz, JCHa,CHb = 13.8 Hz, N-CHb), 3.06 (brs, 1H, OH), 1.54, 1.39 (2s, 3H each, 2CH3), 1.31 (s, 6H, 2CH3); 13C-NMR (75.5 MHz, CDCl3): δ 182.9 (CS), 136.7, 128.9, 127.9, 127.5 (Ar–C), 112.7, 109.9 (2CMe2), 103.8 (C-1), 81.4 (C-4), 80.3 (C-2), 79.9 (C-3), 73.0 (C-5), 68.1 (C-6), 48.1 (CH2Ph), 45.8 (N–CH2), 26.7, 26.6 (×2), 25.2 (4CH3); CIMS m/z 439 ([M+H]+, 37%); HRCI-MS calcd for C21H30N2O6S ([M+H]+): 439.1903, found: 439.1894.
3-C-(3′-Cyclohexylthioureido)methyl-1,2:5,6-di-O-isopropylidene-α-D-allofuranose (15c)
Cyclohexyl isothiocyanate (73 μL, 0.51 mmol) was used. The reaction was stirred for 12 h at 25 °C, and further 2 h at 45 °C. Column chromatography (9:1 → 7:3 hexane–EtOAc) gave 15c (103 mg, 69%); [α]23D +33 (c 0.67, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 6.33 (brs, 2H, 2NH), 5.83 (d, 1H, J1,2 = 3.9 Hz, H-1), 4.49 (d, 1H, H-2), 4.16–4.08 (m, 3H, H-5, H-6a, N–CHa), 3.91 (m, 1H, H-6b), 3.75 (d, 1H, J4,5 = 8.4 Hz, H-4), 3.77–3.71 (m, 1H, H-1′′), 3.69 (dd, 1H, JCHb,NH = 3.3 Hz, JCHa,CHb = 14.4 Hz, N–CHb), 2.07 (brs, 1H, OH), 2.02–1.96 (m, 2H, 2CHcyclohex.), 1.74–1.70 (m, 2H, 2CHcyclohex.), 1.62–1.56 (m, 1H, CHcyclohex.), 1.56, 1.43 (2s, 3H each, 2CH3), 1.34 (s, 6H, 2CH3), 1.37–1.31 (m, 2H, 2CHcyclohex.), 1.27–1.20 (m, 3H, 3CHcyclohex.); 13C-NMR (75.5 MHz, CDCl3): δ 181.4 (CS), 112.8, 109.9 (2CMe2), 103.9 (C-1), 81.5 (C-4), 80.5 (C-2), 80.1 (C-3), 73.2 (C-5), 68.2 (C-6), 52.7 (C-1′′), 45.8 (N–CH2), 32.7 (×2) (2CH2), 26.8, 26.7 (×2) (3CH3), 25.4 (CH2), 25.3 (CH3), 24.7 (×2) (2CH2); CIMS m/z 431 ([M+H]+, 100%); HRCI-MS calcd for C20H35N2O6S ([M+H]+): 431.2216, found: 431.2209; Anal. calcd for C20H34N2O6S: C, 55.79; H, 7.96; N, 6.51; S, 7.45, found: C, 55.62; H, 7.96; N, 6.63; S, 7.74.
General procedure for the preparation of isoureas 8a–c
To a solution of thioureas 15a–c (0.24 mmol) in anhydrous THF (10 mL) was added yellow mercury(II) oxide (468 mg, 2.16 mmol) under an argon atmosphere. The corresponding mixture was stirred at rt in darkness until disappearance of the starting material by TLC, and then it was filtered through a Celite pad. The filtrate was concentrated to dryness and purified as indicated in each case.(5R,6R,8R,9R,4′R)-6-(2′,2′-Dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-2-phenylamino-1,7-dioxa-3-azaspiro[4.4]non-2-ene (8a)
The reaction was stirred during 28 h. Column chromatography (7:3 → 1:1 hexane–EtOAc) gave 8a (59 mg, 63%); [α]22D +60 (c 0.97, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.41 (m, 2H, Ar–H), 7.29 (m, 2H, Ar–H), 7.03–7.00 (t, 1H, JH,H = 7.2 Hz, Ar–H), 5.72 (d, 1H, J8,9 = 3.3 Hz, H-8), 4.45 (d, 1H, H-9), 4.19–4.10 (m, 3H, H-6, H-4′, H-5′a), 4.16 (d, 1H, J4a,4b = 13.2 Hz, H-4a), 4.01–3.98 (m, 1H, H-5′b), 3.62 (d, 1H, H-4b), 1.64, 1.38, 1.37, 1.26 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CDCl3): δ 155.6 (C-2), 138.9, 129.1, 122.7, 118.2 (Ar–C), 114.2, 109.9 (2CMe2), 103.2 (C-8), 87.9 (C-5), 84.3 (C-9), 77.6 (C-6), 73.8 (C-4′), 68.1 (C-5′), 57.3 (C-4), 27.0, 26.8, 26.5, 25.2 (4CH3); CIMS m/z 391 ([M+H]+, 100%); HRCI-MS calcd for C20H27N2O6 ([M+H]+): 391.1869, found: 391.1857.
(5R,6R,8R,9R,4′R)-2-Benzylamino-6-(2′,2′-dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-1,7-dioxa-3-azaspiro[4.4]non-2-ene (8b)
The reaction was stirred for 3 days; removal of the solids and concentration to dryness of the filtrate gave pure 8b (55 mg, 58%); [α]23D +66 (c 0.60, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.33–7.24 (m, 5H, Ar–H), 5.68 (d, 1H, J8,9 = 3.3 Hz, H-8), 4.49 (d, 1H, JH,H = 14.7 Hz, CHaPh), 4.42 (d, 1H, H-9), 4.35 (d, 1H, CHbPh), 4.21–4.09 (m, 3H, H-6, H-4′, H-5′a), 4.03 (d, 1H, J4a,4b = 12.6 Hz, H-4a), 3.99 (dd, 1H, J4′,5′b = 3.3 Hz, J5′a,5′b = 8.4 Hz, H-5′b), 3.50 (d, 1H, H-4b), 1.62 (s, 3H, CH3), 1.37 (s, 6H, 2CH3), 1.33 (s, 3H, CH3); 13C-NMR (75.5 MHz, CDCl3): δ 160.1 (C-2), 138.5, 128.7, 127.6, 127.5, 125.7 (Ar–C), 114.1, 109.9 (2CMe2), 103.2 (C-8), 89.1 (C-5), 84.3 (C-9), 77.5 (C-6), 73.7 (C-4′), 68.0 (C-5′), 56.6 (C-4), 47.1 (CH2Ph), 27.0, 26.9, 26.6, 25.4 (4CH3); CIMS m/z 405 ([M+H]+, 100%); HRCI-MS calcd for C21H29N2O6 ([M+H]+): 405.2026, found: 405.2013.(5R,6R,8R,9R,4′R)-2-Cyclohexylamino-6-(2′,2′-dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-1,7-dioxa-3-azaspiro[4.4]non-2-ene (8c)
The reaction was stirred during 3.5 days. Column chromatography (7:3 hexane–EtOAc → 20:1 EtOAc–Et3N) gave 8c (65 mg, 67%); [α]22D +47 (c 0.95, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 5.62 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.35 (d, 1H, H-9), 4.14–4.03 (m, 3H, H-6, H-4′, H-5′a), 3.94 (d, 1H, J4a,4b = 12.9 Hz, H-4a), 3.92 (dd, 1H, J4′,5′b = 3.9 Hz, J5′a,5′b = 12.0 Hz, H-5′b), 3.42 (d, 1H, H-4b), 3.43–3.33 (m, 1H, H-1′′), 2.03–1.89 (m, 2H, CH2), 1.68–1.60 (m, 2H, CH2), 1.57 (s, 3H, CH3), 1.54–1.46 (m, 1H, CHcyclohex.), 1.37–1.23 (m, 2H, CH2), 1.34, 1.31, 1.26 (3s, 3H each, 3CH3), 1.14–0.99 (m, 3H, CH2, CHcyclohex.); 13C-NMR (75.5 MHz, CDCl3): δ 158.9 (C-2), 114.0, 109.7 (2CMe2), 103.0 (C-8), 88.3 (C-5), 84.3 (C-9), 77.6 (C-6), 73.6 (C-4′), 68.0 (C-5′), 56.7 (C-4), 51.6 (C-1′′), 33.5, 33.3 (2CH2), 27.0, 26.8, 26.4 (3CH3), 25.6 (CH2), 25.4 (CH3), 24.8, 24.7 (2CH2); CIMS m/z 397 ([M+H]+, 100%); HRCI-MS calcd for C20H33N2O6 ([M+H]+): 397.2339, found: 397.2346; Anal. calcd for C20H32N2O6: C, 60.59; H, 8.14; N, 7.07, found: C, 60.31; H, 7.97; N, 6.91.
General procedure for the deprotection of isoureas 8a–c
A solution of O-protected isoureas 8a–c in 9:1 TFA–H2O (10 mL) was stirred at rt for 1 h. Then, the solvent was removed under reduced pressure, co-evaporating with toluene several times. The residue was purified by gel permeation chromatography (Biogel P2) to give the unprotected isoureas 5a–c as α/β mixtures.
(5S,6R,7R and 7S,9R,10S)-6,7,10-Trihydroxy-9-hydroxymethyl-2-phenylamino-1,8-dioxa-3-azaspiro[4.5]dec-2-ene hydrate (5a)
Isourea 8a was used (44 mg, 0.12 mmol) to give compound 5a (36 mg, quant.); [α]23D +3 (c 1.10, MeOH); 1H-NMR (300 MHz, CD3OD): 1:5 α/β ratio, δ 7.49–7.44 (m, 2H, Ar–H), 7.37–7.28 (m, 3H, Ar–H); β-anomer: δ 4.79 (d, 1H, J6,7 = 7.5 Hz, H-7), 4.01 (d, 1H, J4a,4b = 9.9 Hz, H-4a), 3.97 (d, 1H, H-4b), 3.87 (d, 1H, J1′a,1′b = 11.4 Hz, J1′a,9 ≈ 0 Hz, H-1′a), 3.71 (dd, 1H, J1′b,9 = 4.3 Hz, H-1′b), 3.72–3.64 (m, 2H, H-9, H-10), 3.42 (d, 1H, H-6); α-anomer: δ 5.25 (d, 1H, J6,7 = 4.2 Hz, H-7), 4.03–3.95 (m, 1H, H-9), 3.79 (m, 1H, H-1′a), 3.55 (m, 1H, H-1′b), 3.31 (brs, 2H, H-4); 13C-NMR (75.5 MHz, CD3OD): δ 161.6 (C-2), 135.6, 130.9, 129.9, 129.4, 129.1, 128.5 (Ar–C); β-anomer: δ 97.4 (C-5), 96.4 (C-7), 76.7 (C-9), 73.9 (C-6), 69.2 (C-10), 62.4 (C-1′), 47.9 (C-4); α-anomer: δ 93.0 (C-7), 70.5 (C-6), 69.7 (C-9), 62.2 (C-1′); LSIMS m/z 311 ([M–OH]+, 67%), 329 ([M+H]+, 13%); HRLSI-MS calcd for C14H19N2O6 ([M–OH]+): 311.1243, found: 311.1250.
(5S,6R,7R and 7S,9R,10S)-2-Benzylamino-6,7,10-trihydroxy-9-hydroxymethyl-1,8-dioxa-3-azaspiro[4.5]dec-2-ene hydrate (5b)
Isourea 8b was used (57 mg, 0.14 mmol) to give compound 5b (29 mg, 57%); [α]23D +7 (c 0.98, MeOH); 1H-NMR (300 MHz, (CD3)2CO): 1:11 α/β ratio, δ 7.46–7.26 (m, 5H, Ar–H); β-anomer: δ 4.87 (d, 1H, J6,7 = 7.8 Hz, H-7), 4.66–4.55 (m, 2H, CH2Ph), 4.05–3.97 (m, 2H, H-4), 3.83 (d, 1H, J9,10 = 9.4 Hz, H-10), 3.84–3.67 (m, 3H, H-9, H-1′a, H-1′b), 3.53 (d, 1H, H-6); α-anomer: δ 5.27 (d, 1H, J6,7 = 3.6 Hz, H-7); 13C-NMR (75.5 MHz, (CD3)2CO): β-anomer: δ 162.1 (C-2) 129.5, 128.5 (Ar–C), 96.2 (C-5), 96.1 (C-7), 76.0 (C-9), 73.7 (C-6), 69.1 (C-10), 62.1 (C-1′), 46.4 (C-4); α-anomer: δ 92.5 (C-7); CIMS m/z 325 ([M–OH]+, 7%); HRCI-MS calcd for C15H21N2O6, ([M–OH]+): 325.1400, found: 325.1405.
(5S,6R,7R and 7S,9R,10S)-2-Cyclohexylamino-6,7,10-trihydroxy-9-hydroxymethyl-1,8-dioxa-3-azaspiro[4.5]dec-2-ene hydrate (5c)
Isourea 8c was used (70 mg, 0.16 mmol) to give compound 5c (53 mg, quant.); [α]24D+6 (c 1.13, MeOH); 1H-NMR (300 MHz, CD3OD): 1:7 α/β ratio, β-anomer: δ 4.77 (d, 1H, J6,7 = 8.1 Hz, H-7), 4.00–3.83 (m, 2H, H-4a, H-4b), 3.72–3.59 (m, 4H, H-9, H-10, H-1′a, H-1′b), 3.35 (d, 1H, H-6), 3.60–3.31 (m, 1H, H-1′′), 1.96–1.91 (m, 2H, CH2), 1.83–1.79 (m, 2H, CH2), 1.45–1.20 (m, 6H, 3CH2); α-anomer: δ 5.19 (d, 1H, J6,7 = 4.5 Hz, H-7); 13C-NMR (75.5 MHz, CD3OD): β-anomer: δ 161.7 (C-2), 96.2 (C-7), 95.4 (C-5), 76.5 (C-9), 73.9 (C-6), 69.3 (C-10), 62.4 (C-1′), 53.8 (C-1′′), 47.6 (C-4), 33.2 (×2), 25.6 (×3) (5CH2); α-anomer: δ 162.0 (C-2), 93.0 (C-7), 76.7 (C-9), 54.0 (C-1′′), 33.6, 33.4, 26.0 (×3) (5CH2); CIMS m/z 317 ([M–OH]+, 5%); HRCI-MS calcd for C14H25N2O6 ([M–OH]+): 317.1713, found: 317.1707.
General procedure for the preparation of aminothioureas 22a–c
To a solution of diamino 18 (200 mg, 0.69 mmol) in EtOH (10 mL) cooled to −10 °C, was slowly added a solution of the corresponding isothiocyanate in EtOH (2 mL). The solution was stirred during the time and at the temperature indicated in each case. The solvent was then removed under reduced pressure and the residue was purified by column chromatography using the eluant indicated in each case.3-Amino-3-deoxy-1,2:5,6-di-O-isopropylidene-3-C-(3′-phenylthioureido)methyl-α-D-allofuranose (22a)
Phenyl isothiocyanate (29 μL, 0.24 mmol) was used. The reaction was stirred for 30 min at −10 °C. Column chromatography (7:3 → 1:1 hexane–EtOAc) gave compound 22a (104 mg, quant.); [α]24D +13 (c 0.93, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 8.53 (m, 1H, NH), 7.44–7.26 (m, 5H, Ar–H), 5.89 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.38–4.28 (m, 2H, H-2, CHa), 4.17–4.05 (m, 2H, H-5, H-6a), 3.89 (dd, 1H, J5,6b = 4.3 Hz, J6a,6b = 8.3 Hz, H-6b), 3.64 (d, 1H, J4,5 = 9.3 Hz, H-4), 3.43 (brd, 1H, JCHa,CHb = 13.2 Hz, CHb), 1.52, 1.39 1.32, 1.29 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CDCl3): δ 180.6 (CS), 136.6, 130.0, 126.8, 124.6 (Ar–C), 112.3, 109.8 (2CMe2), 104.3 (C-1), 83.3 (C-4), 82.0 (C-2), 73.5 (C-5), 68.9 (C-6), 64.0 (C-3), 46.3 (CH2), 26.8 (×2), 26.4, 25.0 (4CH3); CIMS m/z 424 ([M+H]+, 52%); HRCI-MS calcd for C20H30N3O5S, [M+H]+: 424.1906, found: 424.1895.
3-Amino-3-C-(3′-benzylthioureido)methyl-3-deoxy-1,2:5,6-di-O-isopropylidene-α-D-allofuranose (22b)
Benzyl isothiocyanate (68 μL, 0.51 mmol) was used. The reaction was stirred for 5 h at 25 °C. Column chromatography (7:3 → 1:1 hexane–EtOAc) gave compound 22b (107 mg, 71%); [α]26D +30 (c 1.26, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 7.35–7.23 (m, 5H, Ar–H), 5.83 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.63 (brs, 2H, CH2Ph), 4.31 (m, 1H, H-2), 4.17–4.02 (m, 3H, H-5, H-6a, N–CHa), 3.88 (dd, 1H, J5,6b = 4.8 Hz, J6a,6b = 8.4 Hz, H-6b), 3.61 (d, 1H, J4,5 = 9.6 Hz, H-4), 3.32 (brd, 1H, JH,H = 13.5 Hz, N–CHb), 1.50, 1.41, 1.33, 1.25 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CDCl3): δ 183.0 (CS), 137.2, 128.8, 127.7, 127.5 (Ar–C), 112.7, 109.8 (2CMe2), 104.3 (C-1), 83.4 (C-4), 81.6 (C-2), 73.4 (C-5), 68.9 (C-6), 63.9 (C-3), 60.5 (CH2), 48.0 (CH2Ph), 26.8, 26.7, 26.3, 25.1 (4CH3); CIMS m/z 438 ([M+H]+, 33%); HRCI-MS calcd for C21H32N3O5S ([M+H]+): 438.2063, found: 438.2050.
3-Amino-3-C-(3′-cyclohexylthioureido)methyl-3-deoxy-1,2:5,6-di-O-isopropylidene-α-D-allofuranose (22c)
Cyclohexyl isothiocyanate (73 μL, 0.41 mmol) was used. The reaction was stirred for 17 h at 0 °C. Column chromatography (7:3 → 3:2 hexane–EtOAc) gave compound 22c (175 mg, quant.); [α]25D +22 (c 1.02, CH2Cl2); 1H-NMR (300 MHz, CDCl3): δ 6.98 (brs, 1H, NH), 5.83 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.39 (d, 1H, H-2), 4.11 (dd, 1H, J5,6a = 2.4 Hz, J6a,6b = 8.1 Hz, H-6a), 4.09 (d, 1H, JH,H = 13.8 Hz, N–CHa), 4.08 (ddd, 1H, J4,5 = 9.0 Hz, J5,6b = 4.5 Hz, H-5), 3.86 (dd, 1H, H-6b), 3.94–3.72 (m, 1H, H-1′′), 3.62 (d, 1H, H-4), 3.37 (dd, 1H, JH,NH = 2.4 Hz, N-CHb), 2.00–1.98 (m, 2H, CH2), 1.71–1.67 (m, 2H, CH2), 1.60–1.56 (m, 2H, CH2), 1.50, 1.40 (2s, 3H each, 2CH3), 1.37–1.30 (m, 2H, CH2), 1.30, 1.29 (2s, 3H each, 2CH3), 1.19–1.13 (m, 2H, CH2); 13C-NMR (75.5 MHz, CDCl3): δ 181.3 (CS), 112.3, 109.8 (2CMe2), 104.3 (C-1), 83.3 (C-4), 81.8 (C-2), 73.4 (C-5), 68.9 (C-6), 63.9 (C-3), 52.8 (C-1′′), 45.9 (N–CH2), 32.8, 32.7 (2CH2), 26.8 (×2), 26.4 (3CH3), 25.5 (CH2), 25.0 (CH3), 25.0, 24.7 (2CH2); CIMS m/z 430 ([M+H]+, 81%); HRCI-MS calcd for C20H36N3O5S ([M+H]+): 430.2376, found: 430.2359.
General procedure for the preparation of the guanidines 9a–c
To a solution of thioureas 22a–c (0.24 mmol) in anhydrous THF (10 mL) was added yellow mercury(II) oxide (468 mg, 2.16 mmol) under an argon atmosphere. The mixture was stirred at rt in darkness until the disappearance of the starting material by TLC, and then it was filtered through a Celite pad. The filtrate was concentrated to dryness to give guanidines as pure compounds.(5R,6R,8R,9R,4′R)-6-(2′,2′-Dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-2-phenylamino-7-oxa-1,3-diazaspiro[4.4]non-2-ene (9a)
Thiourea 22a was used, and the reaction was stirred for 30 h to give compound 9a (45 mg, 50%); [α]25D +53 (c 0.95, MeOH); 1H-NMR (300 MHz, CD3OD/DMSO-d6): δ 7.29–7.24 (m, 2H, Ar–H), 7.10–7.07 (m, 2H, Ar–H), 7.01–6.96 (m, 1H, Ar–H), 5.75 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.48 (d, 1H, H-9), 4.27 (dt, 1H, J4′,6 = 7.2 Hz, J4′,5′a = J4′,5′b = 6.0 Hz, H-4′), 4.12 (dd, 1H, J5′a,5′b = 8.4 Hz, H-5′a), 3.95 (d, 1H, H-6), 3.90 (dd, 1H, H-5′b), 3.64 (d, 1H, JH,H = 10.2 Hz, H-4a), 3.29 (d, 1H, H-4b), 1.53, 1.45, 1.35, 1.34 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CD3OD/DMSO-d6): δ 159.4 (C-2), 148.1, 130.1, 123.7, 123.5 (Ar–C), 113.9, 110.6 (2CMe2), 104.5 (C-8), 85.5 (C-9), 80.3 (C-6), 74.7 (C-4′), 70.5 (C-5), 68.3 (C-5′), 48.7 (C-4), 27.0, 26.9, 26.6, 25.6 (4CH3); CIMS m/z 389 (M+, 100%); HRCI-MS calcd for C20H27N3O5 (M+): 389.1951, found: 389.1943.(5R,6R,8R,9R,4′R)-2-Benzylamino-6-(2′,2′-dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-7-oxa-1,3-diazaspiro[4.4]non-2-ene (9b)
Thiourea 22b was used, and the reaction was kept stirring during 6 days to give compound 9b (96 mg, quant.); [α]26D +58 (c 1.17, MeOH); 1H-NMR (300 MHz, CD3OD): [α]26D 7.32–7.18 (m, 5H, Ar–H), 5.67 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.38–4.33 (m, 2H, CH2Ph), 4.33 (d, 1H, H-9), 4.17 (dt, 1H, J4′,6 = 6.6 Hz, J4′,5′a = J4′,5′b = 6.0 Hz, H-4′), 4.03 (dd, 1H, J5′a,5′b = 8.1 Hz, H-5′a), 3.95 (d, 1H, H-6), 3.87 (dd, 1H, H-5′b), 3.58 (d, 1H, JH,H = 11.4 Hz, H-4a), 3.24 (d, 1H, H-4b), 1.50, 1.34, 1.30, 1.27 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CD3OD): δ 163.2 (C-2), 140.5, 129.5, 128.4, 128.2 (Ar–C), 113.8, 110.4 (2CMe2), 104.8 (C-8), 86.4 (C-9), 80.4 (C-6), 74.9 (C-4′), 73.1 (C-5), 67.9 (C-5′), 52.9 (C-4), 47.2 (CH2Ph), 27.0, 26.9, 26.5 (×2) (4CH3); CIMS m/z 404 ([M+H]+, 100%); HRCI-MS calcd for C21H30N3O5 ([M+H]+): 404.2185, found: 404.2172.(5R,6R,8R,9R,4′R)-2-Cyclohexylamino-6-(2′,2′-dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-7-oxa-1,3-diazaspiro[4.4]non-2-ene (9c)
Thiourea 22c was used, and the reaction was stirred during 3 days to give compound 9c (65 mg, 67%); [α]28D +45 (c 1.32, MeOH); 1H-NMR (300 MHz, CD3OD): δ 5.69 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.35 (d, 1H, H-9), 4.19 (dt, 1H, J4′,6 = 6.6 Hz, J4′,5′a = J4′,5′b = 6.0 Hz, H-4′), 4.07 (dd, 1H, J5′a,5′b = 8.3 Hz, H-5′a), 3.94 (d, 1H, H-6), 3.88 (dd, 1H, H-5′b), 3.57 (d, 1H, JH,H = 11.4 Hz, H-4a), 3.83 (tt, 1H, JH,H = 10.3 Hz, JH,H = 3.6 Hz, H-1′′), 3.24 (d, 1H, H-4b), 2.00-1.89 (m, 2H, CH2), 1.78–1.69 (m, 2H, CH2), 1.65–1.50 (m, 2H, CH2), 1.54 (s, 3H, CH3), 1.44–1.31 (m, 2H, CH2), 1.37, 1.33, 1.29 (3s, 3H each, 3CH3), 1.26–1.10 (m, 2H, CH2); 13C-NMR (75.5 MHz, CD3OD): δ 162.3 (C-2), 113.8, 110.4 (2CMe2), 104.6 (C-8), 86.3 (C-9), 80.5 (C-6), 74.9 (C-4′), 72.9 (C-5), 68.0 (C-5′), 53.0 (C-4), 52.1 (C-1′′), 34.6, 34.5 (2CH2), 27.0, 26.9 (2CH3), 26.6 (CH2), 26.5 (CH3), 25.9 (×2) (2CH2), 25.5 (CH3); CIMS m/z 395 (M+, 21%); HRCI-MS calcd for C20H33N3O5 (M+): 395.2420, found: 395.2405; Anal. calcd for C20H33N3O5: C, 60.74; H, 8.41; N, 10.62, found: C, 60.87; H, 8.17; N, 10.28.General procedure for the deprotection of the guanidines 9a–c
A solution of protected guanidines in 9:1 TFA–H2O (10 mL) was stirred at rt for 1 h. Then, the solvent was removed under reduced pressure co-evaporating with toluene several times, and the residue was washed with Et2O (3 × 10 mL) to give unprotected guanidines 6a–c as α/β mixtures.
(5S,6R,7R and 7S,9R,10S)-6,7,10-Trihydroxy-9-hydroxymethyl-2-phenylamino-8-oxa-1,3-diazaspiro[4.5]dec-2-ene hydrate (6a)
Guanidine 9a was used (78 mg, 0.20 mmol) to give compound 6a (65 mg, quant.); [α]29D +44 (c 0.91, MeOH); 1H-NMR (300 MHz, CD3OD) 6.6:1 α/β ratio, α-anomer: δ 7.48–7.40 (m, 2H, Ar–H), 7.34–7.29 (m, 3H, Ar–H), 5.17 (d, 1H, J6,7 = 3.3 Hz, H-7), 3.85–3.76 (m, 5H, H-9, H-1′a, H-1′b, H-4a, H-4b), 3.52–3.44 (m, 2H, H-6, H-10); β-anomer: δ 4.74 (d, 1H, J6,7 = 8.3 Hz, H-7), 3.68 (dd, 1H, J9,1′a = 5.3 Hz, J1′a,1′b = 12.0 Hz, H-1′a), 3.58 (m, 1H, H-9), 3.41 (d, 1H, J9,10 = 10.0 Hz, H-10), 3.17 (d, 1H, H-6); 13C-NMR (75.5 MHz, CD3OD) α-anomer: δ 158.6 (C-2), 136.8, 131.0, 128.1, 124.6 (Ar–C), 94.1 (C-7), 70.6 (C-6), 70.3 (C-9), 69.7 (×2) (C-5, C-10), 62.5 (C-1′), 49.0 (C-4); β-anomer: δ 96.0 (C-7); CIMS m/z 310 ([M–OH]+, 4%); HRCI-MS calcd for C14H20N3O5 ([M–OH]+): 310.1403, found: 310.1405.
(5S,6R,7R and 7S,9R,10S)-2-Benzylamino-6,7,10-trihydroxy-9-hydroxymethyl-8-oxa-1,3-diazaspiro[4.5]dec-2-ene hydrate (6b)
Guanidine 9b was used (138 mg, 0.34 mmol) to give compound 6b (111 mg, 96%); [α]26D +49 (c 1.12, MeOH); 1H-NMR (300 MHz, CD3OD) 5.5:1 α/β ratio, α-anomer: δ 7.43–7.29 (m, 5H, Ar–H), 5.15 (d, 1H, J6,7 = 3.3 Hz, H-7), 4.41 (brs, 2H, CH2Ar), 3.88–3.76 (m, 5H, H-9, H-1′a, H-1′b, H-4a, H-4b), 3.47 (d, 1H, H-6), 3.44 (d, 1H, J9,10 = 10.5 Hz, H-10); β-anomer: δ 4.76 (d, 1H, J6,7 = 8.4 Hz, H-7), 4.46 (brs, 2H, CH2Ar), 3.19 (d, 1H, H-6); 13C-NMR (75.5 MHz, CD3OD): α-anomer: δ 160.8 (C-2), 137.4, 129.9 (×2), 129.0, 128.4 (Ar–C), 94.1 (C-7), 70.6 (C-6), 70.2 (C-9), 69.9 (C-10), 69.5 (C-5), 62.5 (C-1′), 48.7 (C-4), 48.1 (CH2Ar); β-anomer: δ 161.1 (C-2), 137.8, 129.8, 128.8, 128.2 (Ar–C), 96.0 (C-7), 75.0 (C-6), 62.8 (C-1′); CIMS m/z 324 ([M–OH]+, 2%); HRCI-MS m/z calcd for C14H19N2O6 ([M–OH]+): 324.1559, found: 324.1558.
(5S,6R,7R and 7S,9R,10S)-2-Cyclohexylamino-6,7,10-trihydroxy-9-hydroxymethyl-8-oxa-1,3-diazaspiro[4.5]dec-2-ene hydrate (6c)
Guanidine 9c was used (70 mg, 0.18 mmol) to give compound 6c (54 mg, 91%); [α]227D +38 (c 0.71, MeOH); 1H-NMR (300 MHz, CD3OD) 4.2:1 α/β ratio, α-anomer: δ 5.14 (d, 1H, J6,7 = 3.4 Hz, H-7), 3.86–3.73 (m, 5H, H-9, H-1′a, H-1′b, H-4a, H-4b), 3.46 (d, 1H, H-6), 3.42 (d, 1H, J9,10 = 9.8 Hz, H-10), 3.28–3.22 (m, 1H, H-1′′), 2.00 (m, 2H, CH2), 1.77–1.74 (m, 2H, CH2), 1.64–1.56 (m, 1H, CHcyclohex.), 1.44–1.19 (m, 5H, CHcyclohex., 2CH2); β-anomer: δ 4.74 (d, 1H, J6,7 = 8.3 Hz, H-7), 3.68 (dd, 1H, J9,1′a = 5.3 Hz, J1′a,1′b = 12.0 Hz, H-1′a), 3.58 (m, 1H, H-9), 3.41 (d, 1H, J9,10 = 10.0 Hz, H-10), 3.17 (d, 1H, H-6); 13C-NMR (75.5 MHz, CD3OD) α-anomer: δ 159.6 (C-2), 94.1 (C-7), 70.6 (C-6), 70.2 (C-9), 69.8 (C-10), 69.0 (C-5), 62.4 (C-1′), 52.9 (C-1′′), 50.0 (C-4), 33.8, 33.6, 26.2, 25.6, 25.4 (5CH2); β-anomer: δ 159.7 (C-2), 95.9 (C-7), 76.3 (C-9), 74.9 (C-6), 70.5 (C-10), 62.7 (C-1′), 53.2 (C-1′′); CIMS m/z 316 ([M–OH]+, 6%); HRCI-MS m/z calcd for C14H26N3O5 ([M–OH]+): 316.1872, found: 316.1866.
(5R,6R,8R,9R,4′R)-6-(2,2′-Dimethyl-1′,3′-dioxolan-4′-yl)-8,9-dimethylmethylenedioxy-2-phenyl-7-oxa-1,3-diazaspiro[4.4]nonan-2-ene (10a)
To a solution of diamino 18 (81 mg, 0.28 mmol) in EtOH (5 mL) was added benzamidine hydrochloride (57 mg, 0.36 mmol) under an Ar atmosphere. The mixture was kept stirring under reflux for 24 h. Then, the solvent was removed under reduced pressure, the residue was dissolved in CH2Cl2 (20 mL) and washed with water (1 × 10 mL). The aqueous phase was extracted with CH2Cl2 (5 × 20 mL); the combined organic fractions were dried over MgSO4, filtered and the filtrate was concentrated to dryness. The residue was purified by column chromatography (40:1 → 10:1 CH2Cl2–MeOH) to afford 10a (13 mg, 12%); [α]24D +70 (c 1.20, CH2Cl2); 1H-NMR (300 MHz, CDCl3): 7.80–7.77 (m, 2H, Ar–H), 7.49–7.38 (m, 3H, Ar–H), 5.75 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.39 (d, 1H, H-9), 4.23 (dt, 1H, J4′,6 = 8.4 Hz, J4′,5′a = J4′,5′b = 6.0 Hz, H-4′), 4.11 (d, 1H, JH,H = 14.7 Hz, H-4a), 4.11 (dd, 1H, J5′a,5′b = 8.4 Hz, H-5′a), 4.00 (d, 1H, H-6), 3.96 (dd, 1H, H-5′b), 3.64 (d, 1H, H-4b), 1.59, 1.34, 1.27, 1.22 (4s, 3H each, 4CH3); 13C-NMR (75.5 MHz, CDCl3): δ 164.4 (C-2), 131.1, 130.0, 128.6, 127.3 (Ar–C), 113.1, 109.6 (2CMe2), 103.5 (C-8), 85.6 (C-9), 79.6 (C-6), 73.8 (C-4′), 72.6 (C-5), 67.7 (C-5′), 58.0 (C-4), 26.9, 26.8, 26.4, 25.2 (4CH3); CIMS m/z 375 (M+, 82%); HRCI-MS calcd for C20H27N2O5 (M+): 375.1920, found: 375.1915.
(5S,6R,7R and 7S,9R,10S)-6,7,10-Trihydroxy-9-hydroxymethyl-2-phenyl-8-oxa-1,3-diazaspiro[4.5]dec-2-ene hydrate (7a)
A solution of the amidine 10a (13 mg, 0.03 mmol) in 9:1 TFA–H2O (10 mL) was kept at rt for 1 h. Then, the solvent was removed under reduced pressure co-evaporating with toluene several times. The residue was washed with Et2O (3 × 10 mL) to give unprotected amidine 7a (9 mg, quant.); [α]26D +22 (c 0.89, MeOH); 1H-NMR (300 MHz, CD3OD): δ 7.94–7.87 (m, 3H, Ar–H), 7.81–7.75 (m, 2H, Ar–H), 7.66–7.60 (m, 3H, Ar–H), 4.18–4.10 (m, 4H, H-4a, H-4b α/β); pyranose α: δ 5.21 (d, 1H, J6,7 = 3.6 Hz, H-7), 3.89–3.79 (m, 2H, H-1′a, H-1′b), 3.71 (d, 1H, J9,10 = 9.3 Hz, H-10), 3.60 (m, 1H, H-9), 3.57 (d, 1H, H-6); pyranose β: δ 4.90 (d, 1H, J6,7 = 8.1 Hz, H-7), 4.08–4.07 (m, 1H, H-9), 3.89–3.79 (m, 1H, H-1′a), 3.77–3.68 (m, 1H, H-1′b), 3.60 (d, 1H, J9,10 = 10.2 Hz, H-10), 3.30 (d, 1H, H-6); furanose 5.85 (d, 1H, J8,9 = 3.03 Hz, H-8), 4.71 (d, 1H, H-9); 13C-NMR (75.5 MHz, CD3OD): δ 135.8, 130.5, 130.4, 129.7, 129.6, 124.1, 123.9 (Ar–C); pyranose α: δ 168.2 (C-2), 93.9 (C-7), 76.4 (C-10), 73.7 (C-5), 71.6 (C-6), 70.0 (C-9), 62.4 (C-1′), 52.0 (C-4); pyranose β: δ 168.9 (C-2), 95.8 (C-7), 75.2 (C-6), 73.1 (C-5), 70.7 (C-10), 70.0 (C-9), 62.7 (C-1′), 52.3 (C-4); CIMS m/z 295 ([M–OH]+, 6%); HRCI-MS calcd for C14H19N2O5 ([M–OH]+): 295.1294, found: 295.1299.
(5S,6R,7R and 7S,9R,10S)-6,7,10-Trihydroxy-9-hydroxymethyl-2-methyl-8-oxa-1,3-diazaspiro[4.5]dec-2-ene hydrate (7b)
To a solution of diamino 18 (160 mg, 0.55 mmol) in EtOH (10 mL) was added acetamidine hydrochloride (136 mg, 1.44 mmol) under argon. The mixture was kept stirring under reflux for 24 h. Then, the solvent was removed under reduced pressure, and the residue was dissolved in a 9:1 mixture of TFA–H2O (10 mL) and kept at rt for 2 h. After that, the solvent was removed under reduced pressure co-evaporating with toluene several times. The residue was purified by gel permeation chromatography (Biogel P2) to give the amidine 7b as an α/β mixture (20 mg, 14% two steps); [α]26D +19 (c 1.00, MeOH); 1H-NMR (300 MHz, CD3OD): pyranose α: δ 5.15 (d, 1H, J6,7 = 3.6 Hz, H-7), 3.94–3.91 (m, 2H, H-4a, H-4b), 3.83–3.77 (m, 2H, H-1′a, H-1′b), 3.51–3.46 (m, 2H, H-9, H-10), 3.46 (d, 1H, H-6), 2.25 (brs, 3H, CH3); pyranose β: δ 4.70 (d, 1H, J6,7 = 8.1 Hz, H-7), 3.94–3.91 (m, 3H, H-4a, H-4b, H-9), 3.89–3.87 (m, 1H, H-1′a), 3.73–3.65 (m, 1H, H-1′b), 3.75 (d, 1H, J9,10 = 10.5 Hz, H-10), 3.21 (d, 1H, H-6), 2.25 (brs, 3H, CH3); furanose α: 5.89 (d, 1H, J8,9 = 3.6 Hz, H-8), 4.67 (d, 1H, H-9); furanose β: 5.20 (d, 1H, J8,9 = 3.6 Hz, H-8), 3.55 (d, 1H, H-9); 13C-NMR (75.5 MHz, CD3OD): δ pyranose α: δ 170.0 (C-2), 93.7 (C-7), 76.6 (C-10), 72.5 (C-5), 71.2 (C-6), 70.0 (C-9), 62.4 (C-1′), 51.7 (C-4), 12.5 (CH3); pyranose β: δ 170.2 (C-2), 96.1 (C-7), 74.8 (C-6), 73.2 (C-5), 70.4 (C-10), 70.1 (C-9), 62.7 (C-1′), 51.9 (C-4), 12.4 (CH3); CIMS m/z 233 ([M–OH]+, 52%); HRCI-MS m/z calcd for C9H17N2O5 ([M–OH]+): 233.1137, found: 233.1141.
Acknowledgements
We thank the Dirección General de Investigación of Spain (CTQ2008 02813), FEDER funds and Junta de Andalucía (FQM 134) for financial support. P.M.-M. thanks the CONACYT of Mexico and the Fundación Carolina of Spain for the award of a fellowship.References
- T. M. Gloster and G. J. Davies, Org. Biomol. Chem., 2010, 8, 305–320 CAS.
- M. Bols, Ó. López and F. Ortega-Caballero, in Comprehensive Glycoscience-From Chemistry to Systems Biology, ed. J. P. Kamerling, Elsevier, 2007, pp. 815–884 Search PubMed.
- V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Chem. Rev., 2002, 102, 515–553 CrossRef CAS.
- N. Asano, Cell. Mol. Life Sci., 2009, 66, 1479–1492 CrossRef CAS.
- A. D. McNaught, Pure Appl. Chem., 1996, 68, 1919–2008 CrossRef CAS.
- T. M. Gloster, P. Meloncelli, R. V Stick, D. L. Zechel, A. Vasella and G. J. Davies, J. Am. Chem. Soc., 2007, 129, 2345–2354 CrossRef CAS.
- T. Wennekes, A. J. Meijer, A. K. Groen, R. G. Boot, J. E. Groener, M. van Eijk, R. Ottenhoff, N. Bijl, K. Ghauharali, H. Song, T. J. O'Shea, H. Liu, N. Yew, D. Copeland, R. J. van den Berg, G. A. van der Marel, H. S. Overkleeft and J. M. Aerts, J. Med. Chem., 2010, 53, 689–698 CrossRef CAS.
- D. Rejman, A. Rabatinová, A. R. Pombinho, S. Kovačková, R. Pohl, E. Zborníková, M. Kolář, K. Bogdanová, O. Nyč, H. Šanderová, T. Látal, P. Bartůněk and L. Krásný, J. Med. Chem., 2011, 54, 7884–7898 CrossRef CAS.
- T. M. Wrodnigg, A. J. Steiner and B. J. Ueberbacher, Anti-Cancer Agents Med. Chem., 2008, 8, 77–85 CrossRef CAS.
- T. S. Rasmussen, S. Allman, G. Twigg, T. D. Butters and H. H. Jensen, Bioorg. Med. Chem. Lett., 2011, 21, 1519–1522 CrossRef CAS.
- S. A. Yuzwa, M. S. Macauley, J. E. Heinonen, X. Shan, R. J. Dennis, Y. He, G. E. Witworth, K. A. Stubbs, E. J. McEachern, G. J. Davies and D. J. Vocadlo, Nat. Chem. Biol., 2008, 4, 483–490 CrossRef CAS.
- M. Benltifa, J. M. Hayes, S. Vidal, D. Gueyrard, P. G. Goekjian, J.-P. Praly, G. Kizilis, C. Tiraidis, K.-M. Alexacou, E. D. Chrysina, S. E. Zographos, D. D. Leonidas, G. Archontis and N. G. Oikonomakos, Bioorg. Med. Chem., 2009, 17, 7368–7380 CrossRef CAS.
- L. Somsák, V. Nagy, S. Vidal, K. Czifrák, E. Berzsényi and J.-P. Praly, Bioorg. Med. Chem. Lett., 2008, 18, 5680–5683 CrossRef.
- P. K. Kancharla and Y. D. Vankar, J. Org. Chem., 2010, 75, 8457–8464 CrossRef CAS.
- S. N. Richter, I. Menegazzo, M. Nadai, S. Moro and M. Palumbo, Org. Biomol. Chem., 2009, 7, 976–985 CAS.
- J. K. Maity, R. Ghosh, M. G. B. Drew, B. Achari and S. K. Mandal, J. Org. Chem., 2008, 73, 4305–4308 CrossRef CAS.
- S. B. Ferreira, A. C. R. Sodero, M. F. C. Cardoso, E. S. Lima, C. R. Kaiser, F. P. Silva Jr. and V. F. Ferreira, J. Med. Chem., 2010, 53, 2364–2375 CrossRef CAS.
- C. Gasch, P. Merino-Montiel, Ó. López and J. G. Fernández-Bolaños, Tetrahedron, 2010, 66, 9964–9973 CrossRef CAS.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS.
- S. Ogawa, M. Ashiura and C. Uchida, Carbohydr. Res., 1998, 307, 83–95 CrossRef CAS.
- M. Aguilar-Moncayo, M. I. García-Moreno, A. Trapero, M. Egido-Gabás, A. Llebaria, J. M. García Fernández and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 3698–3713 CAS.
- Y. Saito, T. A. Zevaco and L. A. Agrofoglio, Tetrahedron, 2002, 58, 9593–9603 CrossRef CAS.
- J. M. J. Tronchet and J.-M. Bourgeois, Carbohydr. Res., 1973, 29, 373–385 CrossRef CAS.
- C. Gasch, J. M. Illangua, P. Merino-Montiel and J. Fuentes, Tetrahedron, 2009, 65, 4149–4155 CrossRef CAS.
- Ó. López, S. Maza, I. Maya, J. Fuentes and J. G. Fernández-Bolaños, Tetrahedron, 2005, 61, 9058–9069 CrossRef.
- A. N. Van Nhien, L. Domínguez, C. Tomassi, M. R. Torres, C. Len, D. Postel and J. Marco-Contelles, Tetrahedron, 2004, 60, 4709–4723 CrossRef CAS.
- W. B. Whalley, E. L. Anderson, F. Dugan, J. W. Wilson and G. E. Ullyot, J. Am. Chem. Soc., 1955, 77, 745–749 CrossRef CAS.
- H. N. Po, H. Eran, Y. Kim and J. E. Byrd, Inorg. Chem., 1979, 18, 197–201 CrossRef CAS.
- F. S
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czewski, A. Bułakowska and M. Gdaniec, J. Heterocycl. Chem., 2002, 39, 911–915 CrossRef.
czewski, A. Bułakowska and M. Gdaniec, J. Heterocycl. Chem., 2002, 39, 911–915 CrossRef. - M. Bols, R. G. Hazell and I. B. Thomsen, Chem.–Eur. J., 1997, 3, 940–947 CrossRef CAS.
- J. L. Martin, K. Veluraja, K. Ross, L. N. Johnson, G. W. J. Fleet, N. G. Ramsden, I. Bruce, M. G. Orchard, N. G. Oikonomakos, A. C. Papageorgiou, D. D. Leonidas and H. S. Tsitoura, Biochemistry, 1991, 30, 10101–10116 CrossRef CAS.
- H. H. Taussky and E. Shorr, J. Biol. Chem., 1953, 202, 675–685 CAS.
Footnotes |
| † Dedicated to the memory of Dr Consolación Gasch. |
| ‡ Electronic supplementary information (ESI) available: 1H- and 13C-NMR spectra of compounds 5a–c, 6a–c, 7a,b, 8a–c, 9a–c, 10a, 15a–c, 22a–c. See DOI: 10.1039/c2ra21561f |
| This journal is © The Royal Society of Chemistry 2012 |