Synthesis of nanocrystalline solid solutions AlySn1−yO2−y/2 (y = 0.57, 0.4) investigated by XRD, 27Al/119Sn MAS NMR, and Mössbauer spectroscopy†
Ibrahim
Issac
a,
Ralf
Heinzmann
a,
Sebastian M.
Becker
ab,
Thomas
Bräuniger
c,
Zhirong
Zhao-Karger
a,
Christel
Adelhelm
d,
V. S.
Kiran Chakravadhanula
a,
Christian
Kübel
a,
Anne S.
Ulrich
be and
Sylvio
Indris
*a
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, P.O. Box 3640, Karlsruhe, 76021, Germany
bDFG Center for Functional Nanostructures (CFN), Karlsruhe Institute of Technology, Wolfgang-Gaede-Str. 1A, Karlsruhe, 76131, Germany
cMax Planck Institute for Solid-State Research, Heisenbergstrasse 1, Stuttgart, 70569, Germany
dInstitute of Applied Materials, Karlsruhe Institute of Technology, P.O. Box 3640, Karlsruhe, 76021, Germany
eInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Fritz-Haber-Weg 6, Karlsruhe, 76131, Germany
First published on 13th September 2012
Abstract
Nanocrystalline AlySn1−yO2−y/2 (y = 0.57, 0.4) was prepared by a co-precipitation method and subsequent calcination at temperatures of up to 650 °C. Transmission electron microscopy and X-ray diffraction reveal crystallite sizes of about 2 nm and a crystal structure equivalent to that of pure SnO2 cassiterite. The local structure was investigated by 27Al and 119Sn NMR as well as by Sn Mössbauer spectroscopy. The results show the formation of a solid solution with a random distribution of Al and Sn on the cation sites and a random distribution of oxygen vacancies on the anion sites.
1. Introduction
Tin dioxide, SnO2, is an extensively studied material with numerous fields of application like gas sensors,1–4 catalysis,5,6 dye sensitized solar cells,7,8 and electrode materials for lithium-ion batteries.9–18 In an earlier study, we investigated solid solutions of TiySn1−yO2, where Sn4+ is partially replaced by Ti4+.19 Doping of SnO2 with Al can be used to tune the catalytic, electronic, optical, and electrochemical properties.20–24 It could also be shown that doping with Al restricts the grain growth during thermal treatment.25–27 In this paper, we investigate the formation of solid solutions in a nanocrystalline state with high Al content. Thus, by replacing Sn4+ by Al3+, the lower charge has to be compensated by the formation of oxygen vacancies.Magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy as well as Mössbauer spectroscopy are versatile tools to investigate local environments of specific elements in solids and how these change upon going from coarse grained to nano-sized materials.28 We used 27Al MAS NMR to study the coordination state of the Al ions and how it is affected by heating the powders. Additionally, the local structure around the Sn ions was investigated by 119Sn MAS NMR and Sn Mössbauer spectroscopy. The crystal structure was investigated by X-ray diffraction (XRD), and the nanoparticle morphologies were studied by transmission electron microscopy (TEM).
2. Experimental
The starting materials for the synthesis [Al(NO3)3·9H2O (Acros, 99+%), SnCl4·5H2O (Aldrich, 98%), NaOH (Aldrich, 99.998%)] were of reagent grade and were used without further purification. To prepare AlySn1−yO2−y/2 (y = 0.57, 0.4), an appropriate amount of Al(NO3)3·9H2O was dissolved in deionised water and added to solid SnCl4·5H2O. After about 10 min, a colourless solution was formed. An aqueous solution of NaOH was added gradually until a pH value of about 10 was reached. Subsequently, the temperature of the solution was raised to 80 °C, and the mixture was stirred at this temperature for 2 h. Afterwards, the residue was left to settle for about 15 min, filtered, washed with deionised water, and dried at 90 °C for 4 h (about 75% yield of white solid). This sample was divided into portions and calcinated at different temperatures: 450 °C, 550 °C, and 650 °C for 5 h in air. The reaction can be described by the following equations:| y Al(NO3)3·9H2O + (1 − y) SnCl4·5H2O + (4 − y) NaOH → AlySn1−y(OH)4−y + 3y NaNO3 + (4 − 4y) NaCl + (5 + 4y) H2O | (1) |
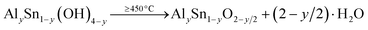 | (2) |
Chemical analysis was performed by various analytical methods. In order to determine the main elements Al and Sn accurately, X-ray fluorescence spectrometry (S4 PIONEER, Bruker AXS) was used. Samples of 10 mg were melted for 4 min at 700 °C and 4 min at 1100 °C (Perl'X 3, PANALYTICAL) together with 5.5 g LiBO2/Li2B4O7 (Spectromelt A12) and 0.5 g Sr(NO3)2 resulting in a homogeneous glassy disk. Calibration samples were prepared by the same technique with known amounts of dried Al2O3 and SnO2 and compositions close to those of the samples.
Oxygen determination has been carried out by the inert gas fusion technique (LECO TC 600). Ni pieces and Sn crucibles with low oxygen content served as fusion aid. The oxygen analysis was calibrated by Al2O3, (99.99% purity, dried at 1100 °C). The calibration range was close to the composition of the sample.
Impurities like Na, besides 40 other elements, have been examined by inductively coupled plasma optical emission spectroscopy (ICP-OES, OPTIMA 4300 DV, Perkin Elmer) after dissolution of 5 mg of sample in a mixture of 3 mL HCl (32%) and 1 mL HNO3 (65%) at 180 °C under pressure by microwave digestion (CEM, MarsXpress).
Simultaneous TGA-DSC-MS measurements were performed under He atmosphere on a Sensys Evo TGA-DSC instrument (Setaram, USA) with a heating rate of 5 °C min−1 from 25 °C to 600 °C using alumina crucibles. It was connected to an Omnistar mass spectrometer (Pfeiffer, Germany) for analysis of the evolved gases. XRD was carried out on a Philips PANalytical X'Pert diffractometer (Cu-Kα1 radiation, Nickel monochromator, Bragg-Brentano geometry). TEM analysis of the samples was performed using an image corrected FEI Titan 80–300 operated at an acceleration voltage of 300 kV and equipped with a Gatan US1000 slow-scan CCD camera providing an information limit of 0.8 Å in TEM mode.
27Al and 119Sn magic-angle spinning (MAS) NMR was performed on a Bruker Avance 600 MHz spectrometer (B0 = 14.7 T) using 2.5 mm zirconia rotors in a dry nitrogen atmosphere and on a Bruker Avance 300 MHz spectrometer (B0 = 7.05 T) using 4 mm zirconia rotors in a dry nitrogen atmosphere. An aqueous 1 M Al(NO3)3 solution was used as a reference for the chemical shift of 27Al (0 ppm), and solid SnO2 powder was used as a reference for the chemical shift of 119Sn (−604.3 ppm29). The typical values for the recycle delay of 27Al and 119Sn were 5 s and 60 s, respectively. All NMR experiments were performed at room temperature (298 K) and with a spinning speed of 25 kHz. 119Sn 1D NMR experiments were performed with a rotor synchronized Hahn-echo sequence (π/2–τ–π–τ–acquisition) using a typical π/2 pulse length of 2 μs on a 600 MHz spectrometer. 27Al 1D NMR experiments were recorded with an ordinary single-pulse experiment (π/18–acq.) on a 600 MHz spectrometer, with the small flip angle (π/18) being employed to avoid nutation effects. 27Al 2D 3QMAS NMR experiments were performed at 298 K on two different magnetic fields, 600 MHz with a spinning speed of 25 kHz and 300 MHz with a spinning speed of 10 kHz, using a split-t1-3QMAS30 pulse sequence (excitation(3Q)–conversion(1Q)–τ–π(selective)–acq.(-1Q)) with appropriate phase cycling.31 Bruker Topspin 2.1 and DMFit (2010)32 were used to analyze the spectra.
119Sn Mössbauer spectroscopic measurements were carried out in transmission mode at room temperature. 119Sn-enriched CaSnO3 was used as a γ-ray source. The velocity scale was calibrated with SnO powder which has an isomer shift of 2.63 mm s−1 and a quadrupole splitting of 1.30 mm s−1.33,34 For each sample, 10 mg of powder were deposited on an area of 1 cm2.
3. Results and discussion
The chemical analysis of two samples, after synthesis and heating to 450 °C, yields the compositions listed in Tab. 1. They are consistent with the two compositions Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8. Thus, the large Al contents of 0.57 and 0.4 lead to an oxygen deficiency of 15% and 10%, respectively. Residual Na could not be removed even by thorough washing.| Al0.57Sn0.43O1.71 | Al0.4Sn0.6O1.8 | |
|---|---|---|
| y = 0.57 | y = 0.4 | |
| mass (%) | mass (%) | |
| Al | 13.305 | 9.61 |
| Sn | 47.48 | 59.346 |
| O | 21.7 ± 0.77 | 25.67 ± 1.04 |
| Na | 2.2 | 2.1 |
Thermogravimetric-differential scanning calorimetry in combination with mass spectrometry (TGA-DSC-MS) was used to determine the calcination temperature required to remove water and other decomposable materials present during the preparation process. It was also used to suggest the correct formula for the decomposition reaction of the as-prepared precursors, in combination with elemental analysis of the materials calcinated at 450 °C for 5 h, and to observe any phase changes or decomposition.
The TG curve of the as-prepared sample Al0.57Sn0.43O1.71 (Fig. 1a) reveals that the dehydration process of the precursor starts at about 80 °C with a maximum weight loss at about 100 °C, as can be seen from the dTG curve. The weight loss is complete at about 420 °C. This is confirmed by the MS signals for masses 17 (OH) and 18 (H2O). The broad nature of the dTG curve may hint at a combination of two overlapping dehydration processes, i.e. loss of adsorbed water and water loss due to dehydration in the mixed hydroxides (see eqn (1)). The overall weight loss is about 9%. No significant weight loss was observed beyond 420 °C. The DSC curve exhibits three endothermic peaks. The first intense and broad peak with a maximum at about 105 °C corresponds to the dehydration process. The second small peak at 520 °C may be attributed to the formation of a solid solution (see eqn (2)) since there is no weight loss observed from the TG curve in this temperature region. The third small peak observed at 560 °C, also with no weight loss, may be attributed to initial grain growth, which is supported by XRD (Fig. 3) of the samples calcinated at 450, 550, and 650 °C. No further decomposition or phase changes were observed up to 600 °C.
 | ||
| Fig. 1 (a) Weight loss (TG), differential weight loss (dTG), differential scanning calorimetry (DSC), and MS signals of Al0.57Sn0.43O1.71 (blue), and Al0.4Sn0.6O1.8 (red). (b) Extended view of the DSC scan at temperatures between 450 °C and 585 °C. | ||
The TGA and the dTG curves of the as-prepared sample Al0.4Sn0.6O1.8 are similar to those of Al0.57Sn0.43O1.71. Here also the thermal decomposition starts at about 80 °C and is complete at about 420 °C. On the DSC curve, again an intense and broad peak appears with a maximum at 130 °C, shifted to slightly higher temperatures. The second small endothermic peak is observed at 500 °C, shifted by about 20 °C to lower temperatures. A third small peak appeared as a shoulder at about 550 °C. The maximum weight loss occurred at about 130 °C, cf. dTG curve. It is also related to the loss of adsorbed and hydrated water as apparent from the broad curve again, which might be due to two overlapping stages of water loss. This is also supported by the broad and intense first endothermic peak at 130 °C. The total weight loss was about 6.7%. Beyond 420 °C no significant weight loss was observed. The second small endothermic peak on the DSC curve at about 500 °C may again be attributed to the formation of mixed oxide solid solution (see eqn (2)). The weak peak at about 550 °C may again be attributed to an increase in crystallinity.
Fig. 2a shows the HRTEM micrograph of the Al0.57Sn0.43O1.71 powder after heating to 450 °C, which reveal highly aggregated nanoparticles with particle size in the range 2–4 nm. The selected area diffraction (SAED) pattern of the associated region indicates d-values of 0.332 nm, 0.263 nm, 0.174 nm, 0.143 nm measured with increasing ring diameters from the center of the SAED (Fig. 2b). These d-values correspond to the (110), (101), (211) and (112) reflections of the SnO2 structure.35 All the samples indicated a uniform distribution of Al, Sn and O through STEM-EDX analysis. The TEM analysis of the images is provided in the supporting information. Only in the case of Al0.57Sn0.43O1.71 heated to 650 °C signatures of segregation were observed by STEM-EDX analysis with regions rich in Sn and O but absence of Al.
 | ||
| Fig. 2 (a) HRTEM micrograph and (b) the associated SAED pattern of Al0.57Sn0.43O1.71 after heating to 450 °C. | ||
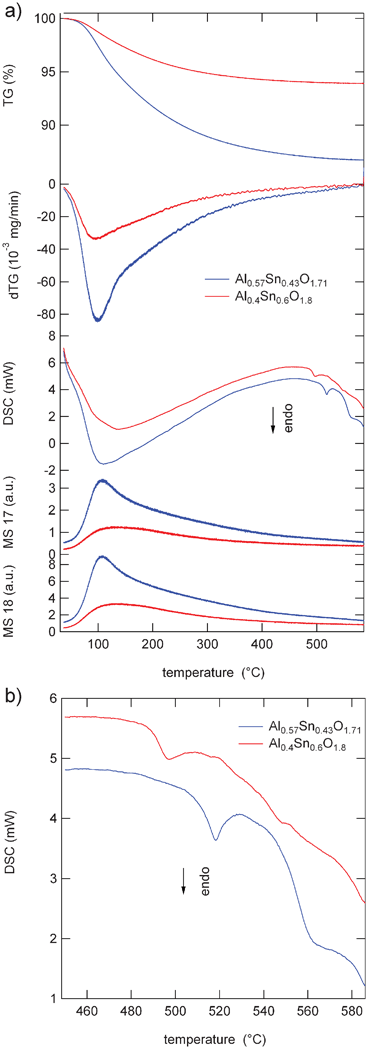
The XRD patterns of Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8 (Fig. 3) reveal peaks that are consistent with the cassiterite structure of pure SnO2 (red lines in Fig. 3). The peaks show strong broadening and evaluation of the peak widths using Scherrer's equation36 indicates crystallite sizes of about 2 nm, in good agreement with the TEM results. In comparison to pure SnO2, the peaks are shifted to higher diffraction angles, indicating that the lattice constants are decreased. This finding can be explained by the smaller ionic radius of Al3+ in an octahedral environment (0.535 Å) compared to Sn4+ (0.69 Å). The very small crystallite sizes might be due to the presence of Al in the lattice.27
 | ||
| Fig. 3 XRD patterns of (a) Al0.57Sn0.43O1.71 and (b) Al0.4Sn0.6O1.8 after heating to different temperatures, in comparison to database values of SnO2 cassiterite (red lines). | ||
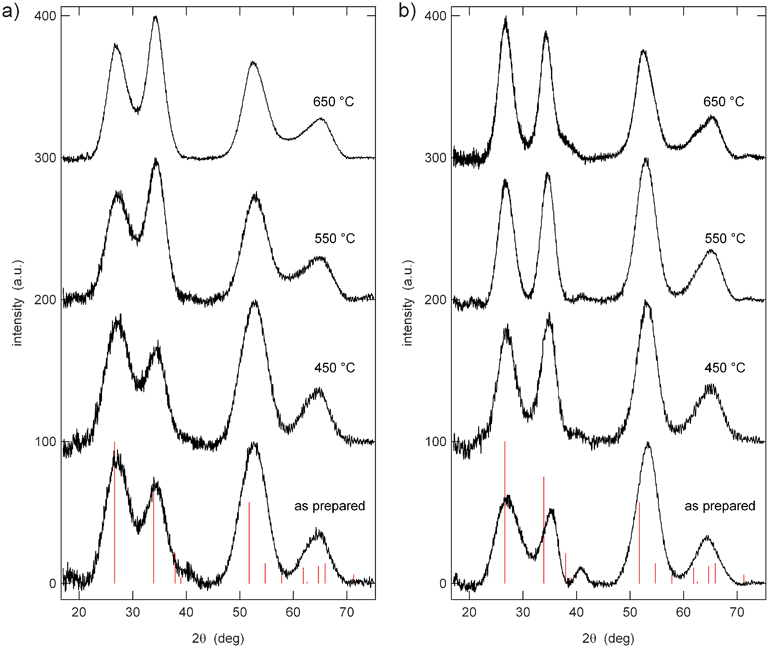
27Al 1D NMR was performed on the two samples Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8 after heating to different temperatures. The spectra show pronounced changes in the range from 0 to 100 ppm (Fig. 4). For the as prepared material Al0.57Sn0.43O1.71 (25 °C), there is only one narrow peak at 7 ppm. After heating to 450 °C, this peak is broadened and two additional signals appeared at 36 ppm and 67 ppm. Further heating steps to 550 °C and 650 °C increased the signal intensity of these two new peaks relative to the first signal at 7 ppm.
 | ||
| Fig. 4 27Al MAS NMR spectra of (a) Al0.57Sn0.43O1.71 and (b) Al0.4Sn0.6O1.8 for different sintering temperatures. | ||
The Al0.4Sn0.6O1.8 sample shows similar results. For the starting material, before heating (25 °C), there is only one signal at 7 ppm. When the sample was heated to 450 °C, two additional signals appeared at 36 ppm and 67 ppm. Compared to the Al0.57Sn0.43O1.71 sample, the increase in intensity of the second and third peak is weaker.
In general, NMR on quadrupolar nuclei like 27Al (nuclear spin I = 5/2), can lead to complex lineshapes with more than one maximum per Al environment. The position of the resonance line for quadrupolar nuclei is determined by both the chemical shift and the magnitude of the quadrupolar coupling, via the induced quadrupolar second-order shift, which scales down with the Larmor frequency.31 Accordingly, all 27Al 1D NMR spectra were measured at a magnetic field of 14.1 T in order to reduce the effect of the second-order quadrupole coupling. To better characterize the line shapes of the 27Al spectra, 3QMAS NMR experiments were performed for Al0.57Sn0.43O1.71 (heated to 550 °C) at different magnetic fields, i.e. 7.0 T and 14.1 T. As can be seen from Fig. 6, three distinct signals are observed in the 3QMAS spectrum. The line shapes displayed in Fig. 4 and 6 are typical for aluminates (see e.g., ref. 37), and derive from the existence of NMR parameter distributions caused by small local structure variations. For a more detailed analysis, the spectra were deconvoluted and fitted, using the DMFIT program.32 Using Czjzek-distributions,38,39 DMFIT is capable of fitting distributions for both the isotropic chemical shift and the quadrupolar parameters, delivering the average values <δiso>, <χ>, and <η> (see Table 2). The resulting values of <δiso> can now be used to assign the environments of the three peaks, as it is well known that the chemical shifts of 27Al give reliable information about the coordination of the aluminium (for a recent listing, see ref. 40 and references therein). Thus, the signal in the region around 7 ppm can be assigned to aluminum in an octahedral site, [AlO6]. The NMR peak in the region of 67 ppm represents aluminum in a tetrahedral site [AlO4], and the NMR signals in between, at 36 ppm, show aluminum with fivefold coordination [AlO5].
| MAS speed | Site | <δiso> [ppm] | <χ> [MHz] | <η> | |
|---|---|---|---|---|---|
| 1D MAS B0 = 14.1 T | 25 kHz | [AlO6] | 11.8 | 5.01 | 0.46 |
| [AlO5] | 44.4 | 5.92 | 0.71 | ||
| [AlO4] | 74.9 | 6.50 | 0.47 | ||
| 3QMAS B0 = 14.1 T | 25 kHz | [AlO6] | 12.0 | 4.31 | 0.55 |
| [AlO5] | 41.7 | 4.69 | 0.66 | ||
| [AlO4] | 72.8 | 5.10 | 0.49 | ||
| 3QMAS B0 = 7.0 T | 10 kHz | [AlO6] | 11.5 | 3.63 | 0.54 |
| [AlO5] | 43.5 | 4.13 | 0.65 | ||
| [AlO4] | 72.1 | 4.57 | 0.52 | ||
| Average | [AlO6] | 11.8 | 4.32 | 0.52 | |
| [AlO5] | 43.2 | 4.92 | 0.67 | ||
| [AlO4] | 73.3 | 5.39 | 0.49 |
These evaluations and the integrated peak areas in the 1D NMR spectra provided further information on the total occupation by aluminum in each of the three crystal sites (Fig. 5). In Al0.57Sn0.43O1.71, the occupation of the octahedral site [AlO6] rapidly decreases upon heating, and the occupation of the tetrahedral site [AlO4] and also the fivefold coordinated site [AlO5] increase correspondingly. In the end, at 650 °C, the occupation of the fivefold coordinated site is even higher than that of the tetrahedral site. For Al0.4Sn0.6O1.8, an evaluation of the occupation shows also an increase of the tetrahedral [AlO4] and the fivefold [AlO5] sites, accompanied by a decrease of the octahedral [AlO6] site. Although the same behavior as in the case of Al0.57Sn0.43O1.71 is found, the level of increase and decrease up to the final temperature of 650 °C is less prominent.
![Relative intensities of the peaks in the 27Al MAS NMR spectra assigned to [AlO4], [AlO5], and [AlO6] environments in (a) Al0.57Sn0.43O1.71 and (b) Al0.4Sn0.6O1.8 for different sintering temperatures.](/image/article/2012/RA/c2ra21540c/c2ra21540c-f5.gif) | ||
| Fig. 5 Relative intensities of the peaks in the 27Al MAS NMR spectra assigned to [AlO4], [AlO5], and [AlO6] environments in (a) Al0.57Sn0.43O1.71 and (b) Al0.4Sn0.6O1.8 for different sintering temperatures. | ||
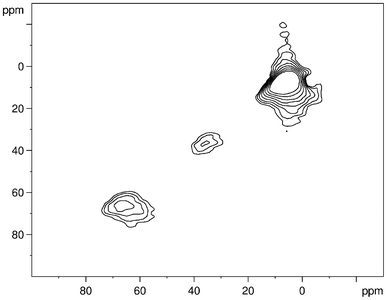 | ||
| Fig. 6 27Al MQ MAS NMR spectrum of Al0.57Sn0.43O1.71 (sintering temperature 550 °C). | ||
A single cation site exists in the cassiterite structure of SnO2. It is located within a surrounding octahedron of oxygen anions. The crystal structure is preserved when the Sn is partially replaced by Al (see XRD results, Fig. 3). The only change is a decrease in the lattice constant. Replacing Sn4+ by Al3+ ions in this structure must therefore result in the formation of oxygen vacancies. Assuming a random distribution of Al and Sn on the cation sites together with a random distribution of oxygen vacancies on the anion lattice, the probability that an Al ion is located in a [AlOn] environment can be described by the binomial distribution
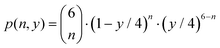 | (3) |
Here, n is the number of oxygen neighbors. The probability to find a vacancy on an anion site in the structure of AlySn1−yO2−y/2 is (y/2)/2 = y/4 and the probability to find an oxygen ion on this anion site is 1 − y/4.
The results of the measured and calculated relative intensities of the [AlOn] environments are listed in Tab. 3. A good overall agreement is found for both samples, corroborating the assumption of a solid solution of Al and Sn on the cation lattice and resulting oxygen vacancies on the anion lattice.
| y = 0.57 | y = 0.4 | |||
|---|---|---|---|---|
| Al0.57Sn0.42O1.71 | Al0.4Sn0.6O1.9 | |||
| obs. | calc. | obs. | calc. | |
| [AlO6] | 0.46 | 0.39 | 0.65 | 0.53 |
| [AlO5] | 0.32 | 0.40 | 0.21 | 0.35 |
| [AlO4] | 0.22 | 0.17 | 0.14 | 0.10 |
119Sn 1D NMR experiments on Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8 reveal a single broad NMR line centered around −600 ppm for all temperatures, with one example shown in Fig. 7. Whereas pure SnO2 in bulk shows a fairly narrow line in the 119Sn spectrum (cf.Fig. 7), it has been documented that the resonance broadens with smaller particle size.41,42 The line of Al0.57Sn0.43O1.71 shows extreme broadening with a width of about 60 ppm, which is much larger than what has been reported for pure SnO2 nanoparticles before.42 Similar strong broadening of the 119Sn spectrum, however, has been observed recently in fluorine-doped SnO2.43 This suggests that a distribution of local environments around the Sn ions, caused by the presence of heteroatoms (F in ref. 43 and Al in our samples) results in additional broadening of the resonance line via chemical shift variations. As explained above, this distribution is also observed in the 27Al-NMR line shapes of our samples. In comparison to bulk SnO2, the 119Sn-NMR line maximum of Al0.57Sn0.43O1.71 is shifted downfield to a smaller negative chemical shift. This may indicate a reduced electron density around the Sn nuclei, which can be caused by the presence of O vacancies.
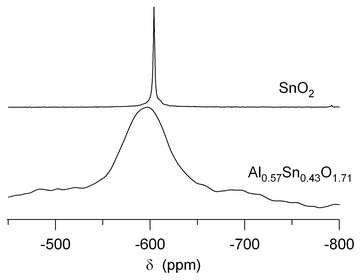 | ||
| Fig. 7 119Sn MAS NMR spectrum of Al0.57Sn0.43O1.71 after heating to 550 °C, in comparison to that of pure bulk SnO2. | ||
The Sn Mössbauer spectra of Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8 for different sintering temperatures are illustrated in Fig. 8. All spectra show a broad peak at about 0 mm s−1 which is characteristic of Sn4+ ions in an octahedral environment. The lineshape has to be described in terms of a doublet with overlapping Lorentzian lines (Table 4). Both the as prepared samples show a moderate quadrupole splitting of 0.53 mm s−1. Heating the samples leads to a strong increase of this quadrupole splitting with values of up to 0.69 and 0.67 mm s−1 for Al0.57Sn0.43O1.71 and Al0.4Sn0.6O1.8, respectively. This increase shows that during the formation of the solid solution, local structural disorder occurs resulting in strong distortions of the oxygen octahedra around the Sn atoms and thus in strong electric field gradients. For Al0.57Sn0.43O1.71, at 650 °C, the quadrupole splitting is reduced again to 0.53 mm s−1. The complete formation of the solid solution and possibly beginning grain growth seem to re-increase the symmetry around the Sn atoms. For Al0.4Sn0.6O1.8, this effect is much less pronounced with a final quadrupole splitting of 0.64 mm s−1.
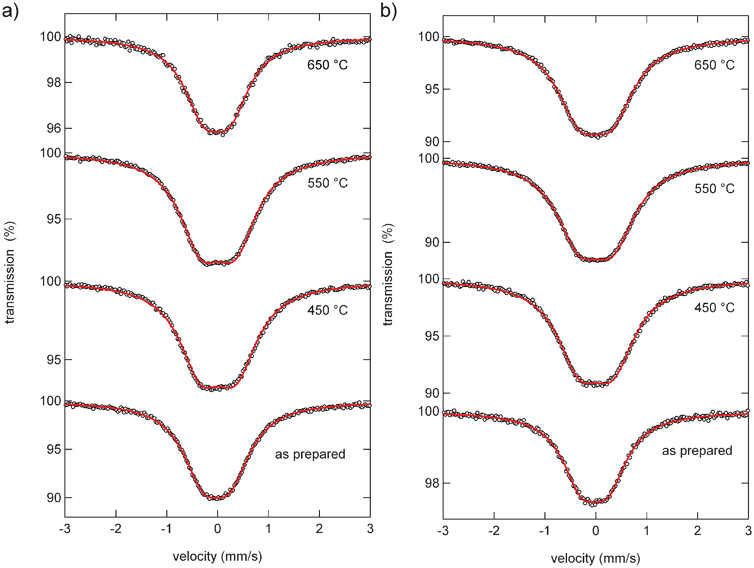 | ||
| Fig. 8 Sn Mössbauer spectra of (a) Al0.57Sn0.43O1.71 and (b) Al0.4Sn0.6O1.8 for different sintering temperatures. | ||
| Temp. | IS | QS | line width | |
|---|---|---|---|---|
| Al0.57Sn0.43O1.71 | as prep. | −0.021 ± 0.002 | 0.534 ± 0.005 | 0.978 ± 0.008 |
| 450 °C | 0.015 ± 0.001 | 0.695 ± 0.003 | 1.027 ± 0.006 | |
| 550 °C | 0.012 ± 0.001 | 0.681 ± 0.003 | 1.033 ± 0.005 | |
| 650 °C | −0.016 ± 0.003 | 0.525 ± 0.008 | 0.934 ± 0.013 | |
| Al0.4Sn0.6O1.8 | as prep. | −0.024 ± 0.003 | 0.527 ± 0.007 | 0.916 ± 0.011 |
| 450 °C | 0.008 ± 0.002 | 0.670 ± 0.004 | 1.065 ± 0.007 | |
| 550 °C | 0.005 ± 0.001 | 0.672 ± 0.003 | 1.099 ± 0.005 | |
| 650 °C | −0.005 ± 0.002 | 0.643 ± 0.004 | 1.070 ± 0.007 |
Conclusions
Nanocrystalline AlySn1−yO2−y/2 (y = 0.57, 0.4) with an average crystallite size of 2 nm was prepared by a co-precipitation method. The presence of a solid solution could be verified by XRD and 27Al MAS NMR. XRD reveals that the crystal structure of pure SnO2 cassiterite is preserved while the lattice constants are decreased due to the smaller cation Al3+. 27Al MAS NMR shows that the Al and Sn ions are randomly distributed on the cation sites and the oxygen vacancies are randomly distributed on the anion sites. This arrangement results in local distortions around the Sn4+ ions, as evidenced by 119Sn MAS NMR and Sn Mössbauer spectroscopy.Acknowledgements
We are grateful to the German Federal Ministry of Education and Research as well as to the DFG Center for Functional Nanostructures (CFN) for financial support.References
- T. Oyabu, J. Appl. Phys., 1982, 53, 2785 CrossRef CAS.
- N. Yamazoe, Sens. Actuators, B, 1991, 5, 7 CrossRef.
- J. Watson, K. Ihokura and G. S. V. Coles, Meas. Sci. Technol., 1993, 4, 711 CrossRef.
- W. Göpel and K. D. Schierbaum, Sens. Actuators, B, 1995, 26, 1 CrossRef.
- M. J. Fuller and M. E. Warwick, J. Catal., 1973, 29, 441 CrossRef CAS.
- P. W. Park, H. H. Kung, D.-W. Kim and M. C. Kung, J. Catal., 1999, 184, 440 CrossRef CAS.
- A. Kay and M. Grätzel, Chem. Mater., 2002, 14, 2930 CrossRef CAS.
- A. N. M. Green, E. Palomares, S. A. Haque, J. M. Kroon and J. R. Durrant, J. Phys. Chem. B, 2005, 109, 12525 CrossRef CAS.
- A. Courtney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045 CrossRef.
- A. C. Bose, D. Kalpana, P. Thangadurai and S. Ramasamy, J. Power Sources, 2002, 107, 138 CrossRef CAS.
- H. J. Ahn, H.-C. Choi, K.-W. Park, S.-B. Kim and Y.-E. Sung, J. Phys. Chem. B, 2004, 108, 9815 CrossRef CAS.
- C. Kim, M. Noh, M. Choi, J. Cho and B. Park, Chem. Mater., 2005, 17, 3297 CrossRef CAS.
- G. X. Wang, Y. Chen, L. Yang, J. Yao, S. Needham, H. K. Liu and J. H. Ahn, J. Power Sources, 2005, 146, 487 CrossRef CAS.
- L. Yuan, Z. P. Guo, K. Konstantinov, H. K. Liu and S. X. Dou, J. Power Sources, 2006, 159, 345 CrossRef CAS.
- L. Yuan, Z. G. Guo, K. Konstantinov, J. Z. Wang and H. K. Liu, Electrochim. Acta, 2006, 51, 3680 CrossRef CAS.
- Y. Liang, J. Fan, X. Xia and Z. Jia, Mater. Lett., 2007, 61, 4370 CrossRef CAS.
- Q.-H. Wu, J. Song, J. Kang, Q. F. Dong, S.-T. Wu and S.-G. Sun, Mater. Lett., 2007, 61, 3679 CrossRef CAS.
- C. Wang, Y. Zhou, M. Ge, X. Xu, Z. Zhang and J. Z. Jiang, J. Am. Chem. Soc., 2009, 132, 46 CrossRef.
- I. Issac, M. Scheuermann, S. M. Becker, E. Gil Bardají, C. Adelhelm, D. Wang, C. Kübel and S. Indris, J. Power Sources, 2011, 196, 9689 CrossRef CAS.
- C. Xu, J. Tamaki, N. Miura and N. Yamazoe, Sens. Actuators, B, 1991, 3, 147 CrossRef.
- I. Sayago, J. Gutiérrez, L. Arés, J. I. Robla, M. C. Horillo, J. Getino, J. Rino and J. A. Agapito, Sens. Actuators, B, 1995, 26, 19 CrossRef.
- M.-M. Bagheri-Mohagheghi and M. Shokooh-Saremi, J. Phys. D: Appl. Phys., 2004, 37, 1248 CrossRef CAS.
- R. Alcántara, F. J. Fernández-Madrigal, C. Pérez-Vicente, J. L. Tirado, J. C. Jumas and J. Olivier-Fourcade, Chem. Mater., 2000, 12, 3044 CrossRef.
- M. Lei, Q. R. Hu, S. L. Wang and W. H. Tang, Mater. Lett., 2010, 64, 19 CrossRef CAS.
- L. Xi, D. Qian, X. Huang and H. E. Wang, J. Alloys Compd., 2008, 462, 42 CrossRef CAS.
- Y. M. Al-Angari, M. W. Kadi, I. M. Ismail and M. A. Gabal, Cent. Eur. J. Chem., 2010, 8, 331 CrossRef CAS.
- T. A. Miller, S. D. Bakrania, C. Perez and M. S. Wooldridge, J. Mater. Res., 2005, 20, 2977 CrossRef CAS.
- S. Indris, M. Scheuermann, S. M. Becker, V. Šepelák, R. Kruk, J. Suffner, F. Gyger, C. Feldmann, A. S. Ulrich and H. Hahn, J. Phys. Chem. C, 2011, 115, 6433 CAS.
- S. Chaudhuri, F. Wang and C. P. Grey, J. Am. Chem. Soc., 2002, 124, 11746 CrossRef CAS.
- S. P. Brown and S. J. Wimperis, Magn. Reson., 1996, 119A, 280 Search PubMed.
- A. Goldbourt and P. K. Madhu, Annu. Rep. NMR Spectrosc., 2004, 54, 1 CrossRef.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70 CrossRef CAS.
- R. H. Herber, Phys. Rev. B, 1983, 27, 4013 CrossRef CAS.
- M. S. Moreno and R. C. Mercader, Phys. Rev. B: Condens. Matter, 1994, 50, 9875 CrossRef CAS.
- W. H. Baur, Acta Crystallogr., 1956, 9, 515 CrossRef CAS.
- H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures, John Wiley & Sons: New York, 1959 Search PubMed.
- A. Düvel, E. Romanova, M. Sharifi, D. Freude, M. Wark, P. Heitjans and M. Wilkening, J. Phys. Chem. C, 2011, 115, 22770 Search PubMed.
- G. Czjzek, J. Fink, F. Gotz, H. Schmidt, J. M. D. Coey, J. P. Rebouillat and A. Lienard, Phys. Rev. B, 1981, 23, 2513 CrossRef CAS.
- J.-B. d'Espinosa de Lacaillerie, C. Fretigny and D. Massiot, J. Magn. Reson., 2008, 192, 244 CrossRef.
- T. Bräuniger, C. Vinod Chandran, U. Wedig and M. Jansen, Z. Anorg. Allg. Chem., 2011, 637, 530 CrossRef.
- S. de Monredon, A. Cellot, F. Ribot, C. Sanchez, L. Armelao, L. Gueneau and L. Delattre, J. Mater. Chem., 2002, 12, 2396 RSC.
- V. Sabarinathan, C. Vinod Chandran, S. Ramasamy and S. Ganapathy, J. Nanoscience and Nanotechnology, 2008, 8, 321 CAS.
- Y. S. Avadhut, J. Weber, E. Hammarberg, C. Feldmann, I. Schellenberg, R. Pöttgen and J. Schmedt auf der Günne, Chem. Mater., 2011, 23, 1526 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21540c |
| This journal is © The Royal Society of Chemistry 2012 |