A mini-review on air-stable organometallic Lewis acids: synthesis, characterization, and catalytic application in organic synthesis
Renhua
Qiu
a,
Yi
Chen
ab,
Shuang-Feng
Yin
*a,
Xinhua
Xu
a and
Chak-Tong
Au
ac
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, China. E-mail: sf_yin@hnu.edu.cn; Fax: +86-731-88821310
bCollege of Basic Medicine, Hunan University of Chinese Medicine, Changsha, 410208, China
cDepartment of Chemistry, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong, China
First published on 29th August 2012
Abstract
Organometallic Lewis acids play an important role in modern organic synthesis. How to design and synthesize highly efficient and recyclable organometallic Lewis acid catalysts that can be conveniently applied in chemical reactions are key issues for sustainable synthetic processes. In general, stronger acidity means higher catalytic activity for organometallic Lewis acids. However, with the rise in acidity, the compound becomes more susceptible to hydrolysis and cannot be recycled. Simultaneous improvement of the hygroscopic character and Lewis acidity/catalytic activity of organometallic Lewis acids is highly desirable from the standpoint of practical applications. In this mini-review, the history of air-stable organometallic Lewis acids is introduced, with emphasis on our research works on metallocene, organobismuth, and organoantimony Lewis acids to the aspects of synthesis, characterization and catalytic application in carbon–carbon bond (Friedel–Crafts acylation, Mukaiyama aldol reactions; allylation, cyclotrimerization, Mannich reactions, cross-condensation reactions) and carbon–heteroatom bond (acylation, S–S bond cleavage, glycosylation) formation reactions. In terms of stability, storage, versatile ability, high catalytic activity and chemo-/stereo-selectivity, the complexes will find broad applications in organic synthesis.
 Renhua Qiu | Renhua Qiu was born in Hunan Province, China. He received a B.Sc. degree from Hunan Normal University in 2004, and received his Ph.D. from Hunan University in 2011. He was a research assistant (2010) with Prof. C.-T. Au at Hong Kong Baptist University and visiting researcher (2011) with Dr L.-B. Han at AIST (Japan). From 2009 to present, he worked as a lecturer and later as an assistant professor (2011) at Hunan University. At present, he is visiting Osaka University (Japan) as a JSPS fellow with Prof. N. Kambe. Dr Qiu's main research interest includes organometallic chemistry, Lewis acid catalysis, and C–H bond activation. |
 Yi Chen | Yi Chen was born in Hunan Province, China. She received her B.Sc. degree from Hunan Normal University in 1996, and received a Master’s Degree from Hunan University of Chinese Medicine in 2003. From 1996 to present, she has worked as a lecturer and later full professor (2011) at Hunan University of Chinese Medicine. At present, she is a visiting researcher with Prof. S. F. Yin. Prof. Chen's main research interest includes basic research of the cardiovascular and cerebrovascular diseases and organobismuth medicine. |
 Shuang-Feng Yin | Shuang-Feng Yin was born in Hunan Province, China. He received a B.Sc. degree from Beijing University of Chemical Technology in 1996, and got his Master’s Degree from the Research Institute of Petroleum Processing in 1999, and received a Ph.D. from Tsinghua University in 2003. He undertook postdoctoral research in HKBU with Prof. C.-T. Au from 2002 to 2004. From 2004 to present, he has worked as lecturer and later full professor (2006) in Hunan University. From 2004 to 2006, he visited Japan as a JSPS fellow with Dr S. Shimada. Prof. Yin's main research interests include organometallic chemistry, CO2 chemistry, catalysis and new energy sources. |
 Xinhua Xu | Xinhua Xu was born in Hunan Province, China. He received his Master’s Degree from Hunan Normal University in 1991, and received his Ph.D. from Zhejiang University with Prof. Xian Huang in 1998. He did postdoctoral research in Nankai University from 1998 to 2000 with Prof. Ruyu Chen. From 2000 to present, he has worked as an associate professor and later full professor (2006) at Hunan University. In 2002, he visited Japan as a visiting researcher with Prof. J. Otera. Prof. Xu's main research interest includes organometallic chemistry, heteroatom chemistry, and homogeneous catalysis. |
 Chak-Tong Au | Chak-Tong Au was born in Hong Kong. In 1981, he received his Ph.D. at the University of Bradford, UK. From 1980 to 1986, he did research at the University College, Cardiff, Wales. In 1986, he joined Xiamen University, China, as an associate professor and was promoted to professor in 1987. From 1990 to present, he worked as lecturer and later full professor in Hong Kong Baptist University. He was awarded a D.Sc. degree by the University of Liverpool in 2003. Prof. Au has published over 380 papers in areas of heterogeneous catalysis and novel materials. At present, he serves as editor of Applied Catalysis A: General. |
1. Introduction
Organometallic Lewis acids play a crucial role in green chemistry and sustainable development.1–6 Hence, how to design and synthesize highly efficient and recyclable organometallic Lewis acid catalysts that can be conveniently applied in various chemical reactions are the key issues for the development of sustainable synthetic processes.7 In general, for enhancement of catalytic activity, an organometallic Lewis acid should be as strongly acidic as possible. The dilemma is that with rise of acidity, the compound becomes more susceptible to hydrolysis and harder to recycled.8 Therefore, simultaneous improvement of the hygroscopic character and the Lewis acidity/catalytic activity of organometallic Lewis acids is highly desirable from the standpoint of practical utilization as catalysts.1.1 Lewis acid theory
In order to conduct a sensible discussion on the issue, it is necessary to define “Lewis acids”. There are three kinds of theories related to Lewis acids. The first one is the chemical bonding theory proposed by Lewis in 1923.9 Lewis suggested that an electron-pair donor can be classified as a Lewis base and an electron-pair acceptor can be classified as Lewis acid. The second theory is the Brønsted–Lowry acid–base theory published in the same year.10,11 The two theories are distinctly different but complementary. A Lewis base is also a Brønsted–Lowry base, but a Lewis acid does not need to be a Brønsted–Lowry acid. A Brønsted–Lowry acid is a proton donor, not an electron-pair acceptor. The third one is the “hard and soft (Lewis) acids and bases” (HSAB) theory published in 1963 (also known as the Pearson acid–base concept).12 Lewis acids and bases are classified according to their hardness or softness: hard implies small and nonpolarizable whereas soft indicates larger atoms that are more polarizable. In this theory, the strength of adduct formation is predicted based on two key concepts: (1) “hard acid–hard base” interactions are stronger than “hard acid–soft base” or “soft acid–hard base” interactions; (2) “soft acid–soft base” interactions are stronger than “soft acid–hard base” or “hard acid–soft base” interactions.The IUPAC definition is that Lewis acid is an electron-pair acceptor and therefore able to react with a Lewis base to form a Lewis adduct by sharing the electron pair furnished by the Lewis base.13 It means that a Lewis acid is an atomic or molecular species with a localized empty atomic or molecular orbital of low energy. This lowest energy molecular orbital (LUMO) can accommodate a pair of electrons. This definition is both more general and more specific—the electron pair need not be a lone pair, but the reaction should give an adduct.
1.2 Lewis acid categorisation
According to the definition of Lewis acid, a metal that contains empty orbital(s) (LUMO) can be considered as a Lewis acid (Fig. 1). However, only the noble metals (e.g. Pd) show significant catalytic activity,14 and most of the metals in the periodic table are inert in terms of acting as a Lewis acid catalyst. Nonetheless, when a metal forms metal halides, there is improvement in Lewis electron deficiency (e.g. AlCl3),15 and consequently an enhancement in catalytic activity. It was reported that there is a further increase in Lewis acidity and catalytic activity when the halide ion is replaced by a trifluoromethyl sulfonate or perfluorooctane sulfonic acid anion with large electron-withdrawing ability. Some of these Lewis acids are air-stable and water-tolerant and can be recycled several times (e.g., Ge(OTf)3).7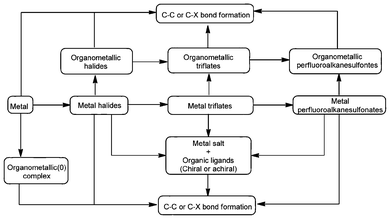 | ||
| Fig. 1 Types of metal Lewis acids and their catalytic applications. | ||
In order to obtain better catalytic activity and stereoselectivity, it is desirable to couple Lewis acids and bases, and such ideas have been investigated extensively. Yamamoto and coworkers16,17 developed the “designed” catalyst systems of Lewis acid/bases and Brønsted acid/bases that showed high synergistic catalytic activity and selectivity in asymmetric synthesis. The chiral and non-chiral organic ligands and the Lewis acid metal salts are also combined to become a catalytic system (e.g., Feng's chiral N,N-dioxides18), but some of these catalytic systems may have more than one stereospecific reactive species with a certain negative influence on stereoselectivity.
Recently, one of the hottest topics in Lewis acid chemistry is the frustrated Lewis pairs (FLPs) developed by Stephan and Erker,19 which are compounds or mixtures containing a Lewis acid and a Lewis base that cannot combine to form an adduct due to steric hindrance. Because of their "unquenched" reactivity, such systems are reactive enough to split dihydrogen heterolytically. The resulting H+/H− pairs serve as active catalysts for the hydrogenation of, for example, bulky imines, enamines, or enol ethers, giving a result similar to that of transition metals. However, it should be noted that most of these catalytic systems are air-sensitive and the manipulation processes have to be done under an argon atmosphere with the Schlenk technology. Furthermore, these systems are two-molecule systems.
In the last 20 years, the development of bifunctional Lewis acid catalysis has been extensively studied.20,21 With the Lewis acid site and Lewis base site in one molecule, the activation of an electrophilic reagent substrate by a Lewis acid center and the activation of a nucleophile substrate by a Lewis base center occur synergistically. With the two activation reactions occurring close to each other in correct stereochemistry, it is reasonable to envisage that there is synergistic effect that is similar to that of an enzyme-catalyzed reaction. However, due to the self-quenching potential, and hard-to-recycle as well as air-sensitive properties, further improvement is necessary for practical applications, especially for the metal complex Lewis acids.
1.3 Development of air-stable Lewis acid metal complexes
Recently, many research groups have made contributions to the development of air-stable Lewis acid metal complexes.22–25 Kobayashi and coworkers developed a series of zirconium Lewis acid complexes with an air-stable Zr–O bond. The storable chiral zirconium catalysts are highly efficient for enantioselective Mannich-type, aza-Diels–Alder, aldol, and hetero Diels–Alder reactions.26–28 Utilizing the stability of the La–O bond, Shibasaki and Matsunaga fabricated a series of linked-BINOL organometallic Lewis acids that can be used in practical asymmetric multifunctional catalysis.29 Boerner et al. constructed a series of zinc complexes that can be used as lactide polymerisation catalysts, and the complex benefits from the fact that the Zn–N bond is air-stable.30 Ishihara and Yamamoto have also made a contribution in this area.31 One of the most important Lewis acids developed by the research group is the air-stable, water-tolerant Lewis acid tris(pentafluorophenyl)borane which is thermally stable (even at 270 °C), and is soluble in many organic solvents.32 It shows high catalytic efficiency in reactions such as Mukaiyama aldol, conjugate, Sakurai–Hosomi allylation, rearrangement of epoxides as well as hydrosilylation reactions. It can be regarded as the first example of the general air-stable Lewis acids of this kind.In the recent years, our group has focused on the synthesis, characterization and catalytic applications of organometallic complexes with air-stable C–M bonds.33–41 Research work on this area is scant. Motoyama et al. developed the air-stable and water-tolerant (Phebox)RhCl(H2O) complex that can be applied in enantioselective allylation of aldehydes.42,43 Dijkstra et al. synthesized the rigid, nanosize multipalladium cartwheel pincer compounds and observed high catalytic efficiency when they were used as Lewis acid catalysts in the double Michael reaction between ethyl α-cyanoacetate and methyl vinyl ketone.44 As for the use of cheap metals in this area of research, there are only limited examples. A typical one is the air-stable organotin Lewis acids developed by Otera and coworkers involving the replacement of triflates by perfluorooctane sulfonates. The Lewis acids can catalyze various carbon–carbon bond formation reactions, indicating that the bulky perfluooctanesulfonate with electron-withdrawing, hydrophobic and antioxidative nature can make an impact on the synthesis of air-stable organotin Lewis acids.45
1.4 Synthesis strategies for air-stable organometallic Lewis acids
We are interested in organometallic Lewis acids with an air-stable carbon–metal (C–M) bond. Our synthesis strategy is as follows:1) Starting from commercially available organometallic halides with stable C–M bonds, we obtained air-stable and water-tolerant organometallic Lewis acids (e.g., metallocene Lewis acids) by replacing the halide with large electron-withdrawing and hydrophobic antioxidant perfluoroalkyl or aryl sulfonic anions.8,40,41
2) A ligand with appropriate spatial structure is synthesized and reacted with MCln to prepare the organometallic halide with stable C–M bonds. Air-stable and water-tolerant organometallic Lewis acids can be obtained through the abstraction of Cl− with large electron-withdrawing and hydrophobic antioxidative anions (e.g., N-bridged organobismuth Lewis acids).46,47
3) A ligand with Lewis basic site and appropriate spatial structure is synthesized and reacted with MCln to prepare the organometallic halide with stable C–M bonds. Air-stable and water-tolerant organometallic Lewis acid–base bifunctional catalysts can be obtained through the abstraction of Cl− with large electron-withdrawing and hydrophobic antioxidative anions (e.g., S-bridged organobismuth Lewis acids).35–37,39
4) Unquenched Lewis acid–base pairs are constructed by modulating the strength of the coordinated bond between metal and coordinated atoms of ligand. The most significant feature of this class of Lewis acid–base pairs is their air-stability (e.g., N-bridged organoantimony Lewis acids).48
5) Constructing Lewis acid catalysts with multi-metal centers that show bimetallic synergistic effects (e.g., S-bridged binuclear organobismuth Lewis acids).33
In this review, we summarize research works on air-stable organometallic Lewis acids, viz. metallocene, organobismuth, and organoantimony Lewis acids, focusing on their fabrication, characterization, and catalytic application in organic synthesis.
2. Air-stable metallocene Lewis acids
Metallocene dichlorides (Cp2MCl2, M = Zr (1a), Hf(1b), Ti(1c)) and their derivatives have been regarded as organometallic compounds that are somewhat air-stable.49 However, Cp2MCl2 has rarely been used as Lewis acid in organic synthesis on account of its weak acidity. To improve Lewis acidity, a triflate group was incorporated into Cp2MCl2 to afford the generation of metallocene bis(triflate) complexes Cp2M(OTf)2 (Cp = C5H5, M = Ti, Zr, Tf = CF3SO2), and the complexes were successfully employed as catalysts for carbon–carbon bond formation reactions.50–53 Unfortunately, the metallocene bis(triflate) complexes must be handled under strictly anhydrous conditions because they suffer from facile hydrolysis in open air.54 As such, improvement of the hygroscopic character of metallocene derivatives is highly desirable from the standpoint of practical utilization as catalysts.In sharp contrast to organotin triflates that are highly hygroscopic, perfluorooctane sulfonate was found to be air-stable and water-tolerant, and was used as an effective counter-anion by Otera et al. to generate cationic organotin species.45 Based on such a finding, it was postulated that longer perfluoroalkanesulfonate groups could be used to overcome the hydrolytic instability of cationic organometallic species in a general sense.
2.1 Synthesis of air-stable metallocene Lewis acids
With the successful synthesis and isolation of air-stable cationic metallocene perfluoroalkyl(phenyl)sulfonate (M = Zr, Ti, Hf), it is possible to conduct an assessment of Lewis acidity and catalytic activity in a convenient manner.8,34,38,40,41,55–58 Shown in Scheme 1 is the synthesis of metallocene perfluoroalkyl(phenyl)sulfonates [2a·xH2O,382b·xH2O·yTHF,403a·xH2O·yTHF,343b·xH2O·yTHF,343c·xH2O,8 and 3d·xH2O8]. Treatment of Cp2MCl2 [M = Zr (1a), Hf(1b), Ti (1c)] with silver perfluorooctane sulfonate (AgOSO2C6F5 or AgOSO2C4F9 or AgOSO2C8F17) (2 equiv) in CH3CN or THF afforded the corresponding perfluoroalkyl(phenyl)sulfonates as hydrates.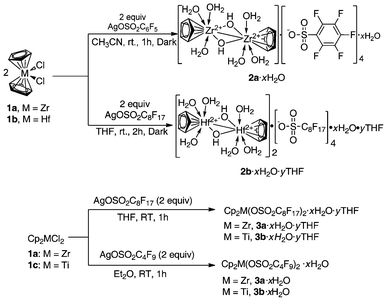 | ||
| Scheme 1 Synthesis of air-stable metallocene Lewis acids.8,34,38,40 | ||
2.2 Characterization of air-stable metallocene Lewis acids
Their composition, structure and physical or chemical properties were characterized by NMR, X-ray single crystal diffraction and TG-DSC technologies.Because the ease of solvate release from the complex is dependent on the strength of Lewis acidic sites, we studied the effect of synthesis conditions on the hydrate number (x) and solvate THF number (y) by 1H NMR spectroscopy (in dry CH3CN) and elemental analysis.8,38,40 It is clear from Table 1, entry 2 that the freshly prepared samples obtained after recrystallization contained 2 to 6 hydrates and 0 to 2 THF solvates. After vacuum treatment at room temperature for a week, solvated THF and hydrates were removed from the metallocene complexes. However, after exposure to air for 2 days, complexes 2a–2b and 3a–3d returned to 6 or 4 hydrates. The results indicate that in catalytic reactions, there is easy removal of solvate so that the organic substrate can have access to the metal centre for activation. After the catalytic action, the solvate can coordinate back to the complex for catalyst recovery.
| Condition | Number d | 2a | 2b | 3a | 3b | 3c | 3d |
|---|---|---|---|---|---|---|---|
| a Condition 1: the sample was kept under vacuum at room temperature for a week. b Condition 2: after recrystallization in organic solvent. c Condition 3: the sample was kept in open air for 2 days. d The hydrate (x) and solvate THF numbers (y) were measured by NMR and elemental analysis. | |||||||
| 1a | x | 0 | 0 | 0 | 0 | 0 | 0 |
| y | 0 | 0 | 0 | 0 | 0 | 0 | |
| 2b | x | 6 | 4 | 3 | 2 | 2 | 2 |
| y | 0 | 2 | 1 | 1 | 0 | 0 | |
| 3c | x | 6 | 6 | 4 | 4 | 4 | 4 |
| y | 0 | 0 | 0 | 0 | 0 | 0 | |
Usually, an organometallic Lewis acid is too air-sensitive to have high air and thermal stability. But with the attachment of perfluoroalkyl(phenyl)sulfonate, the metallocene complexes became air and thermally stable.8 Especially the metallocene complexes remained as dry colorless crystals or white powder in open air and showed no color change after a test period of one month. The complexes 2b, 3a and 3b were kept in the open air for a year and showed no change in color and structure.8,34,38,40
The TG-DSC analysis show that complex 2b is stable up to about 300 °C.40 The metallocene complexes 3a and 3b are also thermally stable at 300 °C and 180 °C, respectively.34 Moreover, no obvious changes were observed after a 3b·2H2O·THF sample was subject to thermal treatment at 180 °C for 2 days.57 Therefore, from the viewpoint of practical operation these metallocene complexes have an advantage over the metallocene triflates and perchlorates.
As characterized by conductivity measurements, complexes 2 and 3 undergo ionic dissociation in aqueous CH3CN (Table 2). The binuclear metallocene complexes 2a–2b were partially dissociated into ionic species, while the mononuclear metallocene complexes 3a–3d were completely dissociated into ionic species.8 The large values of molar conductivity (consistent with complete ionization into a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 electrolyte ([Cp2M]2+[X]2−) in aqueous CH3CN) suggests that the complexes are in cationic format in solution.59
:2 electrolyte ([Cp2M]2+[X]2−) in aqueous CH3CN) suggests that the complexes are in cationic format in solution.59
| Complexes | 2a | 2b | 3a | 3b | 3c | 3d |
|---|---|---|---|---|---|---|
| a In CH3CN (1.0 mmol L−1) at 15 °C. (All complexes were freshly prepared and kept in vacuum at room temperature for two hours after recrystallization). b The value given in the parentheses is the molar conductivity (Λ) [μS cm−1 mol−1]. c The sample was not dissolved completely. | ||||||
| Conductivity | 84.0 | 55.6 | 136.1 | 114.5 | 98.5 | 95.1 |
| (μS cm−1 mol−1)b | (42.0)c | (25.3) c | (136.1) | (114.5) | (98.5) | (95.1) |
Another notable feature of complexes 3a–3d was their unusually high solubility in MeOH, acetone, THF, EtOAc, and CH3CN (Table 3).34 In MeOH, solubility was extraordinarily high (up to 2222 g L−1 in the case of 3c); it is more reasonable to assume that both methanol and complex are miscible with each other so form a slightly viscous liquid. Overall, the unusual solubility reflects the amphiphilic nature of the long fluoroalkyl chain.45 Upon dissolving the metallocene complexes in a polar solvent, a solvent molecule can approach the coordination sphere of the metal atom to replace the hydrated water on account of the compatibility between them, and the resulting solvated species are highly soluble in the same polar solvent.34,45 In Table 3, one can see that in polar organic solvents the solubility of complexes 2a–2b is much lower than that of complexes 3a–3d.34,40 As revealed in X-ray analysis, there is higher steric influence in the dimeric complexes 2a–2b, and the central metal atoms are surrounded front to front by Cp rings.38,40 In such a situation, it is hard to replace the coordinated water with a molecule of organic solvent to generate the highly soluble solvated species.
| Complexb | 2a | 2b | 3a | 3b | 3c | 3d |
|---|---|---|---|---|---|---|
| a All complexes were freshly prepared and kept in vacuum at room temperature for two hours after recrystallization. b The sample formulas were as follows: 2a·6H2O, 2b·4H2O·2THF, 3a·3H2O·THF, 3b·2H2O·THF, 3c·2H2O, 3d·2H2O. | ||||||
| Acetone | 0 | 33 | 957 | 275 | 667 | 1453 |
| THF | 4 | 20 | 244 | 121 | 1776 | 521 |
| EtOAc | 82 | 68 | 729 | 19 | 600 | 260 |
| MeOH | 643 | 400 | 186 | 128 | 2222 | 470 |
| CH3CN | 3 | 4 | 143 | 530 | 524 | 302 |
| Et2O | 0 | 23 | 21 | 11 | 272 | 84 |
| CH2Cl2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Toluene | 0 | 0 | 0 | 0 | 0 | 0 |
| Hexane | 0 | 0 | 0 | 0 | 0 | 0 |
X-ray analysis data (Fig. 2) show that the zirconium and hafnium atoms have a geometry of distorted octahedral coordination with the Cp group being trans to OH in 2a·6H2O and 2b·4H2O·2THF.38,40 The zirconium atom in the cationic ion of 3a·3H2O·THF is coordinated by three water molecules and not by THF.8 The three H2O molecules lie on the plane that bisects the angle between the Cp ring planes. The C6F5SO3− and C8F17SO3− anions as well as the dissociated H2O molecule and solvate THF are packed around the metallocene complex cation of 2a, 2b and 3a in such a way that their oxygen atoms point towards the H2O ligands. The C6F5 and C8F17 sides of the anion, on the other hand, are clustered together to produce hydrophobic domains.8,34,38,40
![ORTEP view of crystal structure of [(CpZr(OH2)3)2(μ2-OH)2]4+ and crystal structure of [(CpZr(OH2)3)2(μ2-OH)2][C6F5SO3]4·6H2O (2a·6H2O) (top) (image reproduced from ref. 38 with permission of the Royal Society of Chemistry); OTREP view of novel cationic structure of [CpHf(H2O)3(μ2-OH)]24+ and ball-and-stick view of crystal structure of [CpHf(μ2-OH)(OSO2C8F17)2]2·10H2O·2THF (2b·4H2O·2THF) (middle); ORTEP view showing 50% probability ellipsoids and packing of Cp2Zr(OSO2C8F17)2·3H2O·THF (3a·3H2O·THF) (bottom) (images reproduced from ref. 34 and 40 with permission of Wiley-VCH).34,38,40](/image/article/2012/RA/c2ra21517a/c2ra21517a-f2.gif) | ||
| Fig. 2 ORTEP view of crystal structure of [(CpZr(OH2)3)2(μ2-OH)2]4+ and crystal structure of [(CpZr(OH2)3)2(μ2-OH)2][C6F5SO3]4·6H2O (2a·6H2O) (top) (image reproduced from ref. 38 with permission of the Royal Society of Chemistry); OTREP view of novel cationic structure of [CpHf(H2O)3(μ2-OH)]24+ and ball-and-stick view of crystal structure of [CpHf(μ2-OH)(OSO2C8F17)2]2·10H2O·2THF (2b·4H2O·2THF) (middle); ORTEP view showing 50% probability ellipsoids and packing of Cp2Zr(OSO2C8F17)2·3H2O·THF (3a·3H2O·THF) (bottom) (images reproduced from ref. 34 and 40 with permission of Wiley-VCH).34,38,40 | ||
Since the metallocene Lewis acid is desired to be as strongly acidic as possible to acquire higher activity, the Lewis acidity of 2a–2b and 3a–3d may satisfy this requirement. Due to the strong complex formation between 10-methylacridone and 3a, the UV-Vis spectra shows a significant red shift illustrating a large Lewis acidity ability of 3a.34,60 Such significant red shift is also observed in the fluorescence spectrum maximum (λem = 476 nm),34 implying that the Lewis acidity of 3a falls between those of Sc3+ (λem = 474 nm) and Fe3+ (λem = 478 nm).34,61,62 The ESR analysis shows that the ΔE value of the titanium complex (O2·−–3b) (Ti4+: gzz = 2.0289, ΔE = 1.06 eV) and the zirconium complex (O2·−–3a) (Zr4+: gzz = 2.0331, ΔE = 0.91 eV) is significantly larger than that of Sc(OTf)3 (gzz = 2.0304, ΔE = 1.00 eV)34 or falls between those of Sc(OTf)3 and Y(OTf)3 (gzz = 2.0349, ΔE = 0.85 eV), respectively.8,34,62 The high Lewis acidity of 3a and 3b is enough to trigger synthetically useful reactions, since the Lewis acids with an ΔE value larger than 0.88 were presumed to be capable of inducing carbon–carbon bond-forming reactions.45 The Hammett indicator method63–65 was also applied to determine the acidity of the complex 2a–2b and 3a–3b, and it was found that both of these Lewis acids have relatively strong acidity with acid strength of 0.8 < Ho ≤ 3.3 (Ho being the Hammett acidity function) except 2b (3.3 < Ho ≤ 4.8).34,38,40
2.3 Catalytic application of air-stable metallocene Lewis acids
The unique characteristics of 2a–2b and 3a–3b motivated us to evaluate its performance as a Lewis acid catalyst for C–C and C–X bond transformation such as acylation,40,41,57 glycosylation,8 S–S bond cleavage,56,58 Friedel–Crafts acylation of alkyl aromatic ethers,34,40 cylcotrimerization of ketones,55 allylation of aldehydes,34,38,40 and Mukaiyama aldol reactions.34,40We investigated the esterification reaction of structurally diverse alcohols, phenols, thiols, and amines with acetic anhydride in the presence of 1.0 mol% of metallocene Lewis acids of 2b, 3a and 3b at room temperature under solvent-free conditions (Scheme 2).34,40,41,57 It was found that these Lewis acids can catalyze acetylation effectively not only for alcohols but also for amines (yields of up to 99%). Phenols and thiols were also acetylated efficiently. Furthermore, in the cases of furan methanol and geraniol with carbon–carbon double bonds, these catalysts showed functional tolerance (yield > 90%). High chemoselectivity was achieved, and tertiary alcohols such as triphenyl methanol was almost unaffected.41,57
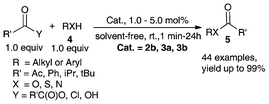 | ||
| Scheme 2 Acylation of alcohol, phenol, thiol and amine catalyzed by air-stable organometallic Lewis acids.40,41,57 | ||
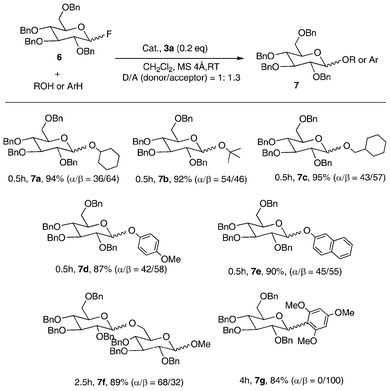 | ||
| Scheme 3 Glycosylation with glycosyl fluoride catalyzed by 3a.8 | ||
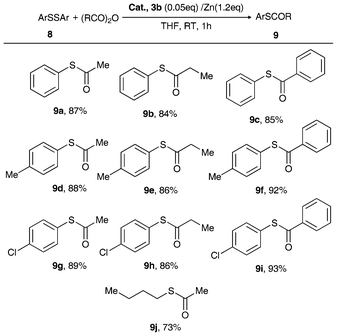 | ||
| Scheme 4 Reductive cleavage of S–S bonds catalyzed by 3b/Zn dust.56,58 | ||
 | ||
| Scheme 5 Friedel–Crafts acylation catalyzed by 3b.34 | ||
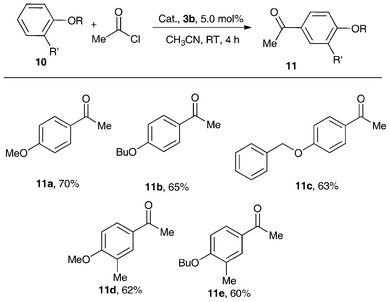
Remarkably, with 5 mol% of the air-stable metallocene Lewis acid catalyst 3b, the Friedel–Crafts acylation of structurally diverse alkyl aromatic ether with 2.0 equivalents of acetic chloride obtained good-to-excellent product yields at RT in CH3CN (Scheme 5).34 The electron-donating groups attached to the aromatic ring enhanced reaction activity, but that of the methyl group in the meta-position decreased the product yield because of the steric effect. It should be noted that this reaction showed high regioselectivity (para-isomer > 99%). This is in sharp contrast to the activity of AlCl3 which was markedly low under the adopted mild reaction conditions. In the absence of the catalyst, no product was obtained. Thus, in terms of good yields and para-regioselectivity, complex 3b can be considered to be an excellent catalyst in Friedel–Crafts acylation.34
Lewis acid 3a (5.0 mol%) was examined as a catalyst for the cyclotrimerization of structurally diverse ketones in refluxing toluene, and good-to-excellent yields (78–98% yield) were obtained (Scheme 6). The acetophenones with electron-donating groups in the para-position of the phenyl plane exhibited higher reactivity than those with electron-withdrawing groups, indicating that the electron-donating groups enhanced the density of super-conjugated π-electrons in the phenyl plane and carbonyl group, leading to stronger interactions of carbonyl group of acetophenones with the zirconium atom of the complex of 3a. The cyclohexanone also gave 85% yields of triannulated benzene (13g), illustrating that 3a was a good catalyst for cyclotrimerization of aliphatic ketones.55
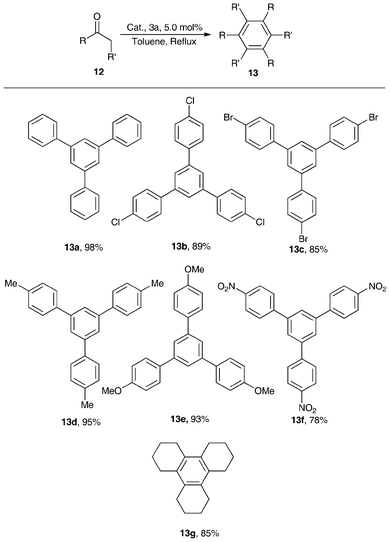 | ||
| Scheme 6 1,3,5-trisubstitutebenzene yields from acetophenones catalyzed by 3a in refluxing toluene.55 | ||
| Entry | RCHO(15) | Yield/% | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 15 | 3a | 3b | 2b | 2a | 16 | 3a | 3b | 2b | ||
|
a RCHO (14), 1.0 mmol; tetraallyltin, 0.3 mmol; 3a or 3b, 2b, 0.05mmol; CH3CN; 3.0 mL; 1h, isolated yield.
b RCHO (14), 1mmol; tetraallyltin, 0.3mmol; Cat., 0.05mmol; RT.; 1h; 3mL solvent (CH3OH:H2O = 4:1).
c RCHO (14), 1.0mmol; ketene silyl acetals, 1.2 mmol; 3a or 3b, 2b, 0.05mmol; Et2O 3.0 mL; RT, 6 h, isolated yield.
|
||||||||||
| 1 | PhCHO | 15a | 94 | 82 | 92 | 97 | 16a | 92 | 80 | 89 |
| 2 | p-MeC6H4CHO | 15b | 91 | 79 | 91 | 91 | 16b | 92 | 81 | 87 |
| 3 | p-MeOC6H4CHO | 15c | 90 | 80 | 89 | 90 | 16c | 93 | 80 | 86 |
| 4 | p-ClC6H4CHO | 15d | 93 | 82 | 93 | 94 | 16d | 94 | 84 | 92 |
| 5 | p-CF3C6H4CHO | 15e | 94 | 83 | 95 | 99 | 16e | 96 | 85 | 94 |
| 6 | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO CHCHO |
15f | 90 | 82 | 92 | 93 | 16f | 90 | 80 | 85 |
| 7 | PhCH2CH2CHO | 15g | 93 | 82 | 94 | 89 | 16g | 87 | 76 | 81 |
| 8 | C7H15CHO | 15h | 87 | 75 | 90 | 90 | 16h | 83 | 70 | 75 |
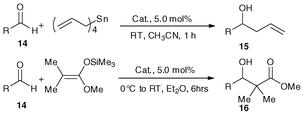
We assessed the metallocene Lewis acids catalysts 3a, 3b, 2a and 2b in the allylation and Mukaiyama aldol reactions of aldehydes (14) with nucleophiles such as tetraallyltin and ketene silyl acetals, and observed high catalytic activity (Table 4).34,38,40 As expected, the reactions resulted in good yields of homoallyl alcohols (15a–15h) and β-hydroxy ester derivatives (16a–16h) in CH3CN or Et2O. A similar tendency of catalytic activity of complexes 2a, 2b, 3a and 3b was observed when aliphatic aldehydes and aromatic aldehydes with electron-donating or -withdrawing groups were investigated. It is worth noting that 3a with appropriate Lewis acidity is better than the other three metallocene Lewis acids 3b, 2a and 2b for these two carbon–carbon bond forming reactions. It is because nucleophiles (such as tetraallyltin and ketene silyl acetals) decompose rather than reacting with the aldehyde over catalysts of high Lewis acidity (15a–15h),34,38,40 while the lower Lewis acidity of 2b will result in somewhat lower catalytic activity. Due to the high tolerance of 3a, 3b, 2a and 2b towards hydrolysis, the solvents adopted in these reactions were used as received and were not subject to any kind of drying procedure. Furthermore, using 2a as a catalyst, reactions of benzaldehyde and tetraallyltin can be conducted in aqueous methanol giving nearly quantitative yield of 1-phenylbut-3-en-1-ol (97%).39 It is noted that without the use of a catalyst, the yield of 1-phenylbut-3-en-1-ol was only 21% under the same conditions.38
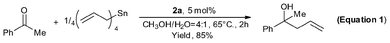 | (1) |
As we know, highly reactive reagents, such as Grignard, lithium and titanium reagents, would usually cause poor chemoselectivity and fail to discriminate between aldehydes and ketones.40 Remarkably however, using 2a good chemoselectivity was observed. As shown in Equation 1, despite the fact that allylation of acetophenone with tetraallyltin does not occur at RT, the homoallyl alcohol was obtained in 85% yield at 65 °C in the presence of 2a.38
In terms of green chemistry and sustainable development, the developed organometallic Lewis acids should possess high catalytic activity as well as good recyclability. To test the reusability of the catalyst and reproducibility of catalytic performance, 3a was subject to cycles of allylation and Mukaiyama aldol reactions (Fig. 3).34 It is clear that the catalyst is stable and suitable for reuse. Therefore, 3a has the advantages of being high in activity, selectivity, stability, and reusability. The catalysts 2a, 2b and 3b were also found to be recyclable in reactions (not shown).34,38,40,41,55,57
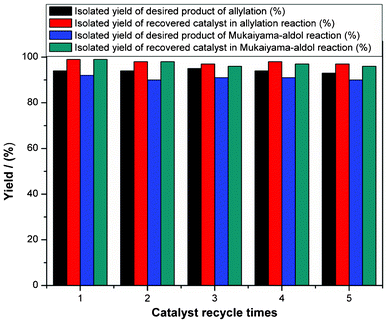 | ||
| Fig. 3 Catalyst recycling in allylation (tetraallyltin and benzaldehyde) and Mukaiyama aldol reactions (ketene silyl acetals and benzaldehyde) with catalyst 3a.34 | ||
3 Air-stable N-bridged organobismuth Lewis acids
Bismuth is relatively inexpensive and low in toxicity, in line with the requirements of green chemistry.83–86 It is hence desirable to use the bismuth resources effectively.87 The use of inorganic bismuth compounds such as BiCl3, BiBr3, Bi(OTf)3, and Bi(NO3)3 as Lewis acid catalysts in organic synthesis has attracted extensive attention.88–90 However, in contrast to the research on transition-metal metallocene Lewis acid, the research on organobismuth Lewis acids is limited.83,84,90 Studies have shown that the introduction of intramolecular electron-donating ligand or bulky organic ligands can improve the stability of organobismuth complexes.87 However, with the increase in stability, there is reduction in Lewis acidity of the bismuth centers. It was envisaged that through the design and synthesis of organobismuth complexes with a stable skeleton structure, there would be enhancement in acidity, hence broadening its scope as efficient catalysts in organic reactions. In this section, we would like to introduce the design, synthesis and characterization of air-stable N-bridged organobismuth Lewis acids with strong acidity, and their catalytic applications in allylation and Mannich reactions.46,473.1 Synthesis of air-stable N-bridged organobismuth Lewis acids
Scheme 7 shows the synthesis routes for N-bridged organobismuth chlorides 18a–18c and organobismuth Lewis acids 19a–19b.46,47,91 The lithiation reaction was conducted at −30 °C and the resulting dianion was treated with anhydrous bismuth chloride to produce compounds 18b–18c.91 The reactions of compound 18b with AgOSO2C8F17 and AgBF4 in THF solution gave compounds 19a and 19b, respectively, in good yields.46,47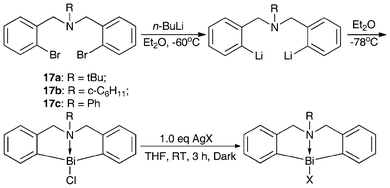 | ||
| Scheme 7 Synthesis of N-bridged organobismuth Lewis acids.46,47,91 | ||
 | ||
| Scheme 8 One-pot three-component Mannich-type reaction catalyzed by N-organobismuth Lewis acid in water. 47 | ||
3.2 Characterization of air-stable N-bridged organobismuth Lewis acids
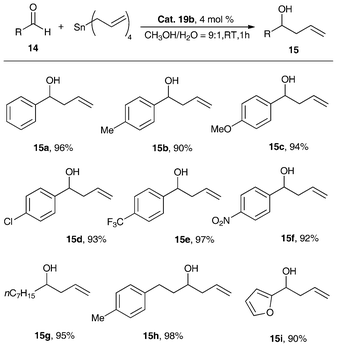
NMR analysis showed that the substituents on the nitrogen atom can affect the strength of N–Bi coordination bond, resulting in precise modulation of the metal center of the Lewis acid. After reacting with AgX (X = BF4, OSO2C8F17), there is modulation in the strength of the Bi–X bonds (X = OSO2C8F17 (19a), BF4 (19b)) as well as that of the N–Bi coordination bond. As shown in Fig. 4, the central bismuth-containing part of the three compounds exhibits a pseudo-trigonal bipyramidal (TBP) structure, where both the C(1) and C(8) atoms of 19a and 19b, C(1) and C(20) atoms of 18b exist in the equatorial position of the TBP structure along with a lone electron pair of bismuth, and the N(1) atoms are at the apical position.46,47,91 These results strongly support the existence of transannular interaction between Bi and N atoms. The Bi–N bond length of the air-stable framework of 5,6,7,12-tetrahydrodibenzo[c,f][1,5]azabismocine can be affected by the electronic properties of the bismuth.46,47,91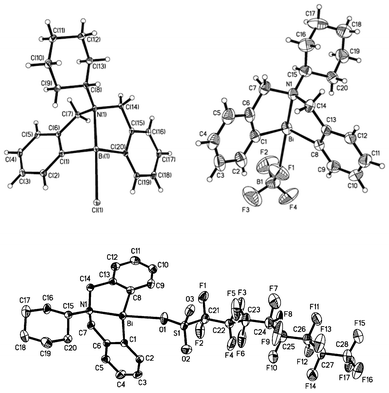 | ||
| Fig. 4 ORTEP view of crystal structure of (C6H11N(C6H4CH2)2Bi(X))(X = Cl (18b, top left); OSO2C8F17 (19a, bottom); BF4 (19b, top right)) (image reproduced from ref. 46,47 and 91 with permission of Elsevier).46,47,91 | ||
The Lewis acidity was determined by the Hammett indicator method, the acid strength of 19a is 4.8 < Ho ≤ 6.8 while that of 19b is 3.3 < Ho ≤ 4.8. Compared to the precursor chloride 18b (4.8 < Ho ≤ 6.8), there is enhancement in acidity.46,47 The compounds 19a–19b stayed as dry colorless crystals or white powder after being kept in open air for six months. The thermal stability of the organobismuth complexes were determined by TG-DSC/DTA (N2); they are thermally stable below 246 °C.46,47
3.3 Catalytic application of air-stable N-bridged organobismuth Lewis acids
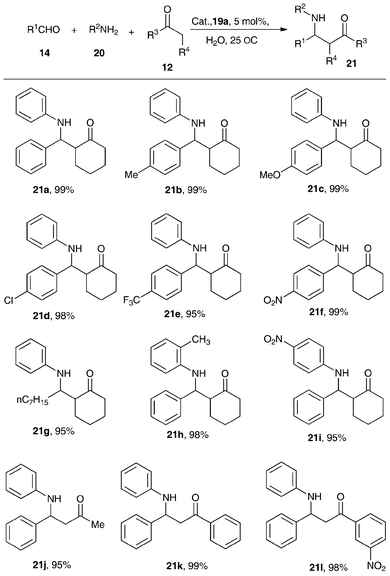
The performance of the air-stable N-bridged organobismuth Lewis acids 19a–19b was evaluated in Lewis acid-catalzyed reactions such as the Mannich reaction in water and allylation of aldehydes in aqueous media.46,47Illustrated in Scheme 8 are the excellent yields of β-amino ketones across the selected aldehydes including those that bear an electron-withdrawing group (21b–21d, yield 98–99%). The use of electron-rich aromatic aldehydes also leads to good yields of products (21e–21f). The aliphatic aldehyde also gives a good result (21g). However, a change of substituted groups in the phenyl plane of aromatic amines results in poor reaction rates (21h–21i). Among the three ketones 21j–21l, m-NO2PhCOCH3 exhibits higher activity than PhCOCH3 due to the electron-withdrawing ability of –NO2.
 | ||
| Scheme 9 Synthesis of homoallyl alcohols catalyzed by 19b.46 | ||
Shown in Table 5 are the catalytic activity of different Lewis acids in the Mannich reaction of benzaldehyde, aniline and cyclohexanone. Without the use of a catalyst (blank test), β-amino ketone yield is only 8% (entry 1). The β-amino ketone yield over the precursor C6H11N(CH2C6H4)2BiCl was 50% (entry 2),47 implying that after abstraction of Cl with the large electron-withdrawing OSO2C8F17 and B(C6F5)4 anions, there is significant increase of catalytic activity (entries 3–4, yield 92–95%).95 It is also superior to the inorganic bismuth compound Bi(OSO2CF3) (entry 5, yield 85%).90 Furthermore, compared with the catalytic activity of the metallocene complexes 2a–2b and 3a–3b in the reaction (entries 6–9, yield 90–99%),34 it is clear that with simple assembling of modified ligands, the bismuth catalysts performs almost as well as the catalysts that are based on transition metals.
| Entry | Catalyst (5 mol %) | Time (h) | Yield (%)b |
|---|---|---|---|
| a Benzaldehyde (1.0 mmol), aniline (1.0 mmol), cyclohexanone (1.0 mmol), cat. (0.05 mol), H2O (2.0 mL), 25 °C. b Isolated yield. | |||
| 1 | No cat. | 5 | 8 |
| 2 | C6H11N(CH2C6H4)2BiCl (5b) | 3 | 50 |
| 3 | C6H11N(CH2C6H4)2Bi(OSO2C8F17) (6a) | 2 | 95 |
| 4 | t BuN(CH2C6H4)2Bi+[B(C6F5)4]− | 2 | 92 |
| 5 | Bi(OTf)3 | 2 | 84 |
| 6 | [(CpZr(OH2)3)2(μ2-OH)2][C6F5SO3]4 (2a) | 2 | 98 |
| 7 | [(CpHf(OH2)3)2(μ2-OH)2][OSO2C8F17]4 (2b) | 2 | 90 |
| 8 | Cp2Zr(OSO2C8F17)2 (3a) | 2 | 99 |
| 9 | Cp2Ti(OSO2C8F17)2 (3b) | 2 | 99 |
To examine the reusability and applicability of the catalyst, the organobismuth Lewis acid 19a was subject to 10 cycles of the Mannich reaction. It was found that the change of product yield is minimal (isolated yield only slightly declined from 95% to 94%), indicating that the catalyst is stable and reusable.47
As shown in Scheme 9, with this N-bridged organobismuth Lewis acid catalytic system, both aromatic and aliphatic aldehydes proceed smoothly to generate the corresponding homoallyl alcohols in good to excellent yields (90%–97%). In addition, the aryl aldehydes with electron-withdrawing and electron-donating groups show similar allylation rates, suggesting that the presence of the electron-withdrawing or electron-donating group of the aldehydes has little effect on the reaction. Compared with the highly catalytic efficient catalysts of the metallocene Lewis acids 2a, 2b, 3a and 3b as shown in Table 4, after simple modification, the main group organobismuth Lewis acids shows the almost the same catalytic activity as that of metallocene Lewis acids based on transition metals, which also broadens the application scope of the organobismuth complexes.
In a test of five cycles, the change of product yield in the allylation reaction of benzaldehyde with tetraallyltin is minimal (isolated yield slightly declined from 96% to 94%), indicating that the catalyst 19b is also stable and reusable.
4 Air-stable S-bridged organobismuth Lewis acid/base bifunctional catalysts
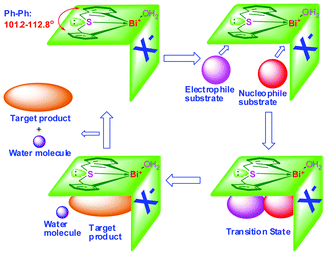
Generally, a single Lewis acid center is not good enough to achieve high stereoselectivity. In the field of asymmetric synthesis, there is a growing emphasis on bifunctional catalysts that have both “hard” Lewis acidity and “soft” Lewis basicity, on which both the electrophilic and nucleophilic partners can be activated simultaneously.96–98 However, recent reports disclosed that the bifunctional catalysts performed poorly in terms of catalytic activity and stereoselectivity due to self-quenching. Moreover, the catalysts with high catalytic activity were unstable in air or water, making storage and utilization of catalysts difficult. Hence, the development of low-cost bifunctional Lewis acid catalysts that do not suffer from self-quenching but show high catalytic activity and stereoselectivity becomes a key issue.With the incorporation of a butterfly-shaped sulfur-bridged ligand into the organobismuth complexes, we successfully synthesized bifunctional organobismuth Lewis acidic/basic complexes which showed high catalytic activity and diastereoselectivity in direct Mannich reactions as well as in the synthesis of α, β-unsaturated ketones.35–37,39
4.1 Synthesis of air-stable S-bridged organobismuth Lewis acid/base bifunctional catalysts
The synthesis of S-bridged organobismuth chloride (24) is the same as that of N-bridged organobismuth chloride (18a–18c): through treatment of the butterfly-shaped sulfur-bridged ligand and n-BuLi with BiCl3 in Et2O (Scheme 10). By incorporating the complex with a counter anion that shows high electron-withdrawing ability, four bifunctional Lewis acidic/basic catalysts (25a–25b) were obtained. These complexes contain a special butterfly-shaped sulfur-bridged ligand in which the sulfur atom has two lone pairs of electrons. One coordinates with the bismuth center while the other is free to function as a Lewis base.35–37,39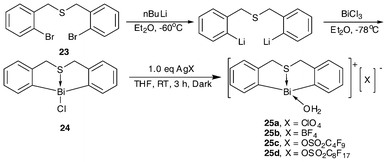 | ||
| Scheme 10 Synthetic routes of butterfly-shaped sulfur-bridged organobismuth complexes 24 and 25a–25d.35–37,39 | ||
4.2 Characterization of air-stable S-bridged organobismuth Lewis acid/base bifunctional catalysts
The samples 25a–25d freshly obtained from recrystallization contain one water molecule. They remained as dry colorless crystals or white powder in ambient environment in a test period of one year, showing good air-stability and water tolerance. The materials also show high thermal stability, especially complexes 25c and 25d with perfluoroalkylsulfonate counter anions (stable up to 230 °C).35–37,39Fig. 5 shows the ORTEP representations of complexes 24 and 25a–25d. It is apparent that the ligands are butterfly-shaped and sulfur-bridged. In the cases of complexes 25a–25d, the organobismuth components are cationic. The oxygen atom of the coordinating water occupies a vacant site of the cationic bismuth centre, making the coordination geometry distorted and equatorially vacant. With four atoms and one lone electron pair, each of them roughly adopts a pseudo-trigonal bipyramidal structure, where the sulfur and the oxygen atoms are at the apical positions and the two carbon atoms are at the equatorial positions. The dihedral angle of the two phenyl planes (ca. 101.2–112.8 degrees) is equal to the C(1)–Bi–C(14) angle (96.8–100.7 degrees), which is similar to the case of having a 1,1′-binaphthol template as the asymmetric catalyst ligand.29 The sulfur atom has two lone pairs of electrons: one coordinates with the bismuth center and the other possibly works as a Lewis base due to its electron-donating property.35–37,39
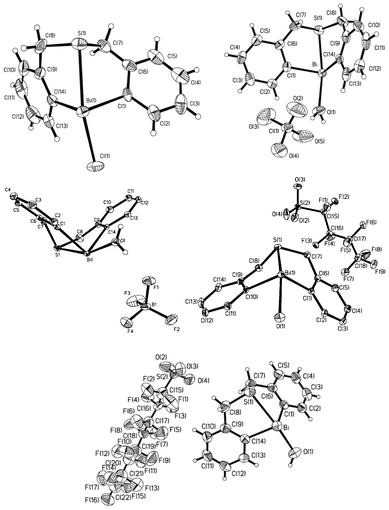 | ||
| Fig. 5 Crystal structures of complexes 24 (X = Cl, top left), 25a (X = ClO4, top right), 25b (X = BF4, middle left), 25c (X = OSO2C4F9, middle right), and 25d (X = OSO2C8F17, bottom). (Images reproduced from ref. 35 with permission of the Royal Society of Chemistry).35–37,39 | ||
To determine acidity and basicity, we also employed the Hammett indicators method.63–65 As shown in Fig. 6 (take 25a as an example),39 the color is changed during the determination process, clearly indicating the moderate acidity with acid strength of 4.8 < Ho ≤ 6.8 for 25a35 and 3.3 < Ho ≤ 4.8 for 25b–25d as well as the basic strength of 7.2 ≤ H− < 8.9 for 24 and 25a–25d.35–37,39 It is worth pointing out that complex 24 shows no acidity but basicity. Furthermore, despite the fact that Lewis acid/base pairs exist in complexes 25a–25d, there is no sign of self-quenching.35
 | ||
| Fig. 6 The upper layer is the indicators themselves in benzene solution, the bottom layer the indicator with the complex 25a in benzene solution (1. black; 2. dimethyl yellow; 3. methyl red; 4. neutral red; 5. bromothymol blue; 6. thymol blue). (Image reproduced from ref. 39 with permission of the Royal Society of Chemistry).39 | ||
4.3 Catalytic application of air-stable S-bridged organobismuth Lewis acid/base bifunctional catalysts
With the steric effect of the butterfly-shaped ligand structure and the presence of bismuth-sulfur bifunctional centers, it is envisaged that complexes 25a–25d are efficient and stereoselective catalysts. Their catalytic performance were examined for the direct Mannich reaction and cross-condensation reaction.35–37,39We used 25a as a catalyst for the Mannich reaction conducted in water (Table 6) and observed high efficiency and stereoselectivity.39 It is noted that despite the recent emphasis on using water as reaction medium, reports on Mannich reaction conducted in water are few.39,47,48,99,100 We also found that the S-bridged organobismuth 25a has the same chemoselectivity in this reaction as that of N-organobismuth complex 19a. For example, the aldehydes with an electron-withdrawing group in the phenyl plane exhibit higher reaction activity than the aldehydes with electron-donating groups (entries 1–5). However, the diastereoselectivity is totally different. Furthermore, the syn-selectivity was independent of the kind of solvents that were employed. The results implies that the strategy of changing from an nitrogen-bridged ligand to a sulfur-bridged ligand works well for making bifunctional Lewis acidic/basic catalysts. Using 25a as catalyst, high diastereoselectivities and high yields were observed in the Mannich reaction of aldehydes with unsaturated double bonds and the enolizable aliphatic aldehydes (entries 6–9). Excellent results were also observed in the two cases of amines, one with a –NO2 group in the para-position and the other with a methyl group in the ortho-position (entries 10–11). All the results indicate the general application potential of 25a.39
| Entry | R1CHO (14) | R2NH2 (20) | Product (21) | Time (h) | Yield (%) b | syn/antic |
|---|---|---|---|---|---|---|
| a R1CHO, 1.0 mmol; R2NH2, 1.0 mmol; cyclohexanone, 1.0 mmol; 25a, 0.05 mmol; 25 °C, H2O, 2.0 mL. b Isolated yield. c Determined by 1H NMR. d 0 °C. | ||||||
| 1 | PhCHO | PhNH2 | 21a | 2 | 98 | 5/95 |
| 2 | p-CH3C6H4CHO | PhNH2 | 21b | 5 | 95 | 7/93 |
| 3 | p-CH3OC6H4CHO | PhNH2 | 21c | 8 | 93 | 4/96 |
| 4 | p-ClC6H4CHO | PhNH2 | 21d | 2 | 98 | 6/94 |
| 5 | p-CF3C6H4CHO | PhNH2 | 21e | 1 | 98 | 4/96 |
| 6d | Cinnamaldehyde | PhNH2 | 21m | 12 | 90 | 15/85 |
| 7d | Furfural aldehyde | PhNH2 | 21n | 12 | 95 | 20/80 |
| 8 | PhCH2CH2CHO | PhNH2 | 21o | 8 | 92 | 9/91 |
| 9 | n-C7H15CHO | PhNH2 | 21g | 8 | 98 | 12/88 |
| 10 | PhCHO | o-CH3C6H4NH2 | 21h | 36 | 98 | 6/94 |
| 11 | PhCHO | p-O2NC6H4NH2 | 21i | 12 | 98 | 5/95 |
In addition, catalyst 25a is superior to its precursor S(CH2C6H4)2BiCl (24), the cationic organobismuth [tBuN(CH2C6H4)2Bi]+[B(C6F5)4]−, and the inorganic bismuth complex Bi(OSO2CF3)3 in catalytic activity and diastereoselectivity (Table 7). Moreover, it was observed that 25a is stable and suitable for reuse.39
| Entry | Cat. | Yield (%) b | syn/antic |
|---|---|---|---|
| a PhCHO, 1.0 mmol; PhNH2, 1.0 mmol; cyclohexanone, 1.0 mmol; cat., 0.05 mmol; 25 °C, H2O, 2.0 mL. b Isolated yield. c Determined by 1H NMR. | |||
| 1 | [S(CH2C6H4)2Bi(OH2)]+[ClO4]− (25a) | 98 | 5/95 |
| 2 | S(CH2C6H4)2BiCl (24) | 20 | 43/57 |
| 3 | [tBuN(CH2C6H4)2Bi]+ [B(C6F5)4]− | 92 | 36/64 |
| 4 | Bi(OSO2CF3)3 | 84 | 14/86 |
In this reaction, the catalyst system shows high efficiency when water is used as solvent (Table 8, entry 1, yield, 95%, E/Z > 99:1). It is the same as that of the Mannich reaction39 during solvent optimization, we also found that the E-selectivity was independent of the kind of solvents that were employed.37 Furthermore, it is clear that the presence of primary amine is indispensable for the occurrence of the reaction.37 To test the versatility of the catalytic system, various aromatic aldehydes with electron-donating and electron-withdrawing groups as well as enolizable aliphatic aldehydes were employed and the desired products were obtained in high yields (Table 8). It should be noted that in the two cases of enolizable aliphatic aldehydes, (E)-α,β-unsaturated ketones are selectively produced in quantitative yields without the formation of aldehyde self-condensation products or any other side-products (entries 7–8).37
| Entry | RCHO | Product (26/E) | Yield (%) b | E/Zc |
|---|---|---|---|---|
| a RCHO, 1.0 mmol; nPrNH2, 1.0 mmol; cyclohexanone, 1.2 mmol; 25d, 0.02 mmol; H2O, 2.0 mL; RT, 3 h. b Isolated yield. c Determined by 1H NMR. d 0 °C. | ||||
| 1 | PhCHO | 26a | 95 | >99/1 |
| 2 | p-CH3C6H4CHO | 26b | 90 | 97/3 |
| 3 | p-CH3OC6H4CHO | 26c | 88 | 97/3 |
| 4 | p-ClC6H4CHO | 26d | 96 | 98/2 |
| 5 | p-CF3C6H4CHO | 26f | 99 | 99/1 |
| 6 d | (E)-Ph–CHCHCHO |
26g | 83 | 93/7 |
| 7 | PhCH2CH2CHO | 26h | 97 | 99/1 |
| 8 | nC7H15CHO | 26i | 99 | 99/1 |
Additionally, in the cases of cyclic and acyclic ketones, (E)-α,β-unsaturated ketones are produced selectively in high yields (Table 9, entries 1–4); the active methylene compounds are efficient substrates as well. Using the adopted method, we obtained almost quantitative yields of the desired products when pentane-2,4-dione, dimethyl and diethyl malonates were used (Table 9, entries 5–7). Moreover, the catalyst is very stable and suitable for reuse.
| Entry | Ketone | Product (E) (26) | Yield (%) b | E/Zc |
|---|---|---|---|---|
| a PhCHO, 1.0 mmol; nPrNH2, 1.0 mmol; ketone, 1.2 mmol; 25d, 0.02 mmol; H2O, 2.0 mL; RT, 3 h. b Isolated yield. c Determined by 1H NMR. d 0 °C to RT. e 24 h | ||||
| 1 |
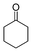
|
 (26a) (26a) |
95 | >99/1 |
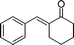
| 2 |

|
 (26j) (26j) |
97 | >99/1 |
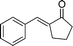
| 3d |
|
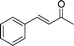 (26k) (26k) |
96 | >99/1 |
| 4e |

|
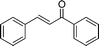 (26l) (26l) |
84 | >99/1 |
| 5 |
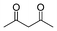
|
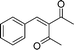 (26m) (26m) |
96 | — |
| 6 |
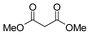
|
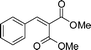 (26n) (26n) |
95 | — |
| 7 |
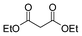
|
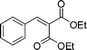 (26o) (26o) |
94 | — |
The reaction probably takes place through a Mannich-type mechanism as shown in Scheme 11.37 It can be deduced that the final product is formed through stereospecific syn-elimination from the intermediate Mannich adduct, as shown in (I). Because the E/Z-selectivity of the current reaction corresponds well to the anti/syn-selectivity of the Mannich reaction when PhNH2 was used rather than n-PrNH2,37,39 it is strongly believed that the change of amines does not affect the diastereoselectivity of the Mannich adducts. Although the structures of the cationic parts of catalysts 25a (and 25d) and [tBuN(CH2C6H4)2Bi]+[B(C6F5)4]−95 are similar, the stereoselectivity of the reaction is rather different, suggesting the importance of the sulfur atom in 25d. As mentioned above, the sulfur atom in 25d can act as a weak Lewis base, and the high stereoselectivity may be explained if the addition reaction step proceeds through a transition state like (II), in which both the Lewis acid and the Lewis base parts of 25d act simultaneously to gather two reacting substrates in a chair-type cyclohexane arrangement with less steric repulsion.37,39
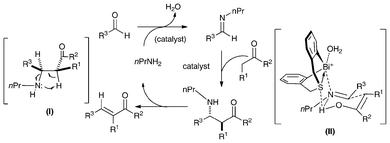 | ||
| Scheme 11 A plausible catalytic cycle for the crossed-condensation reaction of ketones and aldehydes catalyzed by 25d in the presence of n-PrNH2.37 | ||
:0). During the course of reaction, the nature of catalysis switches from homogeneous to heterogeneous (Fig. 7).
![The cross-condensation reaction of benzaldehyde with cyclohexanone over organobismuth complex 25b in the presence of propylamine in [Bmim]BF4: facts and model. (a) At the begin of reaction, the bottom layer is [Bmim]BF4 and catalyst 25b; (b) homogeneous mixture during reaction; (c) the reaction system becomes heterogeneous at completion of reaction: the upper layer is composed of the product (α,β-unsaturated ketones) and unconsumed reactants while the lower layer [Bmim]BF4, complex 25b, and water generated in the reaction. (Image reproduced from ref. 36 with permission of the Royal Society of Chemistry).36](/image/article/2012/RA/c2ra21517a/c2ra21517a-f7.gif) | ||
| Fig. 7 The cross-condensation reaction of benzaldehyde with cyclohexanone over organobismuth complex 25b in the presence of propylamine in [Bmim]BF4: facts and model. (a) At the begin of reaction, the bottom layer is [Bmim]BF4 and catalyst 25b; (b) homogeneous mixture during reaction; (c) the reaction system becomes heterogeneous at completion of reaction: the upper layer is composed of the product (α,β-unsaturated ketones) and unconsumed reactants while the lower layer [Bmim]BF4, complex 25b, and water generated in the reaction. (Image reproduced from ref. 36 with permission of the Royal Society of Chemistry).36 | ||
In this catalytic system, the overall reaction occurs at room temperature and a change in reaction conditions is not needed.106 Moreover, the prominent feature is its excellent solubility in water or polar solvents but immiscibility in apolar α,β-unsaturated ketones. Interactions such as hydrogen bonding and the special phenyl planar geometry (Scheme 12) may also play an important role in this self-separating catalytic system.36 It was observed that the 1H NMR singlet of water coordinated to the Bi center, and those of the methyl and methylene group linked to the nitrogen atom of [Bmim]BF4 shift to high field. The NMR results indicate that there is apparent hydrogen bonding; with the enhancement of electron-withdrawing ability, the diastereoselectivity is hence promoted.36,37 So, at the beginning before adduct formation (between the Bi complex and ILs), benzaldehyde, cyclohexanone, propylamine, complex 25b and [Bmim]BF4 distributed themselves into two phases (Fig. 7a). With adduct formation from the Bi complex and ILs, the catalyst becomes miscible in the ILs, and the system becomes homogeneous (Fig. 7b). At the end of the reaction, with the consumption of reactants, the water generated is absorbed by the hydrophilic ILs, inducing stronger polarity of ILs which is beneficial for the facile separation process, and the system becomes turbid. After 5 min of settling, there is a spontaneous separation of the catalyst system (complex 25b and [Bmim]BF4) and product (Fig. 7c). The upper layer consists of the product and unconsumed reactants while the lower layer consists of [Bmim]BF4, complex 25b, and water (the only side product). In other words, the product can be transferred to the apolar organic phase directly and efficiently, breaking the equilibrium of cross-condensation reaction in a controlled manner.36,37 Eventually, the catalyst system can be easily recovered by simple decantation. It is apparent that the advantages of both homogeneous and heterogeneous catalysis are captured in this method.106
![Proposed interaction of complex 25b and imidazolium cationic ion [Bmim]+ in ionic liquids. (Image reproduced from ref. 35 with permission of the Royal Society of Chemistry).35](/image/article/2012/RA/c2ra21517a/c2ra21517a-s12.gif) | ||
| Scheme 12 Proposed interaction of complex 25b and imidazolium cationic ion [Bmim]+ in ionic liquids. (Image reproduced from ref. 35 with permission of the Royal Society of Chemistry).35 | ||
Furthermore, this catalyst system can be applied to enolizable aliphatic aldehydes as well as to aromatic aldehydes with electron-donating and electron-withdrawing groups (Scheme 13), and there is facile separation as well as high yields and diastereoselectivity.36 The E-selectivity for furfural is consistent with those of the other aldehydes at 0 °C. (E)-α,β-Unsaturated ketones are selectively produced in quantitative yields in the cases of enolizable aliphatic aldehydes, showing no sight of side-products such as those due to aldehyde condensation. The active methylene compounds appear to be efficient substrates in this facile separation catalytic system.
![Synthesis of different α,β-unsaturated ketones catalyzed by cationic organobismuth complex 25b in [Bmim]BF4.36](/image/article/2012/RA/c2ra21517a/c2ra21517a-s13.gif) | ||
| Scheme 13 Synthesis of different α,β-unsaturated ketones catalyzed by cationic organobismuth complex 25b in [Bmim]BF4.36 | ||
In a scale-up (×5) experiment, we found that catalyst loading can be lowered to 0.1 mol% with the facile separation of the catalyst system almost unaffected. Furthermore, the catalyst system can be recycled at least ten times without significant decline in product yield. It should be noted that the total substrate molar ratio (PhCHO:n-PrNH2:cyclohexanone) for ten cycles is 1.0:0.19:1.2, and the TON is up to 9893.36
5 Air-stable N-bridged organoantimony Lewis acids
Stephen and Erker reported that a FLP is a compound or mixture containing a Lewis acid and a Lewis base that, because of steric hindrance, cannot combine to form an adduct.19 Because of steric hindrance, the distance between the basic site and the acidic site is elongated to keep the “unquenched” reactivity. It was envisaged that an effect similar to that of steric hindrance can be induced by weakening the interaction between the acidic site and the basic site. By changing the metal atom from bismuth to antimony (i.e. reducing the electronic deficient ability of metal atom), then fixing the Lewis acidic site as well as the Lewis basic site in the 5,6,7,12-tetrahydrodibenz[c,f][1,5]azostibocine framework, we successfully synthesized the organoantimony Lewis acidic/basic bifunctional catalysts. In such cases, the lone pair electrons of the nitrogen atom can coordinate with the antimony atom to form a stable N–Sb bond as well as functioning as a Lewis basic site for bifunctional catalysis.485.1 Synthesis of air-stable N-bridged organoantimony Lewis acids
As shown in Scheme 14, the synthetic route of organoantimony chlorides 27a–27c is similar to that of organobismuth chlorides 18a–18c.48,91 Using the N-bridged pincer ligand and with the formation of a C–Sb metal bond and a N–Sb coordination bond, one has the air-stable 5,6,7,12-tetrahydrodibenzo[c,f][1,5]azastibocine skeleton that contains a methylene bridge.48 There is a certain degree of flexibility that helps to reduce the inner tension of the complex. The results of NMR analysis showed that the N–Sb coordination bond strength can be modulated by proper selection of the N-bridged pincer ligand. In so doing, the Lewis acidic and Lewis basic conditions of the metal center can be precisely modulated.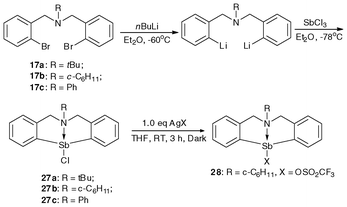 | ||
| Scheme 14 Synthetic routes of air-stable organoantimony Lewis acids.48 | ||
Treatment of organoantimony halide 27b with AgOSO2CF3 afforded the generation of air-stable organoantimony complexes 28. NMR analysis shows an upfield move of proton chemical shift owing to the enhanced N–Sb bond strength resulting from the large electronic-withdrawing ability of OSO2CF3 anion. The upfield shift is a result of N–Sb bond shortening as well as the better shielding effect of the nitrogen lone electron pair over Sb.
5.2 Characterization of air-stable N-bridged organoantimony Lewis acids
Fig. 9 shows the ORTEP view of the structure of complexes 27b and 28. The central antimony-containing part of the complex exhibits a pseudo-trigonal bipyramidal structure.48 The Lewis acidity of 28 and 27b is 3.3 < Ho ≤ 4.8 and 4.8 < Ho ≤ 6.8), respectively. However, the expected Lewis basicity is not observed, plausibly due to the fact that the lone electron pair of the nitrogen atom is partially coordinated with the antimony, and there is restriction in its delocalization.48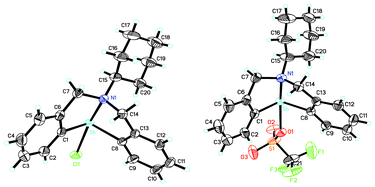 | ||
| Fig. 9 ORTEP view of the crystal structure of (C6H11N(C6H4CH2)2Sb(X))(X = Cl (27b, left); OSO2CF3 (28, right)). (Image reproduced from ref. 48 with permission of Elsevier).48 | ||
After an exposure period of six months in the open air, 27a–27c and 28 remained as dry colorless crystals or white powder. Therefore, these organoantimony compounds are air-stable. What is more, these two organoantimony complexes are thermally stable up to 300 °C (Fig. 10).48
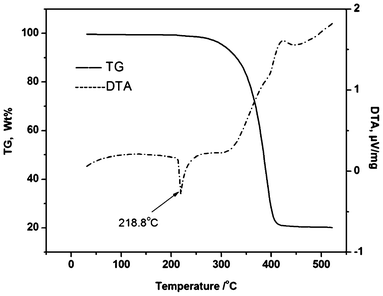 | ||
| Fig. 10 TG-DSC curves of organoantimony Lewis acid (28). (Image reproduced from ref. 48 with permission of Elsevier).48 | ||
5.3 Catalytic application of air-stable N-bridged organoantimony Lewis acids
In our previous study, we synthesized a cationic organobismuth perchlorate complex ([S(CH2C6H4)2Bi(OH2)]+[ClO4]−) that is catalytically highly active, giving anti/syn molar ratio ≤ 95:5 in the direct diastereoselective Mannich reaction.39 It was observed that there was no diastereoselectivity over covalent organobismuth (III) perfluorooctane sulfonate (C6H11N(CH2C6H4)2 Bi(OSO2C8F17)) which showed high catalytic efficiency towards the Mannich-type reactions.47 However, with the same ligand but just changing the metal center from the bismuth to antimony, high catalytic efficiency and selectivity (anti/syn = 99/1, yield, 98%) was observed when the organoantimony complex 28 was adopted as a catalyst for the Mannich reaction of benzaldehyde, aniline, and cyclohexanone in water.[48,107] The results show that our design strategy works well in this catalytic system. Various electron-withdrawing and electron-donating aromatic aldehydes were tested, with high yield and diastereoselectivity being observed across the aldehydes (Table 10). It was also observed that in the cases of acetophenones, the corresponding β-amino ketones were obtained in good yields (Table 10, entries 7–11, 65–81%).
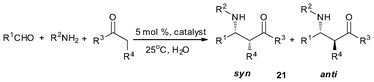
| Entry | R1 | R2 | R3, R4 | Product (21) | Time (h) | anti/synb | Yield (%)c |
|---|---|---|---|---|---|---|---|
| a R1CHO, 1.0 mmol; R2NH2, 1.0 mmol; cyclohexanone, 1.0 mmol; cat., 0.05 mmol; H2O, 2.0 mL, 25 °C. b Determined by 1H NMR. c Isolated yield. | |||||||
| 1 | Ph | Ph | –(CH2)4– | 21a | 4 | 99:1 |
98 |
| 2 | p-CH3C6H4 | Ph | –(CH2)4– | 21b | 5 | 96:4 |
90 |
| 3 | p-CH3OC6H4 | Ph | –(CH2)4– | 21c | 4 | 97:3 |
92 |
| 4 | p-ClC6H4 | Ph | –(CH2)4– | 21d | 4 | 96:4 |
97 |
| 5 | p-CF3C6H4 | Ph | –CH2)4– | 21e | 3 | 97:3 |
95 |
| 6 | Ph | o-CH3C6H4 | –(CH2)4– | 21h | 10 | 95:5 |
90 |
| 7 | Ph | Ph | Ph, H | 21k | 24 | – | 65 |
| 8 | p-CH3C6H4 | Ph | Ph, H | 21p | 24 | – | 81 |
| 9 | p-CH3OC6H4 | Ph | Ph, H | 21q | 24 | – | 78 |
| 10 | p-ClC6H4 | Ph | Ph, H | 21r | 24 | – | 72 |
| 11 | p-CF3C6H4 | Ph | Ph, H | 21s | 24 | – | 70 |
To demonstrate the superiority of the air-stable organoantimony Lewis acid 28, its catalytic performance is compared with that of precursor 27b and antimony trichloride as well as to those of the other air-stable organometallic Lewis acids developed by us and mentioned in this mini-review article. As shown in Table 11, catalyst 28 is superior to the others in catalytic efficiency as well as in diastereoselectivity. Product of single configuration with nearly 100% anti-selectivity was obtained in the case of complex 28, in distinct contrast to the case of complex 25a (anti/syn = 95:5). Also high diastereoselectivity was observed over precursor C6H11N(C6H4CH2)2SbCl (27b). The results imply that there is a correlation between catalyst frameworks and diastereoselectivity to Mannich products. In addition, complex 28 shows good reusability and reproducibility.48
| Entry | Catalyst | Time (h) | Anti/synb | Yield (%)c |
|---|---|---|---|---|
| a PhCHO, 1.0 mmol; PhNH2, 1.0 mmol; cyclohexanone, 1.0 mmol; Cat., 0.05 mmol; H2O (for SbCl3, the solvent is CH3CN), 2.0 mL, 25 °C. b Determined by 1H NMR. c Isolated yield. | ||||
| 1 | No catalyst | 2 | 45:55 |
8 |
| 2 | C6H11N(C6H4CH2)2SbOSO2CF3 (28) | 4 | 99:1 |
98 |
| 3 | C6H11N(C6H4CH2)2SbCl (27b) | 5 | 96:04 |
45 |
| 4 | SbCl3 | 8 | 68:32 |
78 |
| 5 | S(CH2C6H4)2BiCl (24) | 2 | 57:43 |
20 |
| 6 | C6H11N(CH2C6H4)2BiCl(18b) | 2 | 65:35 |
50 |
| 7 | [(CpZr(OH2)3)2(μ2-OH)2][C6F5SO3]4 (2a) | 2 | 72:28 |
98 |
| 8 | [(CpHf(OH2)3)2(μ2-OH)2][OSO2C8F17]4 (2b) | 2 | 76:24 |
90 |
| 9 | Cp2Zr(OSO2C8F17)2 (3a) | 2 | 82:18 |
99 |
| 10 | Cp2Ti(OSO2C8F17)2 (3b) | 2 | 80:20 |
99 |
| 11 | C6H11N(CH2C6H4)2Bi(OSO2C8F17) (19a) | 2 | 78:22 |
95 |
| 12 | [S(CH2C6H4)2Bi(OH2)]+[ClO4]− (25a) | 2 | 95:5 |
98 |
| 13 | [S(CH2C6H4)2Bi(OH2)]+[BF4]− (25b) | 2 | 90:10 |
90 |
| 14 | [S(CH2C6H4)2Bi(OH2)]+[OSO2C4F9]− (25c) | 2 | 99:1 |
98 |
| 15 | [S(CH2C6H4)2Bi(OH2)]+[OSO2C8F17]− (25d) | 2 | >99:1 |
99 |
6 Air-stable binuclear N-bridged organobismuth Lewis acids
Bimetallic catalysis is commonly observed in metalloenzyme activities.108 To achieve cooperative action similar to that of metalloenzymes, the two metal atoms have to be suitably arranged in ligand systems. To the best of our knowledge, reports on the utilization of binuclear organobismuth complexes as catalysts for the CO2 transformation to useful chemicals are scant. This could be partly due to (i) the instability of the Bi–C/Bi bond or the Bi–E–Bi (E = O, S, Br, etc.) bridge,95,109 (ii) weak Lewis acidity of the poorly exposed bismuth centre, and (iii) the fact that cooperative action of the two metal atoms cannot be successfully achieved due to poor design of the ligand system.83,84,110The first example of air-stable binuclear organobismuth sulfides being used as the catalysts in CO2 transformations with the two bismuth atoms exhibiting effective cooperative action was reported by us.33
6.1 Synthesis of air-stable binuclear N-bridged organobismuth Lewis acids
The complexes 29d–29f were quantitatively prepared through treatment of the 5,6,7,12-tetrahydrodibenz[c,f][1,5]aza-bismocine chloride derivatives (18a–18c)47 with sodium sulfide and sodium hydroxide,109 respectively (Scheme 15). In fact, the procedure for generating the complexes is similar to that of binuclear organobismuth oxides (29a–29c),109 The binuclear organobismuth oxides 29a–29c are moisture as well as CO2 sensitive, and the rigorous manipulation requirement limits their utilization in catalysis.109 Interestingly, when the bridging atom is changed from oxygen to sulfur, the binuclear organobismuth sulfides LBi–S–BiL (29d–29f) are air-stable and the compounds could remain as dry crystals or powder (slightly yellow in color) for more than one year in ambient environment.33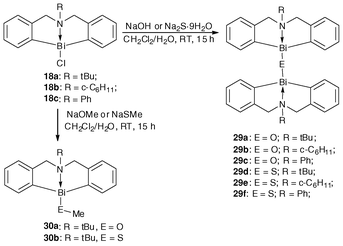 | ||
| Scheme 15 Synthetic routes of organobismuth complexes 29a–29f and 30a–30b.33,47,109 | ||
6.2 Characterization of air-stable binuclear N-bridged organobismuth Lewis acids
The crystal structures of 29d–29f freshly obtained after recrystallization in toluene/hexane (CH2Cl2/hexane for 29d) were determined by X-ray analysis.33 As shown in Fig. 11, the 5,6,7,12-tetrahydrodibenz[c,f][1,5]azabismocine framework of complexes 29d–29f is bridged by a sulfur atom, which occupies a vacant site of the bismuth centre; in the equatorially distorted trigonal bipyramidal geometry, nitrogen and sulfur atoms are in the apical positions and carbon atoms are in the equatorial positions. With sulfur bridging, the bismuth centre is exposed more efficiently than that of their precursors, making the two bismuth atoms of 29d–29f closer to each other in comparison to the case of 29a. Nonetheless, the distance between the two bismuth atoms of 29d–29f is larger than that of a covalent single Bi–Bi bond (3.092(6)–3.2092(8))111 and BiBi double bond (3.0648(2)),112 possibly a favorable parameter for maintaining suitable stability of the bimetallic complexes. In addition, the five linked atoms N(1)–Bi(1)–S(1)–Bi(1A)–N(1A) are almost in the same plane (distortion angle < 0.5°) bisecting the butterfly-shaped tetrahydrodibenz[c,f]-[1,5]azabismocine framework, where the four butterfly-shaped phenyl planes of 29d–29f are cis-geometrically located to make the two bismuth atoms available for cooperative action. The N-substituents of 29d–29f are positioned close to the bismuth centres, protecting the Bi–C bond and Bi–S–Bi bridge from infringers such as water and CO2. Hence, by using a sulfur atom for bridging and varying the N-substituent, one can regulate the state suitability, as well as the ligand system, of the bismuth centres to mimic those of the natural metalloenzymes.
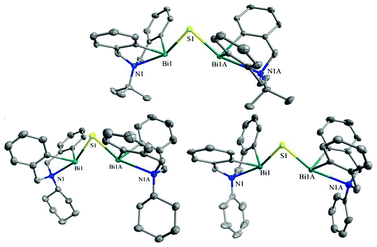 | ||
| Fig. 11 Thermal ellipsoid plots (50% probability level) of 29d (top), 29e (bottom left), 29f (bottom right). Hydrogen atoms on the carbon atoms are omitted for clarity. (Image reproduced from ref. 33 with permission of Wiley-VCH).33 | ||
6.3 Catalytic application of air-stable binuclear N-bridged organobismuth Lewis acids
To examining the cooperative action of the binuclear bismuth centres, complexes 29a–29f were examined for the synthesis of cyclic carbonates from epoxides and CO2 (Table 12), and their performances were compared with those of mononuclear organobismuth complexes 18a–18c and 30a–30b. Most of them showed good catalytic efficiency (entries 1–6, yield up to 100%, selectivity up to 100%). In terms of the catalytic efficiency and air-stability, an N-substituent with a flexible and electron-donating group (tBu, 29d; Cy, 29e) is superior to that with a rigid and electron-withdrawing group (Ph, 29f) (entries 4–6) and organobismuth oxides (29a–29c) (entries 1–3), respectively. The results indicate that through simple modification of the N-substituent and bridging atom in the main framework of the bimetallic organobismuth complexes, one can tune the catalytic activity and air-stability. In contrast to the mononuclear organobismuth complexes (Table 12, entries 7–11), it can be clearly deduced that the binuclear organobismuth complexes show significant cooperative effect on CO2 transformation. In contrast, without catalyst, no reaction take place (entry 12).33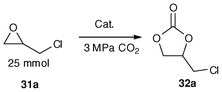
| Entry | Cat. (mol%) | Time (h) | Temp. (°C) | Yield (%) | Sel. (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 2-(chloromethyl)oxirane, 2 mL (25 mmol); initial CO2 pressure, 3.0 MPa, GC yield. b Without catalyst. | |||||
| 1 | 29a (0.5) | 8 | 140 | 99.6 | 99.8 |
| 2 | 29b (0.5) | 8 | 140 | 100 | 100.0 |
| 3 | 29c (0.5) | 8 | 140 | 97.3 | 98.1 |
| 4 | 29d (0.5) | 8 | 140 | 99.4 | 99.7 |
| 5 | 29e (0.5) | 8 | 140 | 94.3 | 98.3 |
| 6 | 29f (0.5) | 8 | 140 | 65.8 | 98.0 |
| 7 | 18a (1.0) | 8 | 140 | 25.9 | 97.7 |
| 8 | 18b (1.0) | 8 | 140 | 41.3 | 98.3 |
| 9 | 18c (1.0) | 8 | 140 | 5.9 | 96.9 |
| 10 | 30a (1.0) | 8 | 140 | 32.5 | 98.3 |
| 11 | 30b (1.0) | 8 | 140 | 14.7 | 92.6 |
| 12b | – | 8 | 140 | 3.7 | 98.2 |
The catalytic performance of 29d was also investigated under the optimized conditions across a diverse range of epoxides (Table 13). In most of the cases, including large steric 2-phenyloxirane and 7-oxabicyclo[4.1.0]heptane, the catalyst is highly effective (entries 1–6). The co-presence of Bu4NI is favorable for CO2 insertion into the non-halide epoxides (entries 2 and 7). Based on the X-ray structures of 29d–29f, the role of the binuclear organobismuth complexes was proposed to be similar to that of the bimetallic salen(Al) complex reported by North and Pasquale.113 Further mechanism research work is being conducted in our laboratory.
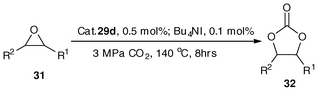
| Entry | R1 | R2 | Product (32) | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 2-(chloromethyl)oxirane, 2 mL; initial CO2 pressure 3.0 MPa, cat. 29d, 0.5 mol%, 8 h, 140 °C. b Without co-catalyst Bu4NI. c Catalyst loading of 29d was 1.0 mol%. d 12 h. | |||||
| 1 | H | H | 32b | 100.0 | 98.2 |
| 2 | CH3 | H | 32c | 99.7 | 99.7 |
| 3b | CH2Cl | H | 32a | 100.0 | 99.8 |
| 4c | CH3 | CH3 | 32d | 76.2 | 76.0 |
| 5d | Ph | H | 32e | 99.5 | 99.5 |
| 6c | –CH2(CH2)2CH2– | 62.3 | 32f | 62.1 | 99.3 |
| 7b | CH3 | H | 32c | 2.3 | 2.3 |
7. Conclusion
In view of the shortcomings of organometallic Lewis acids in their applications as catalysts in organic synthesis, a series of air-stable, water-tolerant organometallic Lewis acid catalysts were synthesized. In view of the special features of metallocene, organobismuth and organoantimony, the adopted strategy is to construct a stable C–M bond and to incorporate electron-withdrawing groups of larger size. Moreover, their applications in carbon–heteroatom and carbon–carbon bond formation were surveyed. Some innovative concepts can be drawn:1) Through the incorporation of longer perfluoroalkanesulfonate groups, metallocene complexes of higher Lewis acidity (catalytic activity) and air-stability can be obtained. The results suggest that the Lewis acidity and air-stability of an organometallic Lewis acid catalyst can be enhanced with the introduction of a long chain of perfluoroalkane that is hydrophobic.
2) By constructing an air-stable carbon–metal (bismuth and antimony) bond and adjusting the coordination atom (N, S), as well as counter anion in organobismuth/organoantimony complexes, significant changes of the Lewis acidity/Lewis basicity as well as catalytic activity and air-stability can be achieved. By bridging two main frameworks of organobismuth together, one can gain significant cooperative catalysis ability.
Acknowledgements
This work was supported by the NSF of Hunan Province (10JJ1003), the NSFC (Grant Nos. 21003040, 20973056), the program for New Century Excellent Talents in Universities (NCET-10-0371), and the Fundamental Research Funds for the Central Universities. Dr C.T. Au thanks the Hunan University for an adjunct professorship. Dr S.F. Yin thanks Dr S. Shimada of AIST (Japan) and Prof. W.-Y. Wong of Hong Kong Baptist University for helpful discussions. Dr R.H. Qiu thanks Prof. N. Kambe and Dr T. Iwasaki of Osaka University for helpful discussions. All authors thank Professors J. Otera and A. Orita of Okayama University of Science for helpful discussions.References
- H. Yamamoto, ed., Lewis acid in organic synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- C. J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307–4365 CrossRef CAS.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3665 CrossRef CAS.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS.
- W. B. Jensen, Chem. Rev., 1978, 78, 1–22 CrossRef CAS.
- G. K. S. Prakash, T. Mathew and G. A. Olah, Acc. Chem. Res., 2012, 45, 565–577 CrossRef CAS.
- R. H. Qiu, G. P. Zhang, X. H. Xu, K. B. Zou, L. L. Shao, D. W. Fang, Y. H. Li, A. Orita, R. Saijo, H. Mineyama, T. Suenobu, S. Fukuzumi, D. L. An and J. Otera, J. Organomet. Chem., 2009, 694, 1524–1528 CrossRef CAS.
- G. N. Lewis, Valence and the Structure of Atoms and Molecules 1923 Search PubMed.
- R. H. Petrucci, W. S. Harwood and F. G. Herring, General Chemistry, Prentice-Hall 2002 Search PubMed.
- G. L. Miessler and D. A. Tarr, Inorganic Chemistry, Prentice-Hall 1998 Search PubMed.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- H. Yamamoto, Lewis acid reagents: a practical approach, Oxford University Press, New York, 1999 Search PubMed.
- M. Benkhaled, S. Morin, C. Pichon, C. Thomazeau, C. Verdon and D. Uzio, Appl. Catal., A, 2006, 312, 1–11 CrossRef CAS.
- Y. H. Yang, F. Diederich and J. S. Valentine, J. Am. Chem. Soc., 1991, 113, 7195–7205 CrossRef CAS.
- H. Yamamoto, P. Jpn Acad. B-Phys., 2008, 84, 134–146 CrossRef CAS.
- H. Yamamoto and S. Saito, Pure Appl. Chem., 1999, 71, 239–245 CrossRef CAS.
- X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS.
- S. S. Kim, Pure Appl. Chem., 2006, 78, 977–983 CrossRef CAS.
- F. Jakle, in, ACS Symp. Ser., 2002, 104–117 CrossRef CAS.
- S. Singh and H. W. Roesky, J. Fluorine Chem., 2007, 128, 369–377 CrossRef CAS.
- R. B. Kargbo and G. R. Cook, Curr. Org. Chem., 2007, 11, 1287–1309 CrossRef CAS.
- C. Ogawa and S. Kobayashi, Curr. Org. Synth., 2011, 8, 345–355 CrossRef CAS.
- S. T. Liddle, I. S. Edworthy and P. L. Arnold, Chem. Soc. Rev., 2007, 36, 1732–1744 RSC.
- S. Kobayashi, M. Ueno, S. Saito, Y. Mizuki, H. Ishitani and Y. Yamashita, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5476–5481 CrossRef CAS.
- M. Ueno, H. Ishitani and S. Kobayashi, Org. Lett., 2002, 4, 3395–3397 CrossRef CAS.
- K. Saruhashi and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 11232–11235 CrossRef CAS.
- M. Shibasaki and S. Matsunaga, Chem. Soc. Rev., 2006, 35, 269–279 RSC.
- J. Boerner, U. Floerke, K. Huber, A. Doering, D. Kuckling and S. Herres-Pawlis, Chem.–Eur. J., 2009, 15, 2362–2376 CrossRef CAS.
- K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527–538 CrossRef CAS.
- K. Ishihara, N. Hanaki, M. Funahashi, M. Miyata and H. Yamamoto, Bull. Chem. Soc. Jpn., 1995, 68, 1721–1730 CrossRef CAS.
- R. Qiu, Z. Meng, S. Yin, X. Song, N. Tan, Y. Zhou, K. Yu, X. Xu, S. Luo, C.-T. Au and W.-Y. Wong, ChemPlusChem, 2012, 77, 404–410 CrossRef CAS.
- R. Qiu, X. Xu, L. Peng, Y. Zhao, N. Li and S. Yin, Chem.–Eur. J., 2012, 18, 6172–6182 CrossRef CAS.
- R. Qiu, S. Yin, X. Song, Z. Meng, Y. Qiu, N. Tan, X. Xu, S. Luo, F.-R. Dai, C.-T. Au and W.-Y. Wong, Dalton Trans., 2011, 40, 9482–9489 RSC.
- R. H. Qiu, Y. M. Qiu, S. F. Yin, X. X. Song, Z. G. Meng, X. H. Xu, X. W. Zhang, S. L. Luo, C. T. Au and W. Y. Wong, Green Chem., 2010, 12, 1767–1771 RSC.
- R. H. Qiu, Y. M. Qiu, S. F. Yin, X. H. Xu, S. L. Luo, C. T. Au, W. Y. Wong and S. Shimada, Adv. Synth. Catal., 2010, 352, 153–162 CrossRef CAS.
- R. H. Qiu, X. H. Xu, Y. H. Li, G. P. Zhang, L. L. Shao, D. L. An and S. F. Yin, Chem. Commun., 2009, 1679–1681 RSC.
- R. H. Qiu, S. F. Yin, X. W. Zhang, J. Xia, X. H. Xu and S. L. Luo, Chem. Commun., 2009, 4759–4761 RSC.
- R. H. Qiu, G. P. Zhang, Y. Y. Zhu, X. H. Xu, L. L. Shao, Y. H. Li, D. L. An and S. F. Yin, Chem.–Eur. J., 2009, 15, 6488–6494 CrossRef CAS.
- R. H. Qiu, Y. Y. Zhu, X. H. Xu, Y. H. Li, L. L. Shao, X. F. Ren, X. T. Cai, D. L. An and S. F. Yin, Catal. Commun., 2009, 10, 1889–1892 CrossRef CAS.
- Y. Motoyama, H. Narusawa and H. Nishiyama, Chem. Commun., 1999, 131–132 RSC.
- Y. Motoyama, M. Okano, H. Narusawa, N. Makihara, K. Aoki and H. Nishiyama, Organometallics, 2001, 20, 1580–1591 CrossRef CAS.
- H. P. Dijkstra, M. D. Meijer, J. Patel, R. Kreiter, G. P. M. van Klink, M. Lutz, A. L. Spek, A. J. Canty and G. van Koten, Organometallics, 2001, 20, 3159–3168 CrossRef CAS.
- D. L. An, Z. H. Peng, A. Orita, A. Kurita, S. Man-e, K. Ohkubo, X. S. Li, S. Fukuzumi and J. Otera, Chem.–Eur. J., 2006, 12, 1642–1647 CrossRef CAS.
- X. W. Zhang, R. H. Qiu, N. Y. Tan, S. F. Yin, J. Xia, S. L. Luo and C. T. Au, Tetrahedron Lett., 2010, 51, 153–156 CrossRef CAS.
- X. W. Zhang, S. F. Yin, R. H. Qiu, J. Xia, W. L. Dai, Z. Y. Yu, C. T. Au and W. Y. Wong, J. Organomet. Chem., 2009, 694, 3559–3564 CrossRef CAS.
- J. Xia, R. H. Qiu, S. F. Yin, X. W. Zhang, S. L. Luo, C. T. Au, K. Xia and W. Y. Wong, J. Organomet. Chem., 2010, 695, 1487–1492 CrossRef CAS.
- I. Marek, ed., Titanium and Zirconium in Organic Synthesis, Wiley-VCH, Weibheim, 2002 Search PubMed.
- W. W. Ellis, A. Gavrilova, L. Liable-Sands, A. L. Rheingold and B. Bosnich, Organometallics, 1999, 18, 332–338 CrossRef CAS.
- G. A. Luinstra, J. Organomet. Chem., 1996, 517, 209–215 CrossRef CAS.
- J. B. Jaquith, C. J. Levy, G. V. Bondar, S. T. Wang and S. Collins, Organometallics, 1998, 17, 914–925 CrossRef CAS.
- S. Q. Lin, G. V. Bondar, C. J. Levy and S. Collins, J. Org. Chem., 1998, 63, 1885–1892 CrossRef CAS.
- T. K. Hollis, N. P. Robison and B. Bosnich, Tetrahedron Lett., 1992, 33, 6423 CrossRef CAS.
- G. P. Zhang, R. H. Qiu, X. H. Xu, H. H. Zhou, Y. F. Kuang and S. H. Chen, Synth. Commun., 2012, 42, 858–864 CrossRef CAS.
- H. Xu, W. Liu, L. Peng, R. Qiu, Y. Zhao, J. Pan and X. Xu, Chin. J. Org. Chem., 2011, 31, 1719–1722 CAS.
- R. H. Qiu, G. P. Zhang, X. F. Ren, X. H. Xu, R. H. Yang, S. L. Luo and S. F. Yin, J. Organomet. Chem., 2010, 695, 1182–1188 CrossRef CAS.
- R. H. Qiu, X. H. Xu, Y. H. Li, L. L. Shao, G. P. Zhang and D. L. An, Synth. Commun., 2010, 40, 3309–3314 CrossRef CAS.
- G. J. Janz and R. P. T. Tomkins, Nonaqueous Electrolytes Handbook, Academic Press, New York, 1972 Search PubMed.
- S. Fukuzumi and K. Ohkubo, J. Am. Chem. Soc., 2002, 124, 10270–10271 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS.
- K. Ohkubo, S. C. Menon, A. Orita, J. Otera and S. Fukuzumi, J. Org. Chem., 2003, 68, 4720–4726 CrossRef CAS.
- K. Tanabe, M. Misono, Y. Ono and H. Hattori, New Solid Acids and Bases: Their Catalytic Properties, Elsevier, Amsterdam, 1989 Search PubMed.
- H. A. Benesi, J. Phys. Chem., 1957, 61, 970–973 CrossRef CAS.
- H. A. Benesi, J. Am. Chem. Soc., 1956, 78, 5490–5494 CrossRef CAS.
- J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, Second Edition, Wiley-VCHVerlag GmbH & Co. KGaA, 2010 Search PubMed.
- H. Tanaka, N. Matoba, H. Tsukamoto, H. Takimoto, H. Yamada and T. Takahashi, Synlett, 2005, 824–828 Search PubMed.
- T. Matsumoto, H. Maeta and K. Suzuki, Tetrahedron Lett., 1988, 29, 3567–3570 CrossRef CAS.
- T. Matsumoto, M. Katsuki and K. Suzuki, Tetrahedron Lett., 1988, 29, 6935–6938 CrossRef CAS.
- P. Bichler and J. A. Love in Top. Organomet. Chem. 201039–64 Search PubMed.
- S. B. Mahato, J. Indian Chem. Soc., 2000, 77, 175–191 CAS.
- P. Tarakeshwar, J. Y. Lee and K. S. Kim, J. Phys. Chem. A, 1998, 102, 2253–2255 CrossRef CAS.
- S. Yous, J. H. Poupaert, I. Lesieur, P. Depreux and D. Lesieur, J. Org. Chem., 1994, 59, 1574–1576 CrossRef CAS.
- S. Kobayashi, Eur. J. Org. Chem., 1999, 15–27 CrossRef CAS.
- I. Hachiya, M. Moriwaki and S. Kobayashi, Tetrahedron Lett., 1995, 36, 409–412 CrossRef CAS.
- J. S. Yadav, D. C. Bhunia, K. V. Krishna and P. Srihari, Tetrahedron Lett., 2007, 48, 8306–8310 CrossRef CAS.
- L. T. Scott, in Modern Arene Chemistry, ed. D. Astruc, Wiley, Berlin, 2002, pp. 20-31 Search PubMed.
- A. W. Amick and L. T. Scott, J. Org. Chem., 2007, 72, 3412–3418 CrossRef CAS.
- L. Ratjen, P. Garcia-Garcia, F. Lay, M. E. Beck and B. List, Angew. Chem., Int. Ed., 2011, 50, 754–758 CrossRef CAS.
- J.-F. Zhao, H.-Y. Tsui, P.-J. Wu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2008, 130, 16492–16493 CrossRef CAS.
- S. E. Denmark, G. L. Beutner, T. Wynn and M. D. Eastgate, J. Am. Chem. Soc., 2005, 127, 3774–3789 CrossRef CAS.
- R. Mahrwald, Chem. Rev., 1999, 99, 1095–1120 CrossRef CAS.
- J. Luan, L. Zhang and Z. Hu, Molecules, 2011, 16, 4191–4230 CrossRef CAS.
- S. Shimada, Curr. Org. Chem., 2011, 15, 601–620 CrossRef CAS.
- X. Zhang, S. Yin, S. Wu, W. Dai, W. Li and X. Zhou, Prog. Chem., 2008, 20, 878–886 CAS.
- N. Yang and H. Sun, Coord. Chem. Rev., 2007, 251, 2354–2366 CrossRef CAS.
- H. Suzuki, Organobismuth chemistry, Elsevier, Amsterdam, 2001 Search PubMed.
- P. A. Evans, A. Grisin and M. J. Lawler, J. Am. Chem. Soc., 2012, 134, 2856–2859 CrossRef CAS.
- T. Ollevier, V. Desyroy and E. Nadeau, Arkivoc, 2007, 10–20 Search PubMed.
- T. Ollevier, in Topics in Current Chemistry, ed. T. Ollevier, Springer, New York, 2011 Search PubMed.
- X.-W. Zhang, J. Xia, H.-W. Yan, S.-L. Luo, S.-F. Yin, C.-T. Au and W.-Y. Wong, J. Organomet. Chem., 2009, 694, 3019–3026 CrossRef CAS.
- U. M. Lindstrøm, Chem. Rev., 2002, 102, 2751–2771 CrossRef.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS.
- M. Bao, T. Hayashi and S. Shimada, Organometallics, 2007, 26, 1816–1822 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2209 CrossRef CAS.
- M. Kanai, N. Kato, E. Ichikawa and M. Shibasaki, Synlett, 2005, 1491–1508 CrossRef CAS.
- C. Pan and Z. Wang, Coord. Chem. Rev., 2008, 252, 736–750 CrossRef CAS.
- S. Bhowmick and K. C. Bhowmick, Tetrahedron: Asymmetry, 2011, 22, 1945–1979 CrossRef CAS.
- K. Majima, R. Takita, A. Okada, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 15837–15845 CrossRef CAS.
- L. Wang, X. Liu, Z. Dong, X. Fu and X. Feng, Angew. Chem., Int. Ed., 2008, 47, 8670–8673 CrossRef CAS.
- J. Mo, X. Chen and Y. R. Chi, J. Am. Chem. Soc., 2012, 134, 8810–8813 CrossRef CAS.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Y. Leng, J. Wang, D. Zhu, X. Ren, H. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS.
- S. Kobayashi, ed., Science of Synthesis: Water in Organic Synthesis., Thieme, 2012 Search PubMed.
- J. A. Ma and D. Cahard, Angew. Chem., Int. Ed., 2004, 43, 4566–4583 CrossRef CAS.
- S.-F. Yin, J. Maruyama, T. Yamashita and S. Shimada, Angew. Chem., Int. Ed., 2008, 47, 6590–6593 CrossRef CAS.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- L. Balazs, H. J. Breunig, E. Lork, A. Soran and C. Silvestru, Inorg. Chem., 2006, 45, 2341–2346 CrossRef CAS.
- S. Shimada, J. Maruyama, Y.-K. Choe and T. Yamashita, Chem. Commun., 2009, 6168–6170 RSC.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |