Design of biocompatible magnetic pectin aerogel monoliths and microspheres
Carlos A.
García-González
*a,
Elisa
Carenza
b,
Muling
Zeng
b,
Irina
Smirnova
*a and
Anna
Roig
*b
aInstitute of Thermal Separation Processes, Hamburg University of Technology, Eißendorferstraße 38, D-21073 Hamburg, Germany. E-mail: carlos.garcia@tuhh.de; irina.smirnova@tuhh.de; Fax: 0049 40 42878 4072; Tel: 0049 40 42878 3642
bInstitut de Ciència de Materials de Barcelona (CSIC), Campus de la UAB, E-08193 Bellaterra, Spain. E-mail: roig@icmab.es; Tel: 0034 935801853
First published on 24th August 2012
Abstract
Novel material formulations for biomedical uses containing magnetic nanoparticles are currently being explored. The synergistic combination of a polymeric matrix and a filler with magnetic properties in hybrid nanoporous materials (aerogels) will confer versatility to the product. In this work, the use of pectin aerogels as a biodegradable matrix containing maghemite nanoparticles (γ-Fe2O3 NPs) is studied. The γ-Fe2O3 NPs-loaded pectin aerogels are developed in two different morphologies (cylindrical monoliths and microspheres) prepared by a combination of sol–gel and supercritical drying methods. In the case of the aerogel microspheres, the sol–gel method was substituted by the emulsion-gelation procedure. The obtained aerogel-based materials were evaluated regarding their physical appearance and stability and their textural and magnetic properties. The particle size distribution and morphology of the aerogel microspheres were additionally analyzed by laser dispersion spectrometry, static image analysis and scanning electron microscopy. Process parameters influenced the distribution of the γ-Fe2O3 NPs within the material. Whereas the γ-Fe2O3 NPs were homogeneously distributed throughout the aerogel monoliths, γ-Fe2O3 NPs were mostly deposited on the outer surface of the aerogel microspheres. The magnetic properties of maghemite were preserved in the end material after overall processing.
A Introduction
Nowadays, there is a great demand for smart products in the form of nanostructured hybrid organic–inorganic materials. The synergistic characteristics of the different components of these materials provide a broad spectrum of selected properties that meet the requirements of many industrial applications, such as lightweight structural materials, catalysis, electronic or optical devices, medical implants or drug delivery systems.1Maghemite nanoparticles (NPs) under the size of ca. 30 nm, have superparamagnetic character at room temperature, high magnetic susceptibility and high saturation magnetization as well as biocompatibility and non-toxicity.2–4 These properties are already being exploited in several biomedical applications (targeted drug delivery, hyperthermia, contrast agents in magnetic resonance imaging or bioseparation).5–7 Additionally, iron oxide nanoparticles are the only magnetic nanoparticle approved for clinical use by the US Food and Drug Administration (USFDA).8 The entrapment of magnetic nanoparticles in polymeric porous matrices will add new functionalities to the polymer and improve the handling of the material. Uses of polysaccharides incorporating iron oxide are being intensively explored because of the non-toxicity and biocompatibility of both materials.3,9–11 Among the polysaccharide family, pectin is particularly effective as a colonic therapeutic delivery agent because of its resistance to protease and amylase in the upper gastrointestinal tract.2,12–15
Supercritical fluid technology, using primarily supercritical carbon dioxide (scCO2), allows the production of nanostructured materials, overcoming some of the limitations of conventional top-down and bottom-up approaches.16,17 Moreover, the use of scCO2 avoids the post-processing steps of purification generally required by conventional methods to remove organic solvents from the material.18 In particular, the supercritical drying of gels is a commonly used technology for the production of lightweight (ρ = 0.05–0.50 g cm−3) mesoporous materials, the so called aerogels. The outstanding porosity and textural properties of aerogels are currently being exploited in thermal insulation, catalysis, drug delivery systems and adsorption of pollutants among other applications.19–21 In addition to these applications, physiologically acceptable microspheres of organic aerogels can be used either for inhalation or oral drug delivery routes and, if composited with a magnetic material, they can be targeted to the appropriate site and tracked by magnetic resonance imaging (MRI).19,22,23
The incorporation of magnetic iron oxide NPs in aerogels has already been reported in the form of pure iron oxide aerogels,24 iron oxide–silica composite aerogels25–27 and CoFe2O4–cellulose composite aerogels.28 In this work, maghemite nanoparticles were incorporated in biocompatible and biodegradable pectin gel matrices in the form of bulk cylinders and microspheres. Supercritical drying was then used to obtain magnetic pectin aerogels. The properties of the final materials were examined with regard to their potential application as magnetic targeting drug delivery vehicles.
B Experimental methods
B.1 Reagents
The following chemicals were purchased from Sigma-Aldrich for γ-Fe2O3 NPs preparation and were used without any purification: iron(III) acetylacetonate (Fe(acac)3, 97%), oleic acid (analytical standard), benzyl alcohol (98%), hexane (anhydrous, 95%), ethanol, water solution (25 wt%) of trimethylammonium hydroxide (TMAOH), sodium citrate and deionised water.Two medium-rapid set HM pectin from apple–citrus (WECJ-1, degree of esterification–DE: 66–68%, Pektowin) and citrus (76280, DE: 63–66%, Fluka) peels were used. Ethanol (99.8% purity) and vegetable (canola) oil were obtained from Omnilab and domestic shops, respectively. Deionized water was used in all gelation experiments.
B.2 Processing
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio were added under magnetic stirring. After this step, the water dispersions were ready to be incorporated into the gels.
:pectin solution emulsions with 3:1 (4:1, 5:1 and 6:1) mass ratios were prepared by mixing (500 rpm) an aqueous phase containing 6% (w/w) of pectin with the corresponding amount of canola oil. For certain 6:1 oil-to-pectin solution emulsions, maghemite NPs were dispersed in water before being added to the oil. Ethanol (50 wt% with respect to water content) was then added to the mixture. The resulting emulsion was heated to 313–333 K for 30 min with agitation (marine type, d = 40 mm, stirring 500 rpm). Then, the mixture was placed in an ice bath and continuously stirred at 1400 rpm. Cooling was continued until the oil dispersion containing the weak pectin gel microspheres reached 293 K, after which additional ethanol (to get 100 wt% total ethanol content with respect to water content) was added drop-wise to the liquid mixture. The dispersion was stirred for 2 additional minutes, and then centrifuged (4500 rpm, 5 min). After centrifugation, the pectin microparticles were separated from the oil phase, immersed in 99.8% (v/v) EtOH and stored at room temperature. After 24 h, the gel microspheres were collected and soaked in fresh 99.8% (v/v) EtOH (i.e. second solvent exchange).
:1 volume ratio were added under magnetic stirring. After this step, the water dispersions were ready to be incorporated into the gels.
:pectin solution emulsions with 3:1 (4:1, 5:1 and 6:1) mass ratios were prepared by mixing (500 rpm) an aqueous phase containing 6% (w/w) of pectin with the corresponding amount of canola oil. For certain 6:1 oil-to-pectin solution emulsions, maghemite NPs were dispersed in water before being added to the oil. Ethanol (50 wt% with respect to water content) was then added to the mixture. The resulting emulsion was heated to 313–333 K for 30 min with agitation (marine type, d = 40 mm, stirring 500 rpm). Then, the mixture was placed in an ice bath and continuously stirred at 1400 rpm. Cooling was continued until the oil dispersion containing the weak pectin gel microspheres reached 293 K, after which additional ethanol (to get 100 wt% total ethanol content with respect to water content) was added drop-wise to the liquid mixture. The dispersion was stirred for 2 additional minutes, and then centrifuged (4500 rpm, 5 min). After centrifugation, the pectin microparticles were separated from the oil phase, immersed in 99.8% (v/v) EtOH and stored at room temperature. After 24 h, the gel microspheres were collected and soaked in fresh 99.8% (v/v) EtOH (i.e. second solvent exchange).
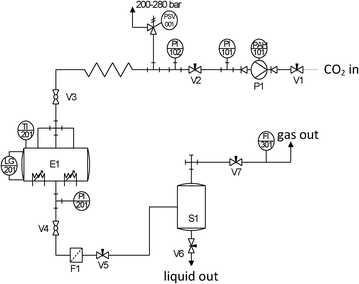 | ||
| Fig. 1 Process flow diagram of the equipment used for the supercritical drying of pectin aerogels. Tags: V1–V7 = valves; P1 = compressor; E1 = autoclave; S1 = separator; F1 = filter; PAH-101 = high-level pressure alarm; PI-xxx = pressure gauges; PSV-001 = safety valve; TI-201 = thermocouple; LG-201 = sapphire window; FI-301 = flow meter. | ||
B.3 Physicochemical characterization of the materials
Powder X-ray diffraction patterns of the nanoparticles were measured with a Rigaku, “rotaflex” RU-200B model using a Cu anode with λKα1 = 1.5406 Å and λKα2 = 1.5444 Å in the 2θ range 20–70°. The diffractogram could be indexed with the maghemite pattern. TEM micrographs for the maghemite nanoparticles were obtained with a JEOL JEM-1210 electron microscope, operating at 120 kV. The samples were prepared by depositing a drop of diluted nanoparticle dispersion in hexane onto a TEM grid and allowing the solvent to evaporate. The mean diameter and polydispersity of each of the nanoparticles were determined by fitting to a Gaussian distribution a particle size histogram of over 200 counts measured from TEM images by the ImageJ software. The hydrodynamic diameters of the maghemite nanoparticles suspended in organic media and water were investigated with a Zetasizer Nano ZS from Malvern Instruments equipped with a He/Ne 633 nm laser. The bulk density (ρbulk) of the pectin aerogel monoliths was determined by measuring the dimensions and weight of the samples after drying with scCO2. The porosity (ε) of the aerogel can be calculated with the obtained ρbulk and skeletal density (ρskeletal) by eqn (1):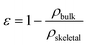 | (1) |
The textural properties of the aerogels were determined by low-temperature N2 adsorption–desorption analysis (Nova 3000e). Prior to measurements, the samples were dried under vacuum (<1 mPa) at 333 K for 20 h. The specific surface area (ABET) was determined by the BET (Brunauer–Emmett–Teller) method. The pore volume (Vp) and mean pore diameter (dp) were estimated using the BJH (Barrett–Joyner–Halenda) method. The iron oxide content in pectin aerogels (6–8 mg) was evaluated thermogravimetrically using a TGA Netzsch TG 209 F1 instrument. Measurements were performed under an oxygen atmosphere with a heating rate of 10 K min−1 in the range between room temperature and 1273 K. Micrographs of the pectin aerogel particles were taken in a scanning electron microscope (SEM), and images were acquired using a Quanta FEI 200F microscope in low vacuum mode with a cone LOW kV P.L.A. type with a 500 μm aperture, equipped with a solid-state backscattered probe for energy dispersive X-ray spectrometry (EDS) analysis. Except for maghemite-loaded materials, samples were gold-sputtered (10 nm thickness) prior to imaging in order to minimise charging and improve the image contrast. Particle size distributions of the pectin wet microspheres dispersed in ethanol were measured using a laser dispersion spectrometer (Beckman Coulter LS1332) and with similar obscuration values. The particle size distributions and shapes of dried aerogel microspheres were evaluated by using static image analysis equipment (Morphologi G3) that allows size measurements between 0.5 μm–3000 μm. Magnetic characterization of the maghemite particles and the nanocomposited microspheres were performed with a quantum interference device (SQUID) magnetometer (Quantum Design MPMS5XL).
C Results and discussion
C.1 Characterization of the γ-Fe2O3 nanoparticles
Maghemite nanoparticles were fabricated by microwave-assisted non-hydrolytic sol–gel synthesis. Oleic acid was added to the initial precursor solution to render the nanoparticles stable as colloids in organic solvents. Subsequently, the nanoparticles were transferred to aqueous solutions through a ligand exchange using TMAOH as an electrostatic stabilizer. Powder X-ray diffraction (Fig. 2a) can be indexed to the maghemite, an inverse spinel structure (ICDD PDF039-1346).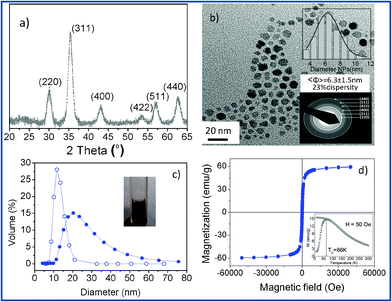 | ||
| Fig. 2 (a) X-Ray diffraction patterns corresponding to maghemite nanoparticles. (b) TEM image of the nanoparticles. Upper inset includes a size distribution histogram fitted to a Gaussian and the lower inset shows a selected area electron diffraction pattern corresponding to maghemite. (c) Dynamic light scattering histogram of the organic stabilized nanoparticles (empty symbols) and the water stabilized dispersion after ligand exchange (full symbols); a cuvette containing the clear nanoparticle dispersion is shown as an inset. (d) Magnetic characterization of the iron oxide nanoparticles. M(H) at 300 K and M(T), ZFC–FC curves (inset) | ||
Size, polydispersity, shape and structural phase of the nanoparticles were studied by TEM (Fig. 2b). The particles have roughly spherical shapes. The size distribution histogram from a 250 particle count (inset of Fig. 2b) gives an average particle size of 6.3 ± 1.5 nm. Nanoparticles transferred in water at pH 7 formed stable aggregates with a 35 nm mean size (Fig. 2c) and a zeta potential value of −40 mV.
The magnetic properties of the material were evaluated by magnetization versus magnetic field at 300 K and zero-field cooled (ZFC)–field cooled (FC) temperature dependent magnetization curves (Fig. 2d). The results at 300 K show a typical magnetization curve of a superparamagnetic system with neither hysteresis nor remanent magnetization. The saturation magnetization is large, 60 emu g−1, pointing to a high crystallinity. Superparamagnetism is also confirmed by the ZFC–FC magnetization curve (inset Fig. 2d). The ZFC magnetization increases with temperature until reaching a maximum, corresponding to the blocking temperature, at 66 K. Above this temperature, the thermal energy becomes larger than the magnetic energy barrier and the nanoparticles enter the superparamagnetic regime.
C.2 γ-Fe2O3-pectin aerogels
Aerogel processing starts with the formation of a gel from an aqueous solution, i.e. a hydrogel. Then, the water filling the pores of the gel structure is replaced by ethanol, leading to an alcogel, followed by the extraction of the ethanol from the gel by supercritical carbon dioxide (scCO2)-assisted drying. The usual method of pectin gelation by the addition of sucrose15 to decrease pectin hydration was not suitable to undergo this aerogel processing. During the needed solvent exchange step of the hydrogel to ethanol, the sucrose precipitated and the gel was no longer mechanically stable (Fig. 3, left). Other gelation mechanisms proposed in the literature33 led to powdery and unstable aerogels by thermal gelation or low textural properties (ABET = 200 m2 g−1; Vp = 0.38 cm3 g−1) by acidic gelation. Therefore, a novel processing method for the preparation of pectin aerogels compatible with the intended life science applications was developed in this work. The thermal gelation of HM-pectin (333–353 K, 30 min) with the addition of ethanol (15–25 wt% to water content) produces mechanically stable pectin hydrogels. The addition of ethanol promotes pectin gelation by means of improving the prevalence of pectin–pectin interactions rather than pectin–solvent interactions. Thus, an increase in the viscosity of the pectin solution and the formation of a stable gel is obtained.34 The use of ethanol as a gelation promoter also effectively reduces the processing time of biocompatible pectin aerogels, either by increasing the solvent exchange rate to ethanol or, if necessary, decreasing the number of solvent exchange steps prior to supercritical drying. Two types of HM-pectin of different origins (citrus and apple–citrus) underwent the same thermal gelation method with equal concentrations of pectin (6 wt% to the water content) followed by direct solvent exchange to ethanol. It was noticed that the immersion of even very soft hydrogel monoliths in EtOH resulted in a rapid increase in hardness after only 1–2 min contact, pointing to a chemical interaction of the pectin with ethanol. Thus, the hydrogels of HM-citrus pectin were generally softer than the gels of apple–citrus pectin but were just as hard after solvent exchange to ethanol. A marked shrinkage in the middle of the gel monolith (Fig. 3, middle) prepared from citrus pectin was observed for different concentrations of pectin, whereas a homogeneous reduction in volume occurred for all samples of apple–citrus pectin (Fig. 3, right). The differences in degree of esterification and molecular structure of pectin depending on the extraction process and the source may lead to the formation of network structures with quite different physical properties.35,36 The differences in solvent diffusion through the HM-citrus and apple–citrus pectin gel networks may also be related to their different segmental rod-like gel structures.33,37 Taking these results into consideration, the HM-pectin from apple–citrus was used for further investigation of pectin aerogel processing. | ||
| Fig. 3 Physical appearance of different pectin gel monoliths after solvent exchange to ethanol: HM-pectin from citrus with (left) and without (middle) sucrose addition during gelation, and HM-pectin from apple–citrus without sucrose addition (right). | ||
Supercritical CO2 drying of pectin alcogels (Fig. 4a) over 4 h produced lightweight (ρ = 0.08 g cm−3 in Table 1), highly porous (ε = 95%) aerogels (Fig. 4b). Shorter supercritical drying times (1 h) resulted in partial solvent (ethanol) removal, leading to dramatic volume shrinkage and poor physicochemical properties (ρ = 0.32 g cm−3; ε = 77.7%; ABET = 129 m2 g−1) (not shown). Gel volume shrinkage due to the direct solvent exchange step to ethanol (after SE shrinkage in Table 1) were small in comparison with those obtained after the supercritical drying step (overall shrinkage). Finally, vacuum drying (60 °C, 4 h) was attempted as an alternative drying method but the wet gel structure was not preserved, resulting in a massive volume shrinkage and pore collapse, leading to xerogel type of materials (Fig. 4c) with poor textural properties (Sa,BET < 5 m2 g−1).
 | ||
| Fig. 4 Drying of (a) pectin hydrogel monoliths by means of (b) supercritical drying, aerogel, and (c) vacuum drying, xerogels. Maghemite nanoparticles can be incorporated in (d) the pectin gel matrix leading to (e) brownish aerogels after supercritical drying. (f) Monolith with 1.5–2.0 wt% in NPs attracted by a magnet. | ||
| Fe NP content | Shrinkage (%) | ρ (g cm−3) | ε (%) | A BET (m2 g−1) | V p (cm3 g−1) | d p (nm) | |
|---|---|---|---|---|---|---|---|
| After SE | Overall | ||||||
| None | 12 ± 3 | 48 ± 28 | 0.08 ± 0.03 | 95 ± 1 | 247 ± 12 | 1.2 ± 0.1 | 17 ± 1 |
| High | 26 ± 3 | 61 ± 2 | 0.11 ± 0.01 | 93 ± 1 | 284 ± 14 | 1.5 ± 0.1 | 22 ± 1 |
The influence of the addition of iron oxide nanoparticles onto the pectin gel and aerogel monoliths was examined in this work. Pectin monoliths were prepared by incorporation of the NPs (1.5–2.0 wt% content as a percentage with respect to the pectin content) to the pectin solution. The pectin solutions turned more viscous with the addition of the NPs. Moreover, such addition interfered with the gelation mechanism, leading to an increase in the gelation time. Translucent dark brownish NP-containing pectin hydrogels were thus obtained. No significant leaching of NPs was observed during solvent exchange to ethanol (Fig. 4d). Upon addition of the NPs to the pectin gel formulation, a higher shrinkage is achieved during the solvent exchange and drying processes (Table 1) resulting in opaque, brownish and mechanically stable aerogels (Fig. 4e). The resulting aerogel density increased, due to both the higher shrinkage of the aerogels and the higher skeletal density of maghemite (4.87 g cm−3)2,38 with respect to raw pectin (1.44 g cm−3). However, the addition of NPs enhanced the textural properties of the aerogels (ABET, Vp and dp in Table 1). Finally, due to the magnetic properties of the maghemite, the pectin aerogel was attracted to a magnet (Fig. 4f).
C.3 Preparation of γ-Fe2O3-pectin aerogels in the form of microspheres
The processing method for the preparation of the maghemite NP-loaded pectin aerogel monoliths described in Section 3.2 was adapted for the preparation of aerogel microspheres. The emulsion-gelation method32,39 was used to produce microspherical gels with the same HM-apple–citrus pectin concentration (6 wt% to the water content) as the monoliths. Previous studies22,32 have shown that the textural properties of aerogel microspheres are mainly influenced by the gelation parameters (e.g. precursor concentration, crosslinking concentration, temperature) and that results obtained for aerogel monoliths can be applied to the development of aerogel microspheres. On the other hand, particle morphology (roundness, mean size, size distribution) is primarily affected by emulsion parameters (e.g. oil-to-aqueous phase ratio, surfactant concentration, agitation speed). Moreover, the ability of HM-apple–citrus pectin to provide the phase inversion of the emulsions at certain pectin concentrations was detected. During the cooling and mixing of the emulsion with a 2:1 (w/w) oil-to-pectin solution ratio, the phase inversion of the emulsion was observed 2–3 min after addition of ethanol. Therefore, the oil-to-pectin solution ratio was increased to 6:1 (w/w) to avoid phase inversion of the emulsion. The increase in the oil-to-pectin solution ratio led to an increase in the mean particle size and a broader particle size distribution (Fig. 5). The formation of larger particles with increasing oil content can be explained by an increase in viscosity of the liquid mixture, resulting in an increase in the aqueous droplet size. The use of different oil-to-water ratios did not significantly affect the textural properties of the resulting pectin aerogel microspheres with BET-specific surface areas in the range 440–480 m2 g−1.
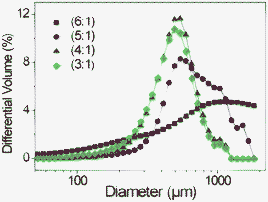 | ||
| Fig. 5 Particle size distribution of pectin particles at different oil to water ratios from pectin raw material. | ||
The addition of maghemite NPs to the formulation of pectin gel microspheres led to a change from a light cream colour to dark brown in both the gel and aerogel microspheres (Fig. 6). No significant leaching of NPs during solvent exchange to ethanol was observed, since the supernatant was colourless and transparent in all cases. The maghemite content of the materials was calculated by TGA as the difference in weight remaining after thermal treatment up to 1263 K of the nanocomposite material (7.6 wt%, Fig. 6c right) compared to the weight loss observed in the pectin aerogel (3.9 wt%, Fig. 6c left). A maghemite content in the aerogel microspheres of 3.7 wt% was obtained. Moreover, the incorporation of the iron oxide-NPs within the pectin aerogel network improved the end textural properties of the microspheres (with 3.7 wt% NPs content: ABET = 501 ± 25 m2 g−1, Vp = 2.8 ± 0.2 cm3 g−1 and dp = 17 ± 1 nm; without NPs: ABET = 442 ± 22 m2 g−1, Vp = 2.4 ± 0.1 cm3 g−1 and dp = 31 ± 2 nm).
 | ||
| Fig. 6 Pectin alcogel microspheres (a) without and (b) with maghemite NPs were dried by supercritical drying to get nanocomposite aerogels. (c) Maghemite nanoparticle addition led to brown-coloured microspheres (without NPs, left, and with NPs, right). (d) Aerogel microspheres with high NP content (3.7 wt%) were attracted to a magnet. | ||
The particle size distribution of wet alcogels and dried aerogels were studied by laser dispersion spectrometry (Fig. 7) and by static image analysis (Fig. 8a), respectively.
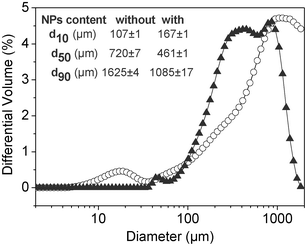 | ||
| Fig. 7 Particle size distribution by laser dispersion spectrometry of pectin gel microspheres with (triangles) and without (circles) maghemite NPs. Inset: percentiles 10 (d10), 50 (d50) and 90 (d90) of the particle size distribution for both samples. | ||
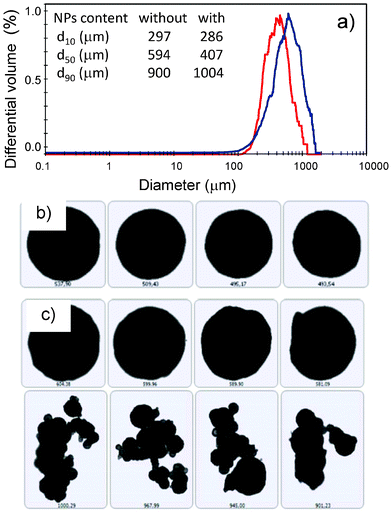 | ||
| Fig. 8 Static image analysis of pectin aerogel microspheres with (red) and without (blue) maghemite NPs: (a) particle size distribution; intake: percentiles 10 (d10), 50 (d50) and 90 (d90) of the particle size distribution for both samples. Microspheres with high sphericity were obtained in both cases (b,c), although the presence of some agglomerates was observed in the case of aerogels without γ-Fe2O3 NPs (c). | ||
Shrinkage of the particle size of pectin microspheres can be observed after supercritical drying when comparing the median particle diameters (d50) of the wet gels and aerogels (insets in Fig. 7 and Fig. 8a). This trend agrees with the volume shrinkage observed for the pectin aerogel monoliths (Section 3.2). High sphericity of the aerogel particles was obtained with circularity values greater than 0.95 (Fig. 8b,c). However, the presence of few aggregates was noticed for the aerogel microspheres without γ-Fe2O3 NPs (Fig. 8c). A shift towards lower particle sizes and coarser particle size distribution was observed when maghemite NPs were incorporated in both the wet and the dry forms.
The SEM pictures of pectin aerogels (Fig. 9a,b) show spherical particles with a porous microstructure formed by a rod-like network. A similar appearance was observed for the maghemite NP-loaded pectin aerogels (Fig. 9c,d). Lighter areas, corresponding to NP addition, were observed in Fig. 9d. SEM pictures of the NP loaded-aerogels without gold sputtering improved the contrast between the iron NPs (conductive) and the pectin matrix (low conductivity) (Fig. 10). It was then observed that the iron NPs formed a layer on the surface of the pectin aerogel microspheres (Fig. 10a). However, the NPs in the aerogel particle were not homogeneously distributed upon the surface but formed islands of a few monolayers thick, which were easily seen when using the electron retrodispersion mode of the SEM (Fig. 10a,b). The maghemite origin of the islands was confirmed by iron content detection with EDS analysis (Fig. 10c).
 | ||
| Fig. 9 SEM pictures of pectin aerogel microspheres without (top) and with the addition of γ-Fe2O3 NPs (bottom). | ||
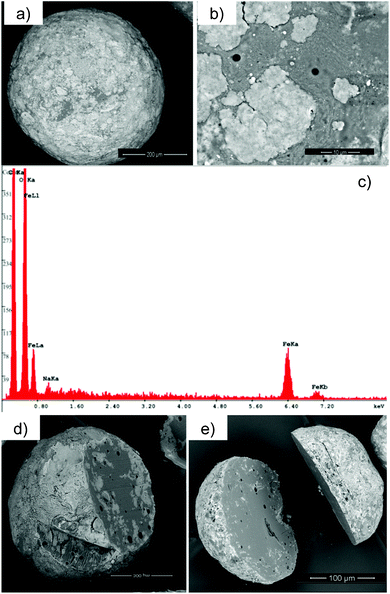 | ||
| Fig. 10 Distribution of γ-Fe2O3 NP loading in pectin aerogel microspheres: (a) γ-Fe2O3 NPs were observed in the outer surface of the aerogel microspheres (b) forming islands. (c) EDS analysis confirmed the γ-Fe2O3 NPs origin of the islands. (d,e) SEM pictures of cut microspheres reveal that γ-Fe2O3 NPs are mainly present in the outer shell of the aerogel microsphere. | ||
The chemical interaction via hydrogen bonding of the hydroxyl groups of the maghemite NPs and the carbonyl groups of the pectin matrix is regarded as a plausible reason for the adsorption of NPs in the aerogel. This NP–pectin interaction also explains the insignificant loss of NPs upon aerogel processing during the removal of the oil phase or the solvent exchange steps to ethanol. Similar chemical interactions between other carbonyl group-containing compounds and inorganic oxides have been previously reported in the literature.40
Cutting of the pectin aerogel microspheres, using a scalpel, revealed that the γ-Fe2O3 NPs were predominantly located on the surface (Fig. 10d,e). The origin of this NP layer on the surface of the pectin aerogel microspheres is thought to arise from the spontaneous location of the NPs in the water–oil interface of the emulsion prior to the gelation of the disperse phase (pectin aqueous solution). This effect was related to the reduction of the surface charge of the NPs due to the competitive adsorption on the NPs of the ethanol added to the system to promote gelation.41 Moreover, the change in surface charge of the NPs is also promoted by the lowering of the pH value in the aqueous solution (γ-Fe2O3 NPs initially stabilized at pH 7 were put in contact with pectin solutions at pH 3.3).42 The reduction of the water–oil interfacial energy by means of the adsorption of the NPs on this region is regarded as the driving force behind this local and spontaneous assembly of the maghemite particles at the interface. Hydrodynamic forces in the emulsion due to the density difference between the NPs and the aqueous solution (e.g. biased Brownian motion of the NPs in a centrifugal force field within the aqueous solution droplets) may also favor the location of the NPs in the water–oil interface.
Moreover, the prevention of droplets from flocculation and coalescence due to the binding of the NPs to the surface of the water droplets (oil/water interphase) in a Pickering-like emulsion might cause the emulsion to be more stable (i.e. fewer aggregates)43 and may explain the reduction in aerogel particle size after the addition of NPs previously observed in Fig. 8.
Finally, the magnetic properties of the microspheres were evaluated by magnetization versus magnetic field at 300 K (Fig. 11). The results show a typical magnetization curve of a superparamagnetic system with neither hysteresis nor remanent magnetization, as was also the case of the original maghemite nanoparticles (Fig. 2). This result indicates that the incorporation of maghemite nanoparticles in the pectin aerogel microspheres does not modify the properties of the initial nanoparticles, even though they formed aggregates. The inset of Fig. 11 contains a zero field cooled–field cooled magnetization curve as a function of temperature. The ZFC curve displays a clear maximum, signaling a blocking temperature at around 40 K, as expected for a superparamagnetic system.
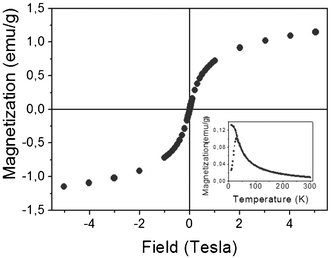 | ||
| Fig. 11 Magnetization versus magnetic field of the maghemite loaded microspheres at 300 K. M(T), ZFC–FC curves are depicted in the inset. | ||
Conclusions
High methoxyl apple–citrus pectin aerogels containing maghemite nanoparticles were produced in the form of cylinders and microspheres. The addition of iron oxide nanoparticles to the aerogel monolith formulation gave the aerogels magnetic properties, changed their color from white to dark brown, and increased the aerogel density (ca. 38% higher) and the specific surface area (ca. 15%). The addition of iron oxide nanoparticles in the emulsion formulation of the same origin resulted in a more viscous solution and prevented the droplets of the dispersed phase of the emulsion from flocculation and coalescence. Maghemite nanoparticles were preferentially adsorbed on the outer surface of the aerogel microspheres in the form of islands, but their magnetic properties were unchanged after being incorporated into the microspherical aerogel matrix. The obtained magnetic nanoporous materials set the basis for a future study regarding their application in targeted delivery and magnetic resonance imaging tracking.Acknowledgements
C.A. García-González acknowledges the Spanish Ministry of Education for the financial support through a postdoctoral fellowship in the frame of the National Program for Staff Mobility from the R&D&i National Plan 2008–2011. M. Zeng acknowledges the CSC scholarship number 2011703001. Authors are grateful to Frank Rimoschat (research group of Prof. Heinrich, Institute of Solids Process Engineering and Particle Technology, Hamburg University of Technology) for his technical support with the measurements of particle size distribution by laser dispersion. The authors want to acknowledge Pektowin company for the supply of the citrus–apple HM-pectin. The work was partially funded by the Spanish Government (MAT2009-08024, CONSOLIDER-Nanoselect-CSD2007-00041) and the Generalitat de Catalunya (2009SGR203). Authors are grateful to Ryan Vargas and Dr Oana Pascu for the synthesis and characterization of some of the nanoparticles. The contributions of Aleksej Vdovin, Jerico J. Uy and Ho Ka Carol Chan in the development of the pectin aerogels are gratefully acknowledged.References
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- S. Sahu and R. K. Dutta, J. Magn. Magn. Mater., 2011, 323, 980–987 CrossRef CAS.
- A. L. Daniel-da-Silva, T. Trindade, B. J. Goodfellow, B. F. O. Costa, R. N. Correia and A. M. Gil, Biomacromolecules, 2007, 8, 2350–2357 CrossRef CAS PubMed.
- B. H. Kenzaoui, M. R. Vilà, J. M. Miquel, F. Cengelli and L. Juillerat-Jeanneret, Nanomedicine, 2012, 7, 1275–1286 CrossRef CAS PubMed.
- E. Taboada, R. Solanas, E. Rodríguez, R. Weissleder and A. Roig, Adv. Funct. Mater., 2009, 19, 2319–2324 CrossRef CAS.
- U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Adv. Mater., 2007, 19, 33–60 CrossRef CAS.
- R. D. Halpert, Gastrointestinal Imaging: The Requisites, Mosby Elsevier, Philadelphia (PA, USA), 2006 Search PubMed.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- F. Llanes, C. Diaz, H. Ryan and R. H. Marchessault, International Journal of Polymeric Materials, 2002, 51, 537–545 CrossRef CAS.
- F. Jones, H. Cölfen and M. Antonietti, Colloid Polym. Sci., 2000, 278, 491–501 CAS.
- S. Dumitriu, Polysaccharides: structural diversity and functional versatility, Marcel Dekker, New York (US), 2005 Search PubMed.
- W. G. T. Willats, J. P. Knox and J. D. Mikkelsen, Trends Food Sci. Technol., 2006, 17, 97–104 CrossRef CAS.
- H. N. Englyst, S. Hay and G. T. Macfarlane, FEMS Microbiol. Lett., 1987, 45, 163–171 CrossRef CAS.
- P. Sriamornsak, Silpakorn University International Journal, 2003, 3, 206–227 Search PubMed.
- S. M. Brejnholt, in Food Stabilisers, Thickeners and Gelling Agents, ed. A. Imeson, Blackwell Publishing Ltd, Singapore, 2010 Search PubMed.
- S. Bozbag, D. Sanli and C. Erkey, J. Mater. Sci., 2012, 47, 3469–3492 CrossRef CAS.
- D. Sanli, S. Bozbag and C. Erkey, J. Mater. Sci., 2012, 47, 2995–3025 CrossRef CAS.
- Y.-P. Sun, Supercritical Fluid Technology in Materials Science and Engineering. Syntheses, Properties and Applications, Marcel Dekker, New York, NY, USA, 2002 Search PubMed.
- C. A. García-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425–1438 CrossRef.
- C. A. García-González, M. C. Camino-Rey, M. Alnaief, C. Zetzl and I. Smirnova, J. Supercrit. Fluids, 2012, 66, 297–306 CrossRef.
- I. Smirnova, S. Suttiruengwong and W. Arlt, J. Non-Cryst. Solids, 2004, 350, 54–60 CrossRef CAS.
- C. A. García-González, J. J. Uy, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2012, 88, 1378–1386 CrossRef.
- A. Berg, M. W. Droege, J. D. Fellmann, J. Klaveness and P. Rongved, EP, 0707474B1, 1994 Search PubMed.
- J. W. Long, M. S. Logan, C. P. Rhodes, E. E. Carpenter, R. M. Stroud and D. R. Rolison, J. Am. Chem. Soc., 2004, 126, 16879–16889 CrossRef CAS PubMed.
- E. Taboada, R. P. del Real, M. Gich, A. Roig and E. Molins, J. Magn. Magn. Mater., 2006, 301, 175–180 CrossRef CAS.
- L. Casas, A. Roig, E. Molins, J. M. Grenèche, J. Asenjo and J. Tejada, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 591–597 CrossRef CAS.
- M. Popovici, M. Gich, A. Roig, L. Casas, E. Molins, C. Savii, D. Becherescu, J. Sort, S. Suriñach, J. S. Muñoz, M. D. Baró and J. Nogués, Langmuir, 2004, 20, 1425–1429 CrossRef CAS PubMed.
- R. T. Olsson, M. A. S. Azizi Samir, G. Salazar Alvarez, L. Belova, V. Strom, L. A. Berglund, O. Ikkala, J. Nogués and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef CAS PubMed.
- I. Bilecka, M. Kubli, E. Amstad and M. Niederberger, J. Sol-Gel Sci. Technol., 2011, 57, 313–322 CrossRef CAS.
- O. Pascu, E. Carenza, M. Gich, S. Estradé, F. Peiró, G. Herranz and A. Roig, J. Phys. Chem. C, 2012, 116, 15108–15116 CAS.
- E. Taboada, E. Rodríguez, A. Roig, J. Oró, A. Roch and R. N. Muller, Langmuir, 2007, 23, 4583–4588 CrossRef CAS PubMed.
- M. Alnaief, M. A. Alzaitoun, C. A. García-González and I. Smirnova, Carbohydr. Polym., 2011, 84, 1011–1018 CrossRef CAS.
- R. J. White, V. L. Budarin and J. H. Clark, Chem.–Eur. J., 2010, 16, 1326–1335 CrossRef CAS PubMed.
- D. Oakenfull and A. Scott, J. Food Sci., 1984, 49, 1093–1098 CrossRef CAS.
- P. Sriamornsak, N. Wattanakorn and H. Takeuchi, Carbohydr. Polym., 2010, 79, 54–59 CrossRef CAS.
- B. R. Sharma, L. Naresh, N. C. Dhuldhoya, S. U. Merchant and U. C. Merchant, Times Food Processing Journal, 2006, 6, 44–51 Search PubMed.
- M. L. Fishman, P. H. Cooke and D. R. Coffin, Biomacromolecules, 2004, 5, 334–341 CrossRef CAS PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory: Preparation and Characterization, Wiley-VCH Verlag GmbH, Weinheim (Germany), 2000 Search PubMed.
- M. Alnaief and I. Smirnova, J. Supercrit. Fluids, 2011, 55, 1118–1123 CrossRef CAS.
- B. P. Binks and C. P. Whitby, Colloids Surf., A, 2005, 253, 105–115 CrossRef CAS.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS PubMed.
- I. T. Lucas, S. Durand-Vidal, E. Dubois, J. Chevalet and P. Turq, J. Phys. Chem. C, 2007, 111, 18568–18576 CAS.
- E. Dickinson, Curr. Opin. Colloid Interface Sci., 2010, 15, 40–49 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |