Incorporation of a high-spin heptanuclear [CuII6Gd] cluster into carboxyl-functionalized mesoporous silica
Ioannis
Kornarakis
a,
George
Sopasis
a,
Constantinos J.
Milios
b and
Gerasimos S.
Armatas
*a
aDepartment of Materials Science and Technology, University of Crete, 71003 Heraklion, Greece. E-mail: garmatas@materials.uoc.gr; Tel: +30 2810 545004; Fax: +30 2810 545197
bDepartment of Chemistry, University of Crete, 71003 Heraklion, Greece
First published on 10th August 2012
Abstract
We report the immobilization of heptanuclear [Cu6Gd] high-spin magnetic clusters, namely [Cu6Gd(aib)6(OAc)3(NO3)3(OH)3] (where, aib = (CH3)2C(NH2)COO− and OAc = CH3COO−), inside the channels of –COOH-functionalized mesoporous SBA-15 silica. X-ray diffraction, TEM and N2 physisorption experiments show that the well-ordered hexagonal structure of the CA/SBA-15 hybrid material is well preserved and that the [Cu6Gd] clusters are located inside the mesopores. The molecular structure of the incorporated [Cu6Gd] was confirmed by elemental X-ray microanalysis, IR and UV-vis-NIR spectroscopy. Magnetic measurements indicate that the polynuclear complexes retain their magnetic properties during the impregnation process. The host–guest nanocomposite displays strong ferromagnetic interactions that result in a high-spin S = 13/2 ground state.
Introduction
The development of molecule-based materials with fine-tuned magnetic behavior is of great importance in materials science and nanotechnology. Such materials represent a significant dimensional reduction of magnetic grain size where single molecules act as tiny magnets. The polynuclear complexes that behave as molecular nanomagnets, referred to as single-molecule magnets (SMMs), have drawn significant attention.1,2 These molecular species display slow relaxation of the magnetization associated with a large spin ground state, S, and high negative magnetic anisotropy, D, under cryogenic conditions. Remarkably, the magnetic hysteresis is due to discrete molecules rather than long-range magnetic ordering as observed in bulk metal and metal oxide magnets.3 Therefore, SMMs are promising for a variety of applications, including quantum computation,4 high-density magnetic recording5 and molecular-based spintronics.6 In addition, in the field of molecular magnetism new species have emerged that can lower the temperature of their surroundings once a magnetic field is applied due to the magnetocaloric effect, and therefore can be used as molecular coolants and refrigerants.7–9 For both single-molecule magnets and molecular refrigerants, the main prerequisites are the low anisotropy, D, and high-spin ground state, S, of the complex.Therefore, it becomes evident that the chemical synthesis of high-spin and/or ferromagnetic complexes is of vital importance for the employment of such species into technological applications. Consequently, the organization of magnetically interesting units in two- and three-dimensional architectures is the next step towards potential applications. For this reason, different methods of integrating molecular clusters (i.e. SMMs) into ordered arrays have been in development in recent years. These include deposition in Langmuir–Blodgett films,10,11 incorporation in ordered nanoporous silica12–14 and polymer matrices,15 and adsorption on self-assembled monolayers16,17 and conducting substrates.18 Among them, the incorporation of complexes into the pore system of organically-modified silica is particularly attractive because of the benefits of ordered pore structure and chemical functionalities of the silica support. As the organic moieties attached to the silica surface allow for immobilization of inorganic complexes, the porous structure can induce fast mass transfer and molecular recognition behaviour. Nanosized arrays of these composites are expected to be of practical applications, especially in the fields of magnetic sensors and magnetic separation. However, examples concerning the synthesis of silica-supported materials are relative scarce as compared with other porous host–guest composites, mainly because the inclusion processes should take place without disturbing the structure of magnetic clusters and host phase. Moreover, the molecular complexes have to incorporate into the host structure in a robust and well-dispersed fashion, avoiding the formation of agglomerates.
Herein, we managed to overcome these difficulties and have successfully immobilized high-spin heptanuclear heterometallic copper(II)–gadolinium(III) molecules of formula [Cu6Gd(aib)6(OAc)3(NO3)3(OH)3] (where, aib = 2-aminoisobutyrate and OAc = acetate), referred to here as [Cu6Gd], into a hexagonal mesoporous –COOH-functionalized silica. The [Cu6Gd] metallic core is formed by a central {GdO9} polyhedron that is surrounded by six Cu(II) ions in an octahedral arrangement. The Cu ions are linked by μ3-O2− bridges to each other and to the central {GdO9} unit. Furthermore, this complex contains six peripheral monoanionic α-aminoisobutyrate ligands able to selectively interact with the anchored carboxyl groups of the silica framework. The metallic core of this complex exhibits dominant ferromagnetic interactions leading to a high-spin ground state of S = 13/2 (vide infra). The results of this work indicate that the [Cu6Gd] polynuclear complexes are successfully embedded in the cavities of mesoporous silica, keeping their molecular structure. Also, these clusters preserve their magnetic properties, maintaining dominant ferromagnetic exchange interactions within the silica matrix.
Experimental
Synthesis of heptanuclear Cu(II)-Gd(III)complex
The [Cu6Gd(aib)6(OAc)3(NO3)3(OH)3] (aib = 2-aminoisobutyrate and OAc = acetate) complex was prepared following the method described in the literature.19Synthesis of –COOH-functionalized mesoporous silica
The mesoporous silica functionalized with carboxyl groups was prepared as previously reported but with some modifications.20 Briefly, 2 g of Pluronic P123 (PEO20PPO70PEO20, where PEO = poly(ethylene oxide) and PPO = poly(propylene oxide), Aldrich) was dissolved in a solution containing 75 ml of water, 11 ml of 37% HCl (Sigma-Aldrich) and 4.5 mmol of 3-(triethoxysilyl)propionitrile (Aldrich, 98%). After stirring for 30 min, 17.5 mmol of tetraethylorthosilicate (TEOS, Merck, ≥99%) was slowly added and the resultant mixture was vigorously stirred for 24 h at 40 °C. After this, the mixture was left to react at 90 °C for 2 days under static conditions and the white powder was recovered by filtration, washed thoroughly with ethanol and acetone, and dried at 90 °C to give cyanide-functionalized silica (CN/SBA-15). To remove the surfactants inside the pores and hydrolyze the cyano groups, 1 g of dried CN/SBA-15 was dispersed in 120 ml of 48 wt% H2SO4 solution (Sigma-Aldrich) and the mixture was refluxed at 95 °C for 18 h. The product was filtered off, washed copiously with ethanol and acetone, and dried at 90 °C for 12 h to obtain carboxyl-functionalized silica (CA/SBA-15).Incorporation of [Cu6Gd] clusters into the CA/SBA-15
The impregnated [Cu6Gd]/CA-SBA hybrid silica was prepared by suspending 55 mg of dried CA/SBA-15 in 12 ml of CH3CN (SDS, Carlo Erba, 99.7% pure) containing 160 mg of [Cu6Gd] compound. The slurry was refluxed at 75 °C for 4 h and the light blue solid was collected by filtration, washed thoroughly with acetonitrile and acetone until the eluent became colorless and dried under vacuum at 60 °C.For comparative studies, the impregnated Cu6Gd/SBA-15 sample was also prepared by suspending 50 mg of mesoporous SBA-15 silica21 in 4 ml of acetonitrile followed by addition of 62 mg of [Cu6Gd]. The slurry was vigorously stirred for 30 min and the solvent was evaporated slowly at 40 °C. During this process, the compound diffused into the pores of silica by capillary action. The solid product was dried at 60 °C to yield the 55% Cu6Gd/SBA-15 sample. The molecular dispersion of [Cu6Gd] in the silica matrix was confirmed by powder X-ray diffraction, which indicated the absence of Bragg peaks (result not shown here).
Physical characterization
Thermogravimetric (TGA) and differential thermal (DTA) analysis experiments were performed in a Perkin-Elmer Diamond thermal analyzer by heating the sample up to 600 °C at heating rate of 5 °C min−1 under nitrogen flow of ∼100 ml min−1. The X-ray diffraction (XRD) patterns were collected on a PANanalytical Xpert Pro MPD X-ray diffractometer operated at 45 kV and 40 mA using Ni-filtered Cu-Kα radiation (λ = 1.5418 Å) in Bragg–Brentano geometry. Elemental microprobe analyses were performed on a JEOL Model JSM-6390LV scanning electron microscope (SEM) equipped with an Oxford INCA PentaFET-x3 energy dispersive X-ray spectroscopy (EDS) detector. The sample was ground and fixed on a carbon-taped aluminium stub for elemental analysis. Data acquisition was performed several times in different areas of sample using an accelerating voltage of 20 kV and acquisition time of 100 s. Transmission electron microscopy (TEM) was performed using a JEOL Model JEM-2100 electron microscope equipped with an EDS detector (Oxford Instruments) and operated at an accelerated voltage of 200 kV with a thermionic (LaB6) emission source. Samples were prepared by dispersing finely ground powders in ethanol using sonication, followed by depositing a drop of solution onto a Ni grid coated with organic (Formvar) film. Nitrogen adsorption and desorption isotherms were measured at −196 °C with a Quantachrome Autosorb 1-MP volumetric analyzer. Before analysis, the samples were degassed overnight at 353 K under vacuum (<10−5 Torr). The specific surface areas were calculated using the Brumauer–Emmett–Teller (BET) method22 on the adsorption data in the 0.05–0.2 relative pressure (p/p0) range. The pore size distributions were derived from the adsorption branch of isotherms, using the Barrett–Joyner–Halenda (BJH) method23 with modified statistical film thickness equation (KJS method).24 Infrared spectra were recorded on a Perkin-Elmer Model Spectrum-BX FT-IR spectrometer. Spectra were obtained on averaging 100 interferograms with resolution of 2 cm−1. Samples were prepared as KBr pellets. Diffuse reflectance UV-vis-near IR spectra were obtained on a Perkin-Elmer Lambda 950 optical spectrophotometer equipped with an integrating sphere. BaSO4 powder was used as a 100% reflectance standard and base material on which the powder sample was coated. The reflectance data were converted to absorption using to the Kubelka–Munk equation:25α/S = (1 − R)2/(2R), where R is the reflectance and α, S are the absorption and scattering coefficients, respectively.Magnetic measurements
Variable-temperature, solid-state direct current (dc) magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet for complexes. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.Results and discussion
Synthesis and chemical composition
The synthesis of a hybrid CA/SBA-15 material, i.e. –COOH-functionalized mesoporous silica, was performed by a two-step grafting and chemical transformation of cyano groups onto the silica matrix. Namely, we utilized direct co-condensation of ethylcyano silane ((EtO)3Si(CH2)2CN) and tetraethyl orthosilicate (Si(OEt)4) monomers in the presence of non-ionic surfactant, followed by chemical treatment with H2SO4 to remove the template and hydrolyze the –CN groups into –COOH functionalities. The carboxylic groups at the surface provide a suitable medium through which the [Cu6Gd] complexes can be anchored via strong chemical bonds. Inclusion of [Cu6Gd] within the silica host was carried out by refluxing a mixture of CA/SBA-15 and [Cu6Cd] in acetonitrile for 4 h (Scheme 1). The reaction conditions were optimized taking into account the solubility and stability of [Cu6Cd] complex. Prolonging the reaction time does not further increase the loading amount of [Cu6Gd], while other solvents like DMSO, DMF and methanol cause progressive disruption of the molecular structure.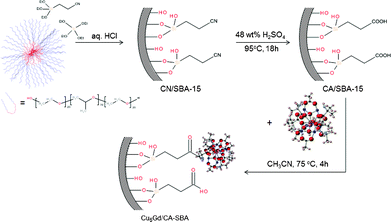 | ||
| Scheme 1 Schematic representation showing the synthesis of Cu6Gd/CA-SBA hybrid material. | ||
The chemical composition of CA/SBA-15 and Cu6Gd/CA-SBA hybrid materials was determined with thermogravimetric/differential thermogravimetric (TG/DTG) and differential thermal (DTA) analysis and energy dispersive X-ray spectroscopy (EDS). The TG curve of CA/SBA-15 shows an appreciable weight loss of ∼11.1 wt% below 320 °C due to release of physisorbed or chemisorbed solvent left in the silica network and possible decomposition of the residual template (Fig. 1a). The rapid weight loss of ∼16.1 wt% observed between 320 and 595 °C, which is accompanied by a broad exothermic peak centered at ∼430 °C in DTA (Fig. 1b), could be attributed mainly to the decomposition of anchored organic groups. This result indicates a concentration of about 2.2 mmol of carboxyl groups per gram of sample, which is considered very high as compared to other values in the literature.26,27 Note that small fluctuations may also contribute to the weight loss in this temperature range due to the hydroxylation of silanol species, but these can be considered minimal.
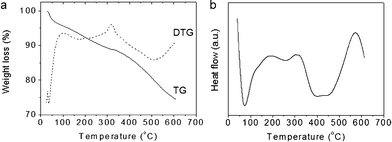
Elemental energy dispersive X-ray (EDS) microanalysis of the Cu6Gd/CA-SBA composite showed the presence of Cu and Cd elements with the expected atomic ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Gd ≈ 6:1. This indicates that the chemical identity of the inserted [Cu6Gd] clusters is retained. The EDS analysis along with the TG/DTA results reveal a ∼55 wt% of [Cu6Gd] content in Cu6Gd/CA-SBA, which corresponds to about one [Cu6Gd] unit per ∼14 carboxyl groups.
:Gd ≈ 6:1. This indicates that the chemical identity of the inserted [Cu6Gd] clusters is retained. The EDS analysis along with the TG/DTA results reveal a ∼55 wt% of [Cu6Gd] content in Cu6Gd/CA-SBA, which corresponds to about one [Cu6Gd] unit per ∼14 carboxyl groups.
Mesoporous structure and surface area
The mesoscopic order of CN/SBA-15, CA/SBA-15 and Cu6Gd/CA-SBA samples was investigated with powder X-ray diffraction (XRD) and transmission electron microscopy (TEM). The diffraction patterns of CN/SBA-15 and CA/SBA-15 show an intense diffraction peak accompanied by a weak feature in the 2θ range of 0.7–2.2°, which according to TEM could be attributed respectively to the (100) and (200) reflections of hexagonal P6mm structure (Fig. 2a). The observation of these peaks constitutes a clear evidence of the long-range hexagonal order in these materials. The XRD pattern of Cu6Gd/CA-SBA resembles that of the pristine CA/SBA-15 solid, indicating that the hexagonal array of the silica host is preserved undisrupted even after [Cu6Cd]-functionalization. The small decrease in intensity of the (100) peak and absence of a high-order (200) reflection for the [Cu6Cd]-loaded sample is attributed to the reduction in X-ray scattering contrast between the silica walls and the pore-filling cluster.28 The mesoporous structures show similar lattice parameters (a0) of ∼11.6–11.8 nm, as calculated from the angular position of the (100) diffraction peak. In wide-angle XRD (2θ > 20°) of Cu6Gd/CA-SBA, the essential absence of Bragg diffractions excludes the possibility of large [Gu6Gd] ensembles within the silica matrix (see Fig. 2b). This means that the [Cu6Gd] clusters are well distributed in the silica mesopores, which is a fascinating property if we consider the very high [Cu6Gd] loading (ca. 55 wt%) of this material.![(a) Low-angle XRD patterns of (i) CN/SBA-15, (ii) CA/SBA-15 and (iii) Cu6Gd/CA-SBA hybrid materials. (b) Wide-angle XRD patterns of (i) mesoporous Cu6Gd/CA-SBA and (ii) polycrystalline [Cu6Gd] compound.](/image/article/2012/RA/c2ra21455e/c2ra21455e-f2.gif) | ||
| Fig. 2 (a) Low-angle XRD patterns of (i) CN/SBA-15, (ii) CA/SBA-15 and (iii) Cu6Gd/CA-SBA hybrid materials. (b) Wide-angle XRD patterns of (i) mesoporous Cu6Gd/CA-SBA and (ii) polycrystalline [Cu6Gd] compound. | ||
Fig. 3, displays representative TEM images and the corresponding fast Fourier transform (FFT) patterns of the mesoporous Cu6Gd/CA-SBA. The TEM images, recorded along the [001] and [110] directions, reveal large domains of hexagonally arranged pore channels. These results along with low-angle XRD data confirm that the long-range order of the pristine CA/SBA-15 material is preserved to a considerable extent after immobilization of the [Cu6Gd] complexes. A detailed analysis of TEM images indicates an average pore-to-pore distance of about 11.2 nm and a pore size of about 7.1 nm, consistent with XRD and N2 physisorption results (see below). Although the [Cu6Gd] clusters inside the mesopore channels are difficult to observe by TEM imaging, their presence was evident by selective area X-ray microanalysis (TEM-EDS). The TEM-EDS spectra acquired across various local areas of mesoporous structure display strong signals from silicon, copper and gadolinium elements with atomic ratios of Si/Cu/Gd ∼ 67.1:28.0:4.8 (Fig. 3d). These values correspond to ∼0.38 mmol of clusters per gram of host material, which is very close to the compositional estimate from scanning electron microscopy (SEM) EDS analysis (∼0.35 mmol g−1). Noteworthy, no visible Gd-rich aggregates were observed on the surface grains, confirming that the molecular clusters are exclusively hosted inside the mesopores.
![(a–c) Typical TEM images and corresponding FFT patterns (insets) of the mesoporous Cu6Gd/CA-SBA taken along the [001] (a) and [110] (b and c) zone axes of hexagonal P6mm symmetry. (d) EDS spectrum obtained on TEM for mesoporous Cu6Gd/CA-SBA. The nickel signal comes from the Ni TEM grid.](/image/article/2012/RA/c2ra21455e/c2ra21455e-f3.gif) | ||
| Fig. 3 (a–c) Typical TEM images and corresponding FFT patterns (insets) of the mesoporous Cu6Gd/CA-SBA taken along the [001] (a) and [110] (b and c) zone axes of hexagonal P6mm symmetry. (d) EDS spectrum obtained on TEM for mesoporous Cu6Gd/CA-SBA. The nickel signal comes from the Ni TEM grid. | ||
Nitrogen physisorption measurements were performed to assess the textural properties of the CN/SBA-15, CA/SBA-15 and Cu6Gd/CA-SBA materials. All the samples display typical type-IV adsorption–desorption isotherms with a pronounced H3 hysteresis loop, according to the IUPAC classification, characteristic for mesoporous solids with ink-bottle pore structure (Fig. 4a).29 The sharp increase in the adsorption branch at relative pressure range p/p0 = 0.6–0.7 is related to the capillary condensation of nitrogen in narrow-sized mesopores. This suggests that despite the presence of organosilanes and [Cu6Gd] clusters onto the silica walls, a significant portion of mesopores are filled with nitrogen, leading to an inflection point in the adsorption curve. As expected, the CN/SBA-15 hybrid sample exhibits a low nitrogen adsorption due to the filling of mesopores with template, while the template-free CA/SBA-15 demonstrates a significant increase in nitrogen uptake which is reflected in the higher surface area and pore volume. Analysis of absorption data with the Brunauer–Emmett–Teller (BET) method indicates that mesoporous CN/SBA-15 and CA/SBA-15 have a surface area of 136 and 578 m2 g−1 and a pore volume of 0.23 and 0.96 cm3 g−1, respectively. After insertion of [Cu6Gd] into the pores of CA/SBA-15, the surface area and pore volume drop to 140 m2 g−1 and 0.21 cm3 g−1, respectively, as a consequence of the pore clogging and the heavier structure due to the added [Cu6Gd] clusters. Nevertheless, the Cu6Gd/CA-SBA composite still possesses a noteworthy surface area and porosity which is accessible on adsorption. All the textural parameters of the CN/SBA-15, CA/SBA-15 and Cu6Gd/CA-SBA hybrid materials are given in Table 1.
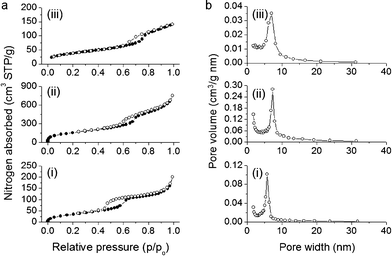 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms at 77 K and (b) corresponding BJH pore size distribution calculated from the adsorption branch of mesoporous materials: (i) CN/SBA-15, (ii) CA/SBA-15 and (iii) Cu6Gd/CA-SBA. | ||
| Sample | Surface area (m2 g−1) | Pore volumea (cm3 g−1) | Pore size (nm) | d 100 spacing (nm) | Unit cellb (nm) |
|---|---|---|---|---|---|
| a Cumulative pore volume at p/p0 = 0.95. b The size of unit cell is given by a0 = (2/√3)d100. | |||||
| CN/SBA-15 | 136 | 0.23 | 5.8 | 10.06 | 11.6 |
| CA/SBA-15 | 578 | 0.96 | 7.4 | 10.25 | 11.8 |
| Cu6Gd/CA-SBA | 140 | 0.21 | 6.9 | 10.02 | 11.6 |
The pore size distributions were calculated from the adsorption branches of the isotherms using the modified Barrett–Joyner–Halenda (BJH) method. All the samples exhibit a quite narrow distribution of pore size with a peak maximum being varied with the treatment process (Fig. 4b). Specifically, after acid treatment of CN/SBA-15, the pore size was significantly increased from ∼5.8 to ∼7.4 nm, owing to the elimination of surfactants. While incorporation of [Cu6Gd] in CA/SBA-15 mesopores led to a reduction of the pore width (∼6.9 nm) due to the pore occupancy by the immobilized clusters.
Spectroscopy
The organic functionalities as well as the molecular structure of [Cu6Gd] complexes on the silica surface were characterized by Fourier transform infrared (FT-IR) and diffuse reflectance ultraviolet-visible-near IR (UV-vis-NIR) spectroscopy. The IR spectra of mesoporous CN/SBA-15, CA/SBA-15 and Cu6Gd/CA-SBA materials and the [Cu6Gd] compound are shown in Fig. 5. The hybrid materials show two intense absorption peaks at ∼974 cm−1 and ∼1045–1215 cm−1 due to the Si–OH and Si–O–Si vibration bands of the silica matrix, respectively.30 The CN/SBA-15 shows a characteristic absorption peak at ∼2257 cm−1, corresponding to the stretching vibration of the cyano (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) groups. The weak absorption peaks at ∼2878 and ∼2932 cm−1 are ascribed to the C–H stretching vibration modes of the ethyl chain. After treatment with sulfuric acid, an intense peak appears at ca. 1723 cm−1 that is related to the carbonyl (C
N) groups. The weak absorption peaks at ∼2878 and ∼2932 cm−1 are ascribed to the C–H stretching vibration modes of the ethyl chain. After treatment with sulfuric acid, an intense peak appears at ca. 1723 cm−1 that is related to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching mode of the carboxylic species. This absorption band is significantly bathochromic/red-shifted compared to the monomeric propanoic acid (ca. 1783 cm−1),31 indicating strong hydrogen-bonding interactions of –COOH groups with other neighboring –COOH groups and/or silanol (SiOH) species at the surface.32 Indeed, the broad absorption in the 3000–3600 cm−1 range is associated with the hydrogen-bonded hydroxyl species. These results together with the strong reduction of the cyano peak imply a complete transformation of the –CN groups into –COOH functionalities.
O) stretching mode of the carboxylic species. This absorption band is significantly bathochromic/red-shifted compared to the monomeric propanoic acid (ca. 1783 cm−1),31 indicating strong hydrogen-bonding interactions of –COOH groups with other neighboring –COOH groups and/or silanol (SiOH) species at the surface.32 Indeed, the broad absorption in the 3000–3600 cm−1 range is associated with the hydrogen-bonded hydroxyl species. These results together with the strong reduction of the cyano peak imply a complete transformation of the –CN groups into –COOH functionalities.
![Infrared spectra of hybrid (i) CN/SBA-15, (ii) CA/SBA-15 and (iii) Cu6Gd/CA-SBA materials and (iv) [Cu6Gd] compound.](/image/article/2012/RA/c2ra21455e/c2ra21455e-f5.gif) | ||
| Fig. 5 Infrared spectra of hybrid (i) CN/SBA-15, (ii) CA/SBA-15 and (iii) Cu6Gd/CA-SBA materials and (iv) [Cu6Gd] compound. | ||
The IR spectrum of Cu6Gd/CA-SBA indicates the presence of [Cu6Gd] compounds, showing a series of absorption peaks in the region of 620–850 cm−1 due to the overlap of N–H and C–H bending vibration bands of α-aminoisobutyrate ligands. The vibrational feature at ∼578 cm−1 is ascribed to the stretching mode of Gd–O bonds in the cluster. The absence of any shift for the Gd–O absorption suggests that the cluster core is fully retained within the mesoporous host structure. Compared to the pristine CA/SBA-15, the Cu6Gd/CA-SBA exhibits a remarkable bathochromic shift (by over 60 cm−1) of the carbonyl stretching band. This suggests that the [Cu6Gd] complexes are strongly anchored to the terminal carbonyl groups possibly via –CO–N amide bonds.
Fig. 6 shows the diffuse reflectance UV-vis-NIR spectra of mesoporous CA/SBA-15 and Cu6Gd/CA-SBA materials and [Cu6Gd] compound. The CA/SBA-15 exhibits a strong absorption at ∼226 nm accompanied by a weak shoulder at ∼273 nm, which are attributed to the π → π* and n → π* electron transitions in the CO bond of carboxylic acids, respectively.33 The analogous absorption bands in the spectrum of the [Cu6Gd]-loaded sample are hard to observe because of the high concentration of complex. The Cu6Gd/CA-SBA exhibits an intense absorption at ∼270 nm, corresponding mainly to the O(−II) → Cu(II) charge transfer transition in Cu–O bonds and a broad absorption peak at low-energy regime (∼700–800 nm), corresponding to the internal d–d electron transition of Cu(II) ions of the polynuclear [Cu6Gd] complex.34,35 The series of absorption peaks in the range of 1400–2000 nm are assigned to the intra-atomic d–d electron transitions of tetrahedral Cu(II) species.36 These absorption bands are slightly bathochromic shifted (about 5 nm) compared to the pure [Cu6Gd] solid, possible due to the binding of [Cu6Gd] clusters to the carboxylic acids.
![Diffuse reflectance UV-vis-NIR spectra of mesoporous CA/SBA-15 and Cu6Gd/CA-SBA materials and [Cu6Gd] compound.](/image/article/2012/RA/c2ra21455e/c2ra21455e-f6.gif) | ||
| Fig. 6 Diffuse reflectance UV-vis-NIR spectra of mesoporous CA/SBA-15 and Cu6Gd/CA-SBA materials and [Cu6Gd] compound. | ||
To investigate the stability of the incorporated clusters in the host material, Cu6Gd/CA-SBA was dispersed in acetonitrile under vigorous stirring for 1 h and the copper and gadolinium content remaining in sample were determined by SEM-EDS analysis. For comparison we also prepared a mesoporous Cu6Gd/SBA-15 composite by impregnating 55 wt% of [Cu6Gd] on the surface of mesoporous SBA-15 silica and this sample was examined for [Cu6Gd] leaching under similar conditions. The analytical data, in Table 2, indicated no appreciable loss of [Cu6Gd] clusters (ca. 3%) from Cu6Gd/CA-SBA, suggesting strong binding of [Cu6Gd] units to the –COOH functional groups. This is in line with the results from IR and UV-vis-NIR spectroscopy. On the contrary, however, the impregnated Cu6Gd/SBA-15 sample suffered a remarkable depletion of [Cu6Gd] (ca. 21%), which could be explained by the weak interactions between the complexes and the surface silanol species. These results demonstrate that the modification of silica surface by carboxyl groups is essential for the strong anchoring of [Cu6Gd] compounds.
| Sample | Atomic ratioa Si:Cu:Gd |
[Cu6Gd] loading (wt%) | [Cu6Gd] lossb (%) |
|---|---|---|---|
| a SEM-EDS elemental data normalized to the six-atom Cu cluster. b Percent of clusters lost determined by reference to the initial [Cu6Gd] content in composite samples (55 wt%). | |||
| Cu6Gd/CA-SBA | 16.1:6.0:1.0 |
53 | 3.2 |
| Cu6Gd/SBA-15 | 34.3:6.0:1.0 |
43 | 21.3 |
Magnetic properties
Direct current magnetic susceptibility measurements were performed on samples of Cu6Gd/CA-SBA material and pure [Cu6Gd] compound in the 5–300 K range under an applied field of 0.1 T. The results are plotted as the χMT product vs. T in Fig. 7. For the pure complex, the χMT value of 10.69 cm3 mol−1 K at 300 K is as expected for six non-interacting Cu(II) ions (g = 2.24) and one Gd(III) centre (gj = 2.0). Upon cooling, the χMT value increases to a maximum value of 17.31 cm3 mol−1 K at 5 K, indicating the presence of dominant ferromagnetic interactions within the metallic cluster, stabilizing a high-spin ground-state of S = 13/2 for the complex. The same situation is observed for the Cu6Gd/CA-SBA material; at room temperature the χMT value is 10.71 cm3 mol−1 K, and upon cooling the χMT value starts increasing as in the case of the pure complex. In the 20–300 K temperature range the two species display identical magnetic behaviour, strongly suggesting that once immobilized within the silica host the [Cu6Gd] unit retains both its structural and magnetic integrity. Below ∼20 K, the magnetic behaviour of the immobilized cluster deviates from the pure complex, most probably due to intermolecular magnetic interactions between neighbouring clusters within the mesoporous material.![Plot of χMT vs. T for Cu6Gd/CA-SBA material and pure [Cu6Gd] complex under an applied dc field of 1000 G.](/image/article/2012/RA/c2ra21455e/c2ra21455e-f7.gif) | ||
| Fig. 7 Plot of χMT vs. T for Cu6Gd/CA-SBA material and pure [Cu6Gd] complex under an applied dc field of 1000 G. | ||
Conclusion
In summary, we have reported the encapsulation of the high-spin heptanuclear copper(II)–gadolinium(III) molecular compound [Cu6Gd] within –COOH-functionalized mesoporous silica. The ordered mesoscopic structure of the hybrid CA/SBA-15 material, as assessed by powder X-ray diffraction and transmission electron microscopy, is maintained undisturbed after incorporation of the [Cu6Gd] compounds. The organic functionalities and molecular structure of incorporated [Cu6Gd] clusters were confirmed by IR and diffuse reflectance UV-vis-NIR spectroscopy. Nitrogen physisorption indicated that the [Cu6Cd]-loaded material preserves the mesoporosity of the pristine CA/SBA-15 sample to a certain extent, but with surface area (from 578 to 140 m2 g−1) and pore size (from 7.4 to 6.9 nm) reduction due to the occupancy of mesopores by the immobilized clusters. The Cu6Gd/CA-SBA nanocomposite exhibits magnetic properties that derive from the incorporated clusters. Magnetic measurements showed that this material has similar magnetic properties to that of pure [Cu6Gd], presenting dominant ferromagnetic interactions within the metallic cluster with high-spin S = 13/2 ground state. We believe that this approach can be extended to other magnetic molecular clusters, allowing the preparation of a diverse set of porous magnetic nanocomposites.Acknowledgements
Financial support by the European Union under the Marie Curie International Reintegration Grant (No. 230868), the Greek Ministry of Education under “THALES” project (MIS 377064) and the University of Crete–Special Account for Research (KA 3475) is kindly acknowledged. The authors also thank P. Trikalitis (University of Crete, Heraklion) for nitrogen physisorption measurements.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Y. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- H. J. Eppley, H. L. Tsai, N. Devries, K. Folting, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1995, 117, 301–317 CrossRef CAS.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS.
- L. Krusin-Elbaum, T. Shibauchi, B. Argyle, L. Gignac and D. Weller, Nature, 2001, 410, 444–446 CrossRef CAS.
- M. H. Jo, J. E. Grose, K. Baheti, M. M. Deshmukh, J. J. Sokol, E. M. Rumberger, D. N. Hendrickson, J. R. Long, H. Park and D. C. Ralph, Nano Lett., 2006, 6, 2014–2020 CrossRef CAS.
- G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285–8288 CrossRef CAS.
- M. Evangelisti and E. K. Brechin, Dalton Trans., 2010, 39, 4672–4676 RSC.
- M. Manoli, A. Collins, S. Parsons, A. Candini, M. Evangelisti and E. K. Brechin, J. Am. Chem. Soc., 2008, 130, 11129–11139 CrossRef CAS.
- M. Clemente-Leon, H. Soyer, E. Coronado, C. Mingotaud, C. J. Gomez-Garcia and P. Delhaes, Angew. Chem., Int. Ed., 1998, 37, 2842–2845 CrossRef CAS.
- L. Bogani, L. Cavigli, M. Gurioli, R. L. Novak, M. Mannini, A. Caneschi, F. Pineider, R. Sessoli, M. Clemente-Leon, E. Coronado, A. Cornia and D. Gatteschi, Adv. Mater., 2007, 19, 3906 CrossRef CAS.
- M. Clemente-Leon, E. Coronado, A. Forment-Aliaga, P. Amoros, J. Ramirez-Castellanos and J. M. Gonzalez-Calbet, J. Mater. Chem., 2003, 13, 3089–3095 RSC.
- S. Willemin, G. Arrachart, L. Lecren, J. Larionova, T. Coradin, R. Clerac, T. Mallah, C. Guerin and M. Sanchez, New J. Chem., 2003, 27, 1533–1539 RSC.
- T. Coradin, J. Larionova, A. A. Smith, G. Rogez, R. Clerac, C. Guerin, G. Blondin, R. E. P. Winpenny, C. Sanchez and T. Mallah, Adv. Mater., 2002, 14, 896–898 CrossRef CAS.
- D. Ruiz-Molina, M. Mas-Torrent, J. Gomez, A. I. Balana, N. Domingo, J. Tejada, M. T. Martinez, C. Rovira and J. Veciana, Adv. Mater., 2003, 15, 42 CrossRef CAS.
- G. E. Poirier, Chem. Rev., 1997, 97, 1117–1127 CrossRef CAS.
- S. A. Levi, P. Guatteri, F. van Veggel, G. J. Vancso, E. Dalcanale and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2001, 40, 1892–1896 CrossRef CAS.
- A. Cornia, A. C. Fabretti, M. Pacchioni, L. Zobbi, D. Bonacchi, A. Caneschi, D. Gatteschi, R. Biagi, U. Del Pennino, V. De Renzi, L. Gurevich and H. S. J. Van der Zant, Angew. Chem., Int. Ed., 2003, 42, 1645–1648 CrossRef CAS.
- G. J. Sopasis, A. B. Canaj, A. Philippidis, M. Siczek, T. Lis, J. R. O'Brien, M. M. Antonakis, S. A. Pergantis and C. J. Milios, Inorg. Chem., 2012, 51, 5911–5918 CrossRef CAS.
- C. M. Yang, B. Zibrowius and F. Schuth, Chem. Commun., 2003, 1772–1773 RSC.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. H. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. Kruk, M. Jaroniec and A. Sayari, J. Phys. Chem. B, 1997, 101, 583–589 CrossRef CAS.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38, 448 CrossRef CAS.
- S. Fiorilli, B. Camarota, D. Perrachon, M. C. Bruzzoniti, E. Garrone and B. Onida, Chem. Commun., 2009, 4402–4404 RSC.
- S. Fiorilli, B. Onida, B. Bonelli and E. Garrone, J. Phys. Chem. B, 2005, 109, 16725–16729 CrossRef CAS.
- W. Hammond, E. Prouzet, S. D. Mahanti and T. J. Pinnavaia, Microporous Mesoporous Mater., 1999, 27, 19–25 CrossRef CAS.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, 2nd edn, Academic Press, London, UK, 1982 Search PubMed.
- R. C. Schroden, C. F. Blanford, B. J. Melde, B. J. S. Johnson and A. Stein, Chem. Mater., 2001, 13, 1074–1081 CrossRef CAS.
- V. Sablinskas, M. Pucetaite, J. Ceponkus and L. Kimtys, J. Mol. Struct., 2010, 976, 263–269 CrossRef CAS.
- K. Takei, R. Takahashi and T. Noguchi, J. Phys. Chem. B, 2008, 112, 6725–6731 CrossRef CAS.
- C. N. R. Rao, Ultra-violet and visible spectroscopy: chemical applications, Butterworths, 1961 Search PubMed.
- M. C. Marion, E. Garbowski and M. Primet, J. Chem. Soc., Faraday Trans., 1990, 86, 3027–3032 RSC.
- L. Bonneviot, O. Legendre, M. Kermarec, D. Olivier and M. Che, J. Colloid Interface Sci., 1990, 134, 534–547 CrossRef CAS.
- A. Le Nestour, M. Gaudon, G. Villeneuve, R. Andriessen and A. Demourgues, Inorg. Chem., 2007, 46, 2645–2658 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |