DOI:
10.1039/C2RA21380J
(Communication)
RSC Adv., 2012,
2, 8243-8249
Highly fluorescent benzofuran derivatives of the GFP chromophore†
Received
6th July 2012
, Accepted 11th July 2012
First published on 13th July 2012
Abstract
Intramolecular cyclization reactions of Green Fluorescent Protein chromophores (GFPc) containing an arylethynyl ortho-substituent at the phenol ring provide new aryl-substituted benzofuran derivatives of the GFPc. Some of these heteroaromatic compounds exhibit significantly enhanced fluorescence at red-shifted emission wavelengths relative to the parent GFPc structure.
Introduction
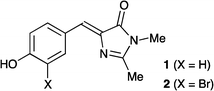
The green fluorescent protein (GFP) is a photoactive protein that has been widely used in molecular and cell biology on account of its fluorescent properties.1 The chromophore itself (GFPc) is a 4-hydroxybenzylideneimidazolinone, which sits at the center of the protein β-barrel via both covalent linkages and a hydrogen-bond network.2 The fluorescent properties have been found to depend strongly on this fixation of the chromophore in the protein binding pocket. Thus, the dimethyl GFPc derivative 1 (Fig. 1) shows a very low fluorescence quantum yield.3 Several derivatives have in recent years been prepared in order to understand the photophysical properties of the chromophore and to enhance the fluorescence in the absence of a protein environment.4 For example, Wu and Burgess5 have obtained strong fluorescence by replacing the phenol with a pyrrole ring followed by bridging of the two rings via complexation of BF2. Also Tolbert, Yampolsky, Solntsev, and co-workers6 have shown that a GFPc analogue in which the phenol ring is locked to the imidazolinone ring via a BF2 bridge becomes highly fluorescent. Another analogue in which the phenol ring is replaced by a hydroxypyridine ring was found to gain fluorescence when the two rings are bridged by Zn2+ or Cd2+.6 As another way to lock the structure, we have recently shown that encapsulation of the chromophore in a covalently linked cyclodextrin cavity results in fluorescence enhancement.7
We became interested to explore the possibility for tuning both the absorption and fluorescence properties of the GFPc by extending the conjugation of the phenol ring rather than hindering rotation about the aryl–alkene bond. In particular, incorporation of an alkyne functionality at an ortho-position to the OH group could provide a precursor for a heteroaromatic benzo[b]furan by an intramolecular 5-endo-dig cyclization. Such cyclizations are known to be promoted by transition metals,8 but can also be accomplished under metal-free conditions.9 The benzofuran ring is a key structural unit in several biologically active compounds and is present in many natural products;10 derivatized benzofurans are therefore in general attractive heteroaromatic targets. In addition, benzo[b]furan is a fluorophore itself,11 and changing the phenol part of the GFPc into a benzofuran could therefore be a particularly attractive and simple way of enhancing the fluorescence. Substitution with either electron-donating or withdrawing groups should allow for additional fine-tuning of the optical properties of the GFPc.
Results and discussion
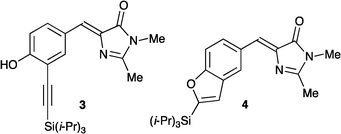
We have recently reported the synthesis of the bromo-functionalized GFPc 2 (Fig. 1),12 which was first attempted as a building block for further functionalization. Treating it with triisopropylsilylacetylene under palladium-catalyzed conditions (Pd(PhCN)2, (t-Bu)3P, CuI, (i-Pr)2NH, DMF) at elevated temperatures does result in some conversion to the ethynyl GFPc 3 and the benzofuran derivative 4 (Fig. 2), most likely resulting from a cyclization of 3 as intermediate. It was, however, difficult to isolate these products pure and in reasonable amounts. We also made several attempts to incorporate a phenyl group by subjecting 2 to a Suzuki cross-coupling with phenyl boronic acid, but all of these attempts failed.
 |
| | Fig. 2 GFPc target molecules. | |
To enhance the reactivity of the arylhalide, we prepared the iodo-derivative 5 according to Scheme 1. The synthesis follows standard procedures13 for preparing GFPc derivatives. First, the known 4-hydroxy-3-iodobenzaldehyde 614 was condensed with N-acetylglycine in the presence of acetic anhydride and sodium acetate to furnish the oxazolinone 7 (previously reported15). The Z-configuration of 7 was confirmed by X-ray crystallography (Fig. 3).† By subjecting this compound to an attack by methylamine, it was converted to a ring-opened and deacetylated product, which was cyclized in refluxing absolute ethanol in the presence of molecular sieves to GFPc 5. Unfortunately, however, when treating this aryliodide with triisopropylsilylacetylene under Sonogashira coupling conditions (using PdCl2(PPh3)2, CuI, NEt3), only traces of desired coupling products seemed to form. Thus, direct palladium-catalyzed reactions on the GFPc do not seem to be a viable and easily reproducible method of functionalization, at least not in our hands.
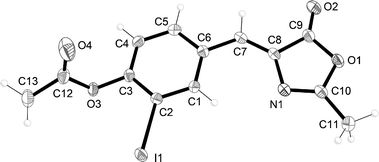 |
| | Fig. 3 Molecular structure of 7 (with displacement ellipsoids at 50% probability for non-H atoms). Crystals were grown from CH2Cl2/heptane. CCDC 890138. | |
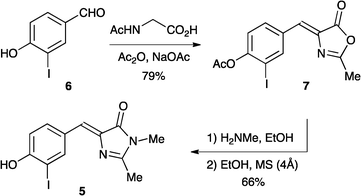 |
| | Scheme 1 Synthesis of GFPc with iodo substituent. | |
As a new strategy, we decided to incorporate the ethynyl substituent earlier in the synthesis according to Scheme 2. The synthesis starts from the known iodobenzaldehyde derivative 8,16 which was treated with four differently substituted terminal alkynes under Sonogashira conditions to provide products 9–12. These aldehydes were then condensed with N-acetylglycine to provide intermediate oxazolinone derivatives, which were found to be rather unstable and hence only purified by pouring them through a short plug of silica before they were used in the next step. Treatment with methylamine followed by refluxing of the intermediate in absolute ethanol containing molecular sieves ultimately gave the alkynylated GFPc 3 and the benzo[b]furan GFPc derivatives 13–15. Thus, the intermediate alkynyl products serving as precursors for 13–15 were found to undergo intramolecular cyclization to the corresponding benzofurans, notably in the absence of a palladium catalyst. This cyclization seems to occur in the final step during reflux of the alkynylphenol in ethanol containing molecular sieves, as supported by successful conversion of a simpler derivative of 2-(phenylethynyl)phenol to a benzofuran under similar conditions. Compound 3 shows two characteristic IR absorptions at 3255 cm−1 (O–H stretch) and 2147 cm−1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch). These absorptions are absent in the IR spectra of compounds 13–15, indicating the formation of cyclized benzofuran products. The structural assignment was also confirmed by 13C-NMR APT experiments, which show two positive signals for compound 3 in the range 80–110 ppm assigned to the internal alkyne. These signals are not present in the spectra of compounds 13–15; instead an additional peak appears around 155 ppm, which is typical for oxygen-bound sp2-hybridized carbon atoms.
C stretch). These absorptions are absent in the IR spectra of compounds 13–15, indicating the formation of cyclized benzofuran products. The structural assignment was also confirmed by 13C-NMR APT experiments, which show two positive signals for compound 3 in the range 80–110 ppm assigned to the internal alkyne. These signals are not present in the spectra of compounds 13–15; instead an additional peak appears around 155 ppm, which is typical for oxygen-bound sp2-hybridized carbon atoms.
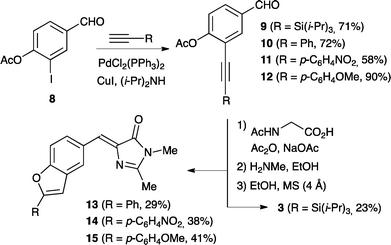 |
| | Scheme 2 Synthesis of benzofuran derivatives of the GFPc. | |
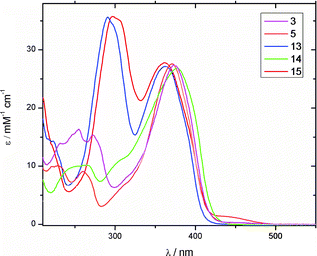
The UV-Vis absorption spectra of the neutral GFP chromophores 3, 5, and 13–15 recorded in methanol are shown in Fig. 4 and data are collected in Table 1 together with the related GFPc 16 and the benzofurans 17 and 1817 (Fig. 5). The compounds have structured absorption spectra and their absorption properties above 350 nm are very similar, consisting of a broad band with a maximum located between 362 and 377 nm depending on the nature of the substituent. GFPc derivatives 3 and 5 also show a shoulder at ∼320 nm and a second, less pronounced peak at ∼260 nm. Addition of NaOMe results in a red-shift in the characteristic GFPc absorption to ca. 430 nm for 1, 3 and 5 (as shown in ESI†), in agreement with generation of the phenolate anion, while the spectra of 13–15 are unaltered by addition of base. Desilylation of 3 is expected to occur upon addition of NaOMe, but, importantly, the red-shifted GFPc absorption supports that this compound has the phenol/phenolate functionality intact. In contrast to the parent GFPc structures, the spectra of the new benzofuran derivatives exhibit a second strong peak at lower wavelength that can be ascribed to the absorption of the benzofuran unit (Fig. 4 and Table 1). GFPc derivatives 13 and 15 display a high value of the extinction coefficient at this lower wavelength (ε ∼ 3.6 × 104 M−1 cm−1) supporting a strong electronic delocalization in these systems. The peak of 14 below 300 nm is blue-shifted relative to 13 and 15 due to the electron-withdrawing effect of the NO2 group.
 |
| | Fig. 5 Reference compounds for spectroscopic studies. | |
Table 1 Photophysical characteristics of compounds in MeOH
| Compound |
λ
absmax/nm (ε/M−1 cm−1) |
λ
emismax/nm |
φ
fl
|
|
1
|
247 (7300), 371 (26900) |
445 |
10−4 |
|
3
|
255 (16400), 377 (27300) |
445 |
10−4 |
|
5
|
261 (9100), 371 (27600) |
441 |
2 × 10−4 |
|
13
|
290 (35600), 362 (27100) |
525 |
2 × 10−3 |
|
14
|
265 (10200), 373 (26800) |
454 |
5 × 10−4 |
|
15
|
297 (35700), 362 (27800) |
612 |
7 × 10−3 |
|
16
|
252 (7400), 376 (27200) |
447 |
10−4 |
|
17
|
240 (9300), 274 (2100), 281 (2400) |
290 |
9 × 10−2 |
|
18
|
262 (30000), 294 (22000) |
495 |
9 × 10−3 |
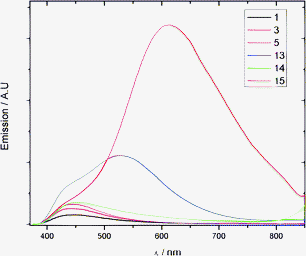
The newly synthesized GFP chromophores also have interesting emission properties as revealed by the fluorescence spectra in Fig. 6 (see also ESI†) and listed in Table 1; thus, the benzofuran derivatives exhibit enhanced fluorescence as clearly visible by the naked eye (Fig. 7), while presence of the alkyne unit in 3 has no significant influence. The fluorescence of 1, 3, 5 and 14 is characterized by a small Stokes shift (∼4000 cm−1) that increases to about 8000 cm−1 and 11000 cm−1 for 13 and 15, respectively. Interestingly, 13 displays both an emission peak (525 nm) very close to the one of the actual protein (GFP) and a shoulder at ∼440 nm close to that of the simple GFPc derivatives 1, 3, 5, and 16. For comparison, the emission of benzofuran (17) occurs at a significantly lower wavelength (290 nm), while the benzofuran derivative 18 incorporating a phenyl substituent and an electron-withdrawing aldehyde group (mimicking the electron-withdrawing methyleneimidazolinone unit) exhibits an emission that is blue-shifted by only 30 nm relative to that of 13. Compounds 3 and 5 exhibit only very weak fluorescence at room temperature (quantum yields (φfl) ∼10−4); the reason for this being a deactivation process from the singlet excited state (cis–trans photoisomerization of the exo-methylene double bond). As for the parent GFP chromophore, the emission spectrum at room temperature originates from the S1-S0 transition and it is approximately the mirror image of the S0-S1 absorption spectrum, pointing out the small differences between the nuclear configuration of the ground and excited electronic states. By contrast, the GFPc derivatives 13–15 show not only a large red-shift in the emission spectra due to the high conjugation of the substituent, but also larger values of φfl (between 5 and 70 times higher than the parent GFPc structure) with the most significant substitution effect exhibited by the methoxyphenyl derivative 15.
 |
| | Fig. 7 Fluorescence of GFPc molecules dissolved in MeOH at a concentration of 0.1 mM. From left to right: neat MeOH, 1, 5, 13, 15, 3, 14. | |
Conclusions
In conclusion, we have devised a simple route to new benzofuran derivatives of the GFP chromophore via intramolecular cyclization of an arylethynylphenol unit. The compounds exhibit significantly enhanced fluorescent properties relative to the parent GFPc and the strategy thus presents a convenient and simple alternative to the previous ones in which fluorescence was enhanced by reducing the flexibility of the chromophore.
Experimental
General experimental conditions and characterization
Dry THF was obtained from a Solvent Purification System, Innovative Technology, Inc. Purification of products was carried out by flash chromatography on silica gel (ROCC 40–63 micron) or by dry column vacuum chromatography on silica gel (ROCC 15–40 micron). Thin-layer chromatography (TLC) was carried out using aluminum sheets pre-coated with silica gel 60F (Merck 5554). The plates were inspected under UV light. NMR spectra were either recorded on a 500-MHz Bruker instrument with cryoprobe or on a 300-MHz Varian instrument using the residual solvent as the internal standard. All chemical shifts are quoted on the δ scale (ppm), and all coupling constants (J) are expressed in Hz. In APT spectra, CH and CH3 correspond to negative signals and C and CH2 correspond to positive signals. Electrospray ionisation (ESP) mass spectra were recorded on a Micromass Q-TOF mass spectrometer with PEG (polyethylene glycol) as internal standard for HRMS. Gas chromatography (GC) mass spectra were recorded on an Agilent 6890A (G1530A) gas chromatograph coupled with a Agilent mass selective detector 5973Network. IR spectra were obtained using a Bruker ALPHA-P spectrometer using the ‘ATR platinum Diamond 1 Refl’ accessory. Melting points are uncorrected. Elemental analyses were obtained at Department of Chemistry, University of Copenhagen.
UV-Visible absorption spectra were recorded at room temperature with a Cary 50 UV-Vis spectrophotometer using quartz cells of 1 cm path. The steady state excitation and emission spectra were measured with a FluoroLog-3 spectrophotometer at room temperature, with 3 nm excitation and emission slit widths, an integration time of 0.5 s and corrected for the instrumental function. The fluorescence quantum yields were determined at room temperature taking fluorescein in 0.1 M NaOH as a standard (φfl = 0.92).18 The solute concentrations were adjusted to achieve an optical density of 0.1 at the excitation wavelength (fixed at 365 nm).
(Z)-4-(4-Acetoxy-3-iodobenzylidene)-2-methyl-1,3-oxazol-5(4H)-one (7)
To a mixture of 4-hydroxy-3-iodobenzaldehyde (614), N-acetylglycine (3.40 g, 29.0 mmol), (6.00 g, 24.2 mmol) and NaOAc (1.64 g, 20.0 mmol) was added Ac2O (14.8 g, 13.7 mL, 145 mmol) under an argon atmosphere, and the suspension was heated to 110 °C for 1.5 h. The mixture was then kept at 5 °C overnight before H2O (10 mL) was added and the brown precipitate was collected. The brown crude was dissolved in EtOAc (100 mL) and poured through a short plug of SiO2, which gave the product as a pale-yellow solid (7.05 g, 79%). MP: 191–193 °C; lit.:15 184–185 °C. 1H NMR (500 MHz, DMSO-d6): δ 8.70 (d, J = 1.9 Hz, 1H), 8.23 (dd, J = 8.4, 1.9 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.21 (s, 1H), 2.41 (s, 3H), 2.35 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 168.2, 167.5, 167.1, 152.6, 142.0, 133.3, 133.0, 132.8, 127.0, 123.6, 92.3, 20.9, 15.5. MS (FAB+): m/z = 372.2 (M+H+). EA (C13H10INO4, 371.13): calcd. C 42.07, H 2.72, N 3.77; found C 42.18, H 2.48, N 3.59.
(Z)-4-(4-Hydroxy-3-iodobenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one (5)
(Z)-4-(4-Acetoxy-3-iodobenzylidene)-2-methyl-1,3-oxazol-5(4H)-one (7) (2.00 g, 5.39 mmol) was dissolved in absolute ethanol (275 mL) under argon, and methylamine (33% wt. in ethanol, 10 mL, 80.8 mmol) was added. The orange solution was left for 40 min at rt before the solvent was removed under reduced pressure, giving an orange solid, which was re-dissolved in absolute ethanol (135 mL); molecular sieves (6.5 g) were added and the mixture was refluxed 18 h under N2. The solution was then concentrated on Celite and transferred directly to a dry column vacuum chromatography setup (SiO2, CHCl3:MeOH; MeOH 0–10%), which gave a pale-yellow non-crystalline powder. Trace amounts of N-methylacetamide were removed by trituration with boiling diethylether (~50 mL/2 g) to furnish the pure product (1.22 g, 66%). MP: 195 °C (decomp.). 1H NMR (500 MHz, DMSO-d6) δ 10.98 (s, 1H), 8.66 (d, J = 2.0 Hz, 1H), 8.02 (dd, J = 8.4, 2.0 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.85 (s, 1H), 3.08 (s, 3H), 2.34 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 169.7, 163.2, 158.4, 142.4, 137.0, 133.8, 127.5, 123.6, 114.9, 85.2, 26.2, 15.4. MS (ESP-): m/z = 341.2 (M−). EA (C12H11IN2O2, 341.99): calcd. C 42.13, H 3.24, N 8.19; found C 42.16, H 3.19, N 8.05.
4-Acetoxy-3-(triisopropylsilylethynyl)benzaldehyde (9)
To a mixture of 4-acetoxy-3-iodobenzaldehyde 816 (500 mg, 1.72 mmol), CuI (16 mg, 0.086 mmol, 5% cat.) and PdCl2(PPh3)2 (60 mg, 0.086 mmol, 5% cat.) under an inert atmosphere of argon were added dry argon-degassed THF (10 mL) and diisopropylamine (2.5 mL). Triisopropylsilylacetylene (471 mg, 0.58 mL, 2.58 mmol) was added after 5 min of stirring and the dark reaction mixture was stirred for additional 3 h. The solution was then poured into sat. aqueous NH4Cl (100 mL) and extracted with diethylether (150 mL). The organic phase was then washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure to a dark yellow oil. Purification of the product by flash column chromatography (SiO2, CH2Cl2) yielded the product as a pale-yellow oil (424 mg, 71%). 1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 8.04 (d, J = 2.0 Hz, 1H), 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 2.34 (s, 3H), 1.14 (s, 21H). 13C NMR (125 MHz, CDCl3): δ 190.5, 168.2, 156.3, 135.6, 134.1, 130.4, 123.4, 119.0, 100.2, 98.4, 21.0, 18.8, 11.3. MS (GC): m/z = 301.2 (M+ -OCCH3), 259.2, 231.2. EA (C20H28O3Si, 344.18): calcd. C 69.72, H 8.19; found C 69.96, H, 8.26.
4-Acetoxy-3-phenylethynylbenzaldehyde (10)
To a mixture of 4-acetoxy-3-iodobenzaldehyde 8 (300 mg, 1.03 mmol), CuI (10 mg, 0.05 mmol, 5% cat.) and PdCl2(PPh3)2 (36 mg, 0.05 mmol, 5% cat.) under an inert atmosphere of argon were added dry argon-degassed THF (5 mL) and diisopropylamine (1 mL). Phenylacetylene (157 mg, 0.17 mL, 1.55 mmol) was added after 5 min of stirring and the dark reaction mixture was stirred for additional 2 h. The solution was then poured into sat. aqueous NH4Cl (100 mL) and extracted with diethylether (150 mL). The organic phase was then washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure to a dark oil. Purification of the product by flash column chromatography (SiO2, CH2Cl2) yielded the product as an off-white solid (196 mg, 72%). MP: 81–82 °C. 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 7.89 (dd, J = 8.3, 2.0 Hz, 1H), 7.55–7.48 (m, 2H), 7.40–7.36 (m, 3H), 7.32 (d, J = 8.3 Hz, 1H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 190.4, 168.3, 156.0, 134.9, 134.3, 131.8, 130.3, 129.2, 128.7, 123.4, 122.5, 118.9, 95.8, 83.1, 21.0. MS (GC): m/z = 264.2 (M+), 222.2, 165.2. EA (C17H12O3, 264.08): calcd. C 77.26, H 4.58; found C 77.08, H 4.15.
4-Acetoxy-3-(4-nitrophenylethynyl)benzaldehyde (11)
To a mixture of 4-acetoxy-3-iodobenzaldehyde 8 (200 mg, 0.69 mmol), CuI (7 mg, 0.035 mmol, 5% cat.) and PdCl2(PPh3)2 (24 mg, 0.035 mmol, 5% cat.) under an inert atmosphere of argon were added dry argon-degassed THF (5 mL) and diisopropylamine (1 mL). After 5 min of stirring, 4-nitrophenylacetylene (152 mg, 1.04 mmol) was added and the dark reaction mixture was stirred for additional 17 h. The solution was then poured into sat. aqueous NH4Cl (100 mL) and extracted with CH2Cl2 (150 mL). The organic phase was then washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure to a dark solid. Purification of the product by flash column chromatography (SiO2, CH2Cl2) yielded a pale-yellow solid (128 mg, 58%). Recrystallization from EtOAc gave the product as yellow needles. MP: 201–202 °C. 1H NMR (500 MHz, CDCl3): δ 10.01 (s, 1H), 8.26 (d, J = 8.9 Hz, 2H), 8.14 (d, J = 2.0 Hz, 1H), 7.96 (dd, J = 8.4, 2.0 Hz, 1H), 7.66 (d, J = 8.9 Hz, 2H), 7.37 (d, J = 8.4 Hz, 1H), 2.42 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 190.2, 168.2, 156.0, 147.6, 135.0, 134.7, 132.6, 131.5, 129.2, 124.0, 123.7, 117.7, 93.3, 88.1, 21.1. EA (C17H11NO5, 309.06): calcd. C 66.02, H 3.58, N 4.53; found C 66.04, H 3.04, N 4.46. IR (ART, cm−1): ν = 3113 w, 3047 w, 2930 w, 2841 vw (HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2219 vw, 1749 m, 1698 m, 1588 m, 1510 m (NO2), 1337 s (NO2), 1195 s, 1104 s, 856 s.
O), 2219 vw, 1749 m, 1698 m, 1588 m, 1510 m (NO2), 1337 s (NO2), 1195 s, 1104 s, 856 s.
4-Acetoxy-3-(4-methoxyphenylethynyl)benzaldehyde (12)
To a mixture of 4-acetoxy-3-iodobenzaldehyde 8 (200 mg, 0.69 mmol), CuI (7 mg, 0.035 mmol, 5% cat.) and PdCl2(PPh3)2 (24 mg, 0.035 mmol, 5% cat.) under an inert atmosphere of argon were added dry argon-degassed THF (5 mL) and diisopropylamine (1 mL). After 5 min of stirring, 4-methoxyphenylacetylene (110 mg, 0.83 mmol) was added and the dark reaction mixture was stirred for additional 3 h. The solution was then poured into sat. aqueous NH4Cl (100 mL) and extracted with diethylether (150 mL). The organic phase was then washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure to a dark yellow oil. Purification of the product by flash column chromatography (SiO2, CH2Cl2) yielded a pale-yellow solid (183 mg, 90%). MP: 79–80 °C. 1H NMR (500 MHz, CDCl3): δ 9.99 (s, 1H), 8.07 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 8.3, 2.0 Hz, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.6 Hz, 2H), 3.84 (s, 3H), 2.40 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 190.5, 168.4, 160.4, 155.9, 134.8, 134.3, 133.3, 130.0, 123.4, 119.2, 114.5, 114.3, 96.1, 82.0, 55.5, 21.0. MS (GC): m/z = 294.2 (M+), 252.2, 237.2. EA (C18H14O4, 294.09): calcd. C 73.46, H 4.79; found C 73.26, H, 4.57.
4-[4-Hydroxy-3-(triisopropylsilylethynyl)benzylidene]-1,2-dimethyl-1H-imidazol-5(4H)-one (3)
N-Acetylglycine (172 mg, 1.47 mmol), 4-acetoxy-3-(triisopropylsilylethynyl)benzaldehyde 9 (422 mg, 1.22 mmol) and NaOAc (100 mg, 1.22 mmol) were placed under an argon atmosphere. To this mixture was added Ac2O (750 mg, 0.69 mL, 7.33 mmol) and the suspension was heated to 110 °C for 1.5 h. The mixture was cooled to rt before H2O (1 mL) was added and the brown precipitate was collected. The brown crude was dissolved in CH2Cl2 (100 mL) and poured through a short plug of SiO2 (CH2Cl2), which after evaporation of the solvent in vacuo gave a pale-yellow solid. The solid was then dissolved in absolute ethanol (75 mL) under argon, and methylamine (33% wt. in ethanol, 2.5 mL, 21.0 mmol) was added. The orange solution was left for 45 min at rt before the solvent was removed under reduced pressure, giving an orange solid, which was re-dissolved in absolute ethanol (60 mL); then molecular sieves (1.0 g) were added and the mixture was refluxed for 12 h under N2. The solution was filtered, the solvent was evaporated and the product purified by column chromatography (SiO2, CH2Cl2:MeOH, 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to yield pale-yellow non-crystalline powder (113 mg, 23%). MP: 161–162 °C. 1H NMR (500 MHz, CDCl3): δ 8.17 (dd, J = 8.7, 2.1 Hz, 1H), 8.08 (d, J = 2.1 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 7.00 (s, 1H), 6.17 (s, 1H), 3.18 (s, 3H), 2.38 (s, 3H), 1.33–0.92 (m, 21H). 13C NMR (125 MHz, CDCl3): δ 170.8, 162.0, 158.7, 137.6, 135.8, 134.8, 127.1, 126.5, 115.2, 110.7, 100.2, 99.9, 26.7, 18.8, 15.8, 11.3. HRMS (ESP+): m/z = 419.2148 [M+Na+] (calcd. for C23H32N2NaO2Si+: 419.2125). IR (ART, cm−1): ν = 3255br (OH), 2940 m, 2863 m, 2147 m (CC), 1691 m, 1637 s, 1594 s, 1283 s, 660 s.
:2) to yield pale-yellow non-crystalline powder (113 mg, 23%). MP: 161–162 °C. 1H NMR (500 MHz, CDCl3): δ 8.17 (dd, J = 8.7, 2.1 Hz, 1H), 8.08 (d, J = 2.1 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 7.00 (s, 1H), 6.17 (s, 1H), 3.18 (s, 3H), 2.38 (s, 3H), 1.33–0.92 (m, 21H). 13C NMR (125 MHz, CDCl3): δ 170.8, 162.0, 158.7, 137.6, 135.8, 134.8, 127.1, 126.5, 115.2, 110.7, 100.2, 99.9, 26.7, 18.8, 15.8, 11.3. HRMS (ESP+): m/z = 419.2148 [M+Na+] (calcd. for C23H32N2NaO2Si+: 419.2125). IR (ART, cm−1): ν = 3255br (OH), 2940 m, 2863 m, 2147 m (CC), 1691 m, 1637 s, 1594 s, 1283 s, 660 s.
1,2-Dimethyl-4-[(2-phenylbenzofuran-5-yl)methylene]-1H-imidazol-5(4H)-one (13)
N-Acetylglycine (104 mg, 0.89 mmol), 4-acetoxy-3-phenylethynylbenzaldehyde 10 (196 mg, 0.74 mmol) and NaOAc (61 mg, 0.74 mmol) were placed under an argon atmosphere. To this mixture was added Ac2O (454 mg, 0.42 mL, 4.45 mmol) and the suspension was heated to 110 °C for 1.5 h. The mixture was cooled to rt before H2O (1 mL) was added and the brown precipitate was collected. The brown crude was dissolved in CH2Cl2 (100 mL) and poured through a short plug of SiO2 (CH2Cl2), which after evaporation of the solvent in vacuo gave a pale-yellow solid. The solid was then dissolved in absolute ethanol (20 mL) under argon, and methylamine (33% wt. in ethanol, 0.55 mL, 4.43 mmol) was added. The orange solution was left for 2 h at rt before the solvent was removed under reduced pressure, giving an orange solid, which was re-dissolved in absolute ethanol (15 mL); then molecular sieves (1.0 g) were added and the mixture was refluxed 12 h under N2. The solution was filtered, the solvent was evaporated and the product purified by column chromatography (SiO2, CH2Cl2:MeOH, 98:2) to yield a pale-yellow non-crystalline powder (67 mg, 29%). MP: 242 °C (decomp.). 1H NMR (500 MHz, CDCl3): δ 8.43 (d, J = 1.6 Hz, 1H), 8.09 (dd, J = 8.7, 1.6 Hz, 1H), 7.87 (dd, J = 8.4, 1.2 Hz, 2H), 7.55 (d, J = 8.7 Hz, 1H), 7.50–7.43 (m, 2H), 7.41–7.33 (m, 1H), 7.23 (s, 1H), 7.07 (s, 1H), 3.21 (s, 3H), 2.42 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.0, 162.1, 157.0, 156.0, 137.7, 130.2, 130.0, 129.5, 129.2, 129.0, 128.3, 125.4, 125.4, 125.1, 111.7, 101.7, 26.8, 15.9. IR (ART, cm−1): ν = 3135 vw, 1706 s, 1641 s, 1465 m, 1119 s. MS (ESP+): m/z = 317 [M+H+].
1,2-Dimethyl-4-{[2-(4-nitrophenyl)benzofuran-5-yl]methylene}-1H-imidazol-5(4H)-one (14)
N-Acetylglycine (55 mg, 0.47 mmol), 4-acetoxy-3-(4-nitrophenylethynyl)benzaldehyde 11 (120 mg, 0.39 mmol) and NaOAc (33 mg, 0.40 mmol) were placed under an argon atmosphere. To this mixture was added Ac2O (613 mg, 0.57 mL, 6.00 mmol) and the suspension was heated to 110 °C for 1.5 h. The mixture was cooled to rt before H2O (1 mL) was added and the brown precipitate was collected. The brown crude was dissolved in CH2Cl2 (100 mL) and poured through a short plug of SiO2 (CH2Cl2), which after evaporation of the solvent in vacuo gave a pale-yellow solid. The solid was then dissolved in absolute ethanol (30 mL) under argon, and methylamine (33% wt. in ethanol, 0.72 mL, 5.85 mmol) was added. The red solution was left for 120 min at rt before the solvent was removed under reduced pressure, giving a red solid, which was re-dissolved in absolute ethanol (25 mL); then molecular sieves (0.7 g) were added and the mixture was refluxed for 12 h under N2. The solution was filtered, the solvent evaporated off and the product purified by column chromatography (SiO2, CH2Cl2:MeOH, 98:2) to yield a yellow non-crystalline powder (53 mg, 38%). MP: 264 °C (decomp.). 1H NMR (500 MHz, CDCl3): δ 8.49 (d, J = 1.6 Hz, 1H), 8.33 (d, J = 9.0 Hz, 2H), 8.18 (dd, J = 8.7, 1.6 Hz, 1H), 8.01 (d, J = 9.0 Hz, 2H), 7.59 (d, J = 8.7 Hz, 1H), 7.29 (s, 1H), 7.22 (s, 1H), 3.22 (s, 3H), 2.42 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 170.9, 162.2, 156.4, 154.4, 147.6, 138.2, 136.1, 130.5, 130.3, 129.4, 127.5, 126.0, 125.5, 124.5, 112.0, 105.5, 26.8, 15.9. HRMS (ESP+): m/z = 362.1170 [M+H+] (calcd. for C20H16N3O4+: 362.1135). IR (ART, cm−1): ν = 3109 w, 2932 w, 2150 vw, 1710 s, 1650 m, 1503 s (NO2), 1345 s (NO2), 855 m.
1,2-Dimethyl-4-{[2-(4-methoxyphenyl)benzofuran-5-yl]methylene}-1H-imidazol-5(4H)-one (15)
N-Acetylglycine (118 mg, 1.00 mmol), 4-acetoxy-3-(4-methoxyphenylethynyl)benzaldehyde 12 (246 mg, 0.84 mmol) and NaOAc (69 mg, 0.84 mmol) were placed under an argon atmosphere. To this mixture was added Ac2O (513 mg, 0.47 mL, 5.02 mmol) and the suspension was heated to 110 °C for 1.5 h. The mixture was cooled to rt before H2O (1 mL) was added and the brown precipitate was collected. The brown crude was dissolved in CH2Cl2 (100 mL) and poured through a short plug of SiO2 (CH2Cl2), which after evaporation of the solvent in vacuo gave a pale-yellow solid. The solid was then dissolved in absolute ethanol (50 mL) under argon, and methylamine (33% wt. in ethanol, 1.4 mL, 11.3 mmol) was added. The orange solution was left for 120 min at rt before the solvent was removed under reduced pressure, giving an orange solid, which was re-dissolved in absolute ethanol (40 mL); then molecular sieves (1.7 g) were added and the mixture was refluxed for 12 h under N2. The solution was filtered, the solvent evaporated off and the product purified by column chromatography (SiO2, CH2Cl2:MeOH, 98:2) to yield a pale-yellow non-crystalline powder (119 mg, 41%). MP: 240–241 °C (decomp.). 1H NMR (500 MHz, CDCl3): δ 8.39 (d, J = 1.6 Hz, 1H), 8.07 (dd, J = 8.6, 1.6 Hz, 1H), 7.87–7.75 (m, 2H), 7.52 (d, J = 8.6 Hz, 1H), 7.23 (s, 1H), 7.06–6.96 (m, 2H), 6.92 (s, 1H), 3.87 (s, 3H), 3.21 (s, 3H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.0, 162.0, 160.4, 157.2, 155.9, 137.7, 130.3, 129.5, 128.8, 128.4, 126.7, 125.1, 123.1, 114.5, 111.5, 100.0, 55.5 26.8, 15.9. HRMS (ESP+): m/z = 347.1404 [M+H+] (calcd. for C21H19N2O3+: 347.1390). IR (ART, cm−1): ν = 2930 vw, 1698 s, 1641 s, 1251 s, 1117 m, 1022 s.
(Z)-4-(4-Hydroxy-3-methylbenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one (16)
First the oxazolone was prepared: 4-Hydroxy-3-methylbenzaldehyde19 (4.70 g, 30.0 mmol), N-acetylglycine (2.21 g, 18.8 mmol), and sodium acetate (0.462 g, 6.3 mmol) were suspended in Ac2O (36 mL, 33.8 mmol). The reaction mixture was then heated to 110 °C for 1.5 h, cooled to rt and allowed to stand at 5 °C overnight. The product precipated as yellow needles, which were recrystallized from boiling carbontetrachloride (200 mL) to yield (Z)-4-(4-acetoxy-3-methylbenzylidene)-2-methyl-1,3-oxazol-5(4H)-one (1.57 g, 32%) as yellow crystals. 1H-NMR (300 MHz, DMSO-d6) δ 8.08 (d, J = 8.3 Hz, 1H), 8.06 (s, 1H), 7.21 (d, J = 8.3 Hz, 1H), 7.17 (s, 1H), 2.39 (s, 3H), 2.32 (s, 3H), 2.16 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 168.7, 167.3, 166.7, 151.1, 134.6, 132.4, 130.9, 130.8, 130.5, 129.0, 122.9, 20.6, 15.8, 15.4. HR-MS (ESP+): m/z = 282.0746 [M+Na+]; calcd. for C13H13NO4Na+: m/z = 282.0737. Conversion to imidazolone 16: (Z)-4-(4-Acetoxy-3-methylbenzylidene)-2-methyl-1,3-oxazol-5(4H)-one (1.01 g, 3.91 mmol) was dissolved in absolute EtOH (200 mL) under argon at rt and treated with MeNH2 (33% in EtOH, 7.2 mL, 5.46 g, 58.7 mmol), which slowly turned the mixture red. After 30 min, the mixture was concentrated in vacuo and redissolved in absolute EtOH (100 mL). Then 4-Å molecular sieves (ca. 5 g) were added to the solution, which was heated to reflux overnight. The mixture was then concentrated in vacuo onto Celite and purified using dry column vacuum chromatography (SiO2, gradient from 0% to 10% MeOH in CHCl3). Recrystallization from 1,2-dichloroethane (170 mL) finally gave the product as a yellow non-crystalline solid (0.64 g, 71%). 1H NMR (300 MHz, DMSO-d6) δ 10.01 (br s, 1H), 7.96–7.89 (m, 2H), 6.87–6.81 (m, 2H), 3.08 (s, 3H), 2.33 (s, 3H), 2.13 (s, 3H) ppm. 13C-NMR (75 MHz, DMSO-d6) δ 169.8, 162.0, 157.8, 136.1, 134.8, 131.7, 125.7, 125.2, 124.3, 114.9, 26.2, 15.9, 15.3 ppm. HR-MS (ESP+): m/z = 231.1120 [M+H+] (calcd. for C13H15N2O2+: m/z = 231.1128).
Crystal structure determination of compound 7
Crystal data: C13H10INO4, M = 371.12, triclinic, a = 7.6448(2), b = 7.9573(2), c = 11.2132(2) Å, α = 85.282(1), β = 81.158(1), γ = 72.499(1)°, U = 642.35(3) Å3, T = 150 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no.2), Z = 2, 6830 reflections measured, 2973 unique (Rint = 0.019) which were used in all calculations. The final wR(F2) was 0.053 (all data).
(no.2), Z = 2, 6830 reflections measured, 2973 unique (Rint = 0.019) which were used in all calculations. The final wR(F2) was 0.053 (all data).
Acknowledgements
We acknowledge financial support from The Danish Council for Independent Research|Natural Sciences (#10-082088). We are grateful to Mr Dennis Larsen and Mr Micke Lisbjerg for recording of HR-MS.
References
-
(a) R. Y. Tsien, Annu. Rev. Neurosci., 1989, 12, 227 CrossRef CAS;
(b) M. Chalfie, Y. Tu, G. Euskirchen, W. W. Bard and D. C. Prasher, Science, 1994, 263, 802 CAS;
(c) R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509 CrossRef CAS;
(d) M. Zimmer, Chem. Rev., 2002, 102, 759 CrossRef CAS;
(e) J. M. Sanders and S. E. Jackson, Chem. Soc. Rev., 2009, 38, 2821 RSC;
(f) W. B. Frommer, M. W. Davidson and R. E. Campbell, Chem. Soc. Rev., 2009, 38, 2833 RSC;
(g) H. E. Seward and C. R. Bagshaw, Chem. Soc. Rev., 2009, 38, 2842 RSC;
(h) T. D. Craggs, Chem. Soc. Rev., 2009, 38, 2865 RSC.
-
(a) M. Ormö, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392 Search PubMed;
(b) F. Yang, L. G. Moss and G. N. J. Philips, Nat. Biotechnol., 1996, 14, 1246 CrossRef CAS;
(c) K. Brejc, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2306 CrossRef CAS;
(d) K. Suhling, J. Siegel, D. Philips, P. M. W. French, S. Leveque-Fort, S. E. D. Webb and D. M. Davis, Biophys. J., 2002, 83, 3589 CrossRef CAS.
- S. R. Meech, Chem. Soc. Rev., 2009, 38, 2922 RSC.
-
(a) M. B. Nielsen, T. Rocha-Rinza and L. H. Andersen, Monatsh. Chem., 2010, 142, 709 CrossRef;
(b) M. Tolbert, A. Baldridge, J. Kowalik and K. M. Solntsev, Acc. Chem. Res., 2012, 45, 171 CrossRef.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089 CrossRef CAS.
-
(a) M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025 CrossRef CAS.
- M. Cacciarini, M. B. Nielsen, E. M. de Castro, L. Marinescu and M. Bols, Tetrahedron Lett., 2012, 53, 973 CrossRef CAS.
-
(a) N. G. Kundu, M. Pal, J. S. Mahanty and D. De, J. Chem. Soc., Perkin Trans. 1, 1997, 2815 RSC;
(b) T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1999, 2849 RSC;
(c) A. Fürstner and P. W. Davies, J. Am. Chem. Soc., 2005, 127, 15024 CrossRef;
(d) D. Saha, R. Dey and B. C. Ranu, Eur. J. Org. Chem., 2010, 6067 CrossRef CAS;
(e) R. Cano, M. Yus and D. J. Ramón, Tetrahedron, 2012, 68, 1393 CrossRef CAS.
-
(a) F. Colobert, A.-S. Castanet and O. Abillard, Eur. J. Org. Chem., 2005, 3334 CrossRef CAS;
(b) M. Jacubert, A. Hamze, O. Promot, J.-F. Peyrat, J.-D. Brion and M. Alami, Tetrahedron Lett., 2009, 50, 3588 CrossRef CAS.
-
(a) G. E. Schneiders and R. Stevenson, J. Org. Chem., 1979, 44, 4710 CrossRef CAS;
(b) S. Yoo, S.-K. Kim, S.-H. Lee, N.-J. Kim and D.-W. Lee, Bioorg. Med. Chem., 2000, 8, 2311 CrossRef CAS;
(c) M. S. Malamas, J. Sredy, C. Moxham, A. Katz, W. Xu, R. McDevitt, F. O. Adebayo, D. R. Sawick, L. Seestauer, D. Sullivan and J. R. Taylor, J. Med. Chem., 2000, 43, 1293 CrossRef CAS;
(d) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS.
- M. Zander and G. Kirsch, Z. Naturforsch., 1989, 44a, 205 Search PubMed.
- K. Lincke, T. Sølling, L. H. Andersen, B. Klærke, D. B. Rahbek, J. Rajput, C. Oehlenslæger, M. Å. Petersen and M. B. Nielsen, Chem. Commun., 2010, 46, 734 RSC.
-
(a) H. D. Dakin, J. Biol. Chem., 1929, 82, 439 CAS;
(b) H. N. C. Wong, Z. L. Xu, H. M. Chang and C. M. Lee, Synthesis, 1992, 793 CrossRef CAS;
(c) S. Kojima, H. Ohkawa, T. Hirano, S. Maki, H. Niwa, M. Ohashi, S. Inouye and F. I. Tsuji, Tetrahedron Lett., 1998, 39, 5239 CrossRef CAS;
(d) X. He, A. F. Bell and P. J. Tonge, Org. Lett., 2002, 4, 1523 CrossRef CAS;
(e) B. Hager, B. Schwarzinger and H. Falk, Monatsh. Chem., 2005, 137, 163 CrossRef;
(f) T. Stafforst and U. Diederichsen, Eur. J. Org. Chem., 2007, 899 CrossRef CAS;
(g) B. Prüger and T. Bach, Synthesis, 2007, 1103 Search PubMed;
(h) M. Å. Petersen, P. Riber, L. H. Andersen and M. B. Nielsen, Synthesis, 2007, 3635 CAS.
- E. Defranq, T. Zesiger and R. Tabacchi, Helv. Chim. Acta, 1993, 76, 425 CrossRef CAS.
- T. Shiba and H. J. Cahnmann, J. Org. Chem., 1964, 29, 1652 CrossRef.
- L. R. Smith, N. Mahoney and R. J. Molyneux, J. Nat. Prod., 2003, 66, 169 CrossRef CAS.
- A. Bochicchio, L. Chiummiento, M. Funicello, M. T. Lopardo and P. Lupattelli, Tetrahedron Lett., 2010, 51, 2824 CrossRef CAS.
- G. Weber and F. W. Teale, Trans. Faraday Soc., 1958, 54, 640 RSC.
- Prepared according to: R. Villagómez-Ibarra, C. Alvarez-Cisneros and P. Joseph-Nathan, Tetrahedron, 1995, 51, 9285 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.