DOI:
10.1039/C2RA21353B
(Paper)
RSC Adv., 2012,
2, 11801-11807
Preparation of gold nanoparticles using reactive species produced in room-temperature ionic liquids by accelerated electron beam irradiation†
Received
4th July 2012
, Accepted 27th September 2012
First published on 1st October 2012
Abstract
Au nanoparticles that are expected to be a key material for supporting future technologies were produced with brief accelerated electron beam irradiation to various 1-alkyl-3-methylimidazolium cation ([R1MeIm]+)-based room-temperature ionic liquids (RTILs) with aurate. The resulting Au nanoparticles were able to remain in a nearly-monodispersed state in the RTILs for several weeks at least although the surface was not covered with a stabilizing agent, which is usually employed so as to prevent aggregation behaviour. The particle size increased with increasing the irradiation dose as well as decreasing the alkyl chain length of the [R1MeIm]+ if the RTIL consisted of the same anion species. The shapes of the Au nanoparticles obviously altered with the anionic species in the RTILs used for the preparation.
Introduction
It is well-known that Au nanoparticles, which can be synthesized by a wide variety of chemical and physical methods,1–4 have notable features including surface plasmon resonance, fluorescent emission etc.5–14 Due to these features, a great number of applications using Au nanoparticles, e.g., sensors, bio-imaging, drug delivery, catalysts, etc., have been proposed so far.3,5,15,16 However, Au nanoparticles are usually covered with self-assembled monolayers in order to prevent their aggregation behaviour. It is difficult to yield Au nanoparticles without a stabilizing agent.
After the discovery of room-temperature ionic liquids (RTILs), which are often called ionic liquids or room-temperature molten salts, having a favourable stability in air,17 they have been widely used as a novel functional solvent since RTILs show several interesting characteristics, such as negligible vapour pressure, wide electrochemical potential, relatively-high ionic conductivity, and wide liquidus temperature.18,19 RTILs are just beginning to be employed to yield metal nanoparticles. Many different approaches including chemical reduction,20 magnetron sputtering,21 plasmaelectrochemistry22etc. have been proposed to date. It is interesting to note that an RTIL works as both an excellent medium for metal nanoparticle formation and a stabilizing agent for the nanoparticles produced regardless of the types of preparation methods. But the stabilizing mechanism and the nanoparticle growth process in the RTIL are not entirely clear. Readers interested in nanoparticle preparation in RTILs can consult recent excellent reviews.23 From a practical application standpoint of nanoparticle preparation in RTILs, we are keeping an eye on γ-ray24 and accelerated electron beam irradiation24,25 methods, which can be conducted at the existing industrial plant for sterilizing medical kits, to establish a mass production method for metal nanoparticles in RTILs without any stabilizing and reducing agents. We have already succeeded in the preparation of various kinds of metal nanoparticles by means of the accelerated electron beam irradiation method. However, the effect of the metal salt concentration in the RTIL and the RTIL species used in the approach on the nanoparticles is not yet fully understood although we have revealed that the imidazolium-based RTIL is suitable for this purpose24,25 and several research groups have reported the radiochemical stability of RTILs.26 In this article, we investigated the effect of the aurate concentration added to the RTIL and of the RTIL species on the particle size and shapes of the Au nanoparticle obtained by accelerated electron beam irradiation to various types of RTILs with Na[AuCl4]·2H2O. The resulting Au nanoparticles were examined with a UV-vis spectrometer, a fluorescence spectrometer and a transmission electron microscope (TEM) equipped with an energy dispersive X-ray spectrometer (EDX).
Experimental
Preparation of RTILs
The organic salts, 1-alkyl-3-methylimidazolium chloride ([R1MeIm]Cl, (R1 stands for the ethyl group (Et); butyl group (Bu); hexyl group (Hex); octyl group (Oct); decyl group (Dec)), were purchased from Kanto Chemical Co., Inc. or IoLiTec GmbH except [EtMeIm]Cl synthesized in our lab. They were purified by precipitation from dry acetonitrile with dry ethyl acetate before use.27 Bis(trifluoromethane)sulfonamide lithium salt (Li[Tf2N], Aldrich, >99.95%), sodium tetrafluoroborate (Na[BF4], Aldrich, 98%) and potassium trifluoro(trifluoromethyl)borate (K[BF3(C2F5)], Tokyo Chemical Industry Co., Ltd., >98.0%) were used without purification for RTIL synthesis. The 1-alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide ([R1MeIm][Tf2N]) was prepared by mixing exactly equal molar amounts of the purified [R1MeIm]Cl and Li[Tf2N] in ultrapure water with dichloromethane. The solution was agitated at room temperature for 12 h. The dichloromethane phase containing the RTIL was washed repeatedly with reverse osmosis (RO) water until the water phase was found to contain no chloride as determined by the addition of a few drops of a silver nitrate solution. Finally the dichloromethane and remaining water were removed by stirring under vacuum conditions (1 × 10−4 Torr) at 373 K for 24 h.27 The prepared RTILs were clear and colourless. The [R1MeIm][BF4] and [EtMeIm][BF3(C2F5)] were prepared by mixing exactly equal molar amounts of the purified [R1MeIm]Cl and Na[BF4] and Li[BF3(C2F5)], respectively, in dry dichloromethane. These solutions were stirred at room temperature for 24 h. After filtration of the solutions containing unreacted and by-product salts, they were purified using the aforementioned method. 1-Ethyl-3-methylimidazolium fluorohydrogenate ([EtMeIm][(FH)2.3F])28 was provided by Professor Rika Hagiwara, Kyoto University (Japan). Specially-manufactured 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)amide ([EtMeIm][(FSO2)2N], Kanto Chemical Co., Inc., ultrapure grade) and 1-butyl-3-methylimidazolium bis(fluorosulfonyl)amide ([BuMeIm][(FSO2)2N], Kanto Chemical Co., Inc., ultrapure grade) were used as received. Other RTILs were purchased from and Merck & Co., Inc. or IoLiTec GmbH. They were dried at 373 K for 24 h in vacuo before use.
Based on the fact that metal nanoparticles can be obtained in [R1MeIm][Tf2N] RTIL with metal salts by γ-ray24 and accelerated electron beam irradiation,24,25 in this investigation only RTILs having the [R1MeIm]+ cation were employed as the reaction medium. In each case, after dissolving Na[AuCl4]·2H2O (Wako Pure Chemical Industries, Ltd.) in the RTILs, the solutions were poured into sample vials and the vials were capped in a VAC NEXUS II argon-filled glove box with an inert gas purification system (O2, H2O < 1 ppm). The irradiation experiments were carried out at an existing common industrial plant for sterilizing medical kits in Osaka, Japan. The sample vials were placed on a stainless steel wagon and carried to the irradiation area by an automatic conveyor system. The absorbed dose was controlled by the conveyor speed. The ionizing radiation source was an accelerated electron beam (accelerated energy: 4.8 MeV; beam current: 10 mA; irradiation dose: 6 or 20 kGy). The times for the 6 and 20 kGy irradiation doses were ca. 2 and 7 s, respectively. The irradiation dose was less than a common sterilization dose for usual medical instruments. By using a film dosemeter we confirmed that the accelerated electron beam can penetrate the vials with ease before the irradiation experiments.
The UV-vis spectra of the RTILs after irradiation experiments were examined by using a Shimadzu MultiSpec-1500 spectrometer or a JASCO V-670 UV-Vis-NIR spectrometer. The fluorescence intensity was estimated using a Horiba FluoroMax-4 fluorescence spectrometer. The morphology of the deposited Au nanoparticles was observed using a Hitachi H-7650 transmission electron microscope (TEM). The accelerated voltage for the TEM observation was 100 kV. Specimens for TEM observation were prepared by dropping the irradiated RTIL onto a TEM grid (∅ 3.0 mm, copper, 400 mesh) coated with an amorphous carbon thin layer, and the grid was rinsed with dry acetonitrile repeatedly to remove the RTIL and to prevent further unexpected reaction. TEM observation was conducted after two weeks of the accelerated electron beam irradiation experiments. The composition of the Au nanoparticles was analysed using an EDAX Genesis XM2 energy dispersive X-ray spectroscopy (EDX) system mounted on the TEM.
Results and discussion
Effect of aurate concentration
Although we have already succeeded in Au nanoparticle preparation using γ-ray and accelerated electron beam irradiation of [BuMeIm][Tf2N] with 0.5 mmol L−1 Na[AuCl4]·2H2O,24 there is no information about the effect of aurate concentration on the resultant Au nanoparticles so far. Here we examined the effect using the [BuMeIm][Tf2N] with different NaAuCl4·2H2O concentrations. Fig. 1 and Fig. S1 (ESI†) show TEM images of Au nanoparticles, which are identified by EDX, prepared in [BuMeIm][Tf2N] with 0.10–20 mmol L−1 Na[AuCl4]·2H2O at 6 and 20 kGy irradiation, respectively. The relationship between the mean particle size of the Au nanoparticles prepared in this investigation and the added Na[AuCl4]·2H2O concentration is exhibited in Fig. 2. At 6 kGy irradiation, spherical Au nanoparticles were yielded, and also partially-coalesced nanoparticles appeared at a concentration over 10 mmol L−1 Na[AuCl4]·2H2O. As for 20 kGy irradiation (Fig. S1, ESI†), spherical Au nanoparticles were produced only when the concentration is below 1.0 mmol L−1, but once the concentration exceeds 10 mmol L−1, the Au nanoparticles formed secondary particles in the solution through nanoparticle coalescence at 2.5–5.0 mmol L−1. The mean particle size estimated from these TEM images is summarized in Table 1. In both irradiation dose conditions, the mean particle size became larger with increasing the concentration, suggesting that the increase in the aurate concentration dissolved in the RTILs leads directly to further Au(III) reduction onto the Au nanoparticles. As we discuss later, the increase in the aurate concentration contributes to increasing the number of Au nanoparticles, too. It is noteworthy that there seems to be a threshold particle size at ca. 8 nm. When exceeding that size, the secondary particle growth behaviour was enhanced. The secondary particle formation would be caused by insufficient stabilization of the Au nanoparticles having a mean particle size over ca. 8 nm. Such secondary particle growth behaviour is also observed if a similar approach using an electron gun of a scanning Auger microprobe is applied to the Au nanoparticle synthesis.29
![TEM images of Au nanoparticles prepared by accelerated electron beam irradiation in [BuMeIm][Tf2N] with different NaAuCl4·2H2O concentrations. The irradiation dose was 6 kGy. The concentrations were (a) 0.10, (b) 0.25, (c) 0.50, (d) 1.0, (e) 2.5, (f) 5.0, (g) 10 and (h) 20 mmol L−1.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f1.gif) |
| | Fig. 1 TEM images of Au nanoparticles prepared by accelerated electron beam irradiation in [BuMeIm][Tf2N] with different NaAuCl4·2H2O concentrations. The irradiation dose was 6 kGy. The concentrations were (a) 0.10, (b) 0.25, (c) 0.50, (d) 1.0, (e) 2.5, (f) 5.0, (g) 10 and (h) 20 mmol L−1. | |
![Mean particle size of Au nanoparticle prepared by accelerated electron beam irradiation in [BuMeIm][Tf2N] with various NaAuCl4·2H2O concentrations. The irradiation dose was (●) 6 or (▲) 20 kGy.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f2.gif) |
| | Fig. 2 Mean particle size of Au nanoparticle prepared by accelerated electron beam irradiation in [BuMeIm][Tf2N] with various NaAuCl4·2H2O concentrations. The irradiation dose was (●) 6 or (▲) 20 kGy. | |
Table 1 Mean particle size and standard deviation of Au nanoparticles prepared in [BuMeIm][Tf2N] with Na[AuCl4]·2H2O
| NaAuCl4·2H2O conc./mmol L−1 |
Irradiation dose |
| 6 kGy |
20 kGy |
|
d
a/nm |
SDb/nm |
d
a/nm |
SDb/nm |
|
Mean diameter.
Standard deviation.
Coalesced Au nanoparticles.
Secondary particles appeared.
|
| 0.10 |
3.16 |
0.67 |
5.58 |
1.16 |
| 0.25 |
3.24 |
0.52 |
6.36 |
1.07 |
| 0.50 |
3.70 |
0.86 |
6.81 |
1.06 |
| 1.0 |
4.01 |
1.12 |
7.32 |
1.89 |
| 2.5 |
5.00 |
0.65 |
7.85c |
1.19 |
| 5.0 |
5.44 |
0.96 |
8.21c |
1.18 |
| 10 |
6.14c |
1.31 |
—d |
—d |
| 20 |
6.84c |
1.49 |
—d |
—d |
From the examination of the Au nanoparticle preparation, we found one unexpected result that larger Au nanoparticles are produced at higher irradiation doses. In the case of the accelerated electron beam irradiation method using an aqueous solution as the reaction medium, it is known that the mean particle size increases at low-dose irradiation given that other things are equal.30 A plausible mechanism has been proposed by several research groups.30,31 It is not easy to understand the mechanism precisely because many chemical reactions occur simultaneously during the accelerated electron beam irradiation. Key reactions for the metal nanoparticle preparation are:
where M
+, e
solv, M, H˙ and H stand for metal
cation, solvated electron, deposited metal atom, hydrogen radical and hydrogen atom, respectively. The
m and
n represent the number of atoms contained in a metal cluster yielded by the irradiation. In addition to these reactions,
eqn (1)–(4), we have to take into account the following association processes between clusters or atoms and unreacted metal ions.
| | | Mm+xx+ + Mm+yy+ → M2m+x+y(x+y)+ | (8) |
where
x and
y show the number of associated ions. A schematic illustration of these reaction processes is depicted in
Fig. 3. Under higher dose irradiation conditions, the crystal growth process of the metal
nanoparticle mainly occurs by the coalescence of the metal deposited in
eqn (1)–(4). At lower dose irradiation, the association processes,
eqn (5)–(7), and the ionic coalescence process,
eqn (8), are predominant reactions because the production of the reactive species that can reduce M
+ is insufficient; that is, many M
+ ions are reduced after the formation of the associated ions,
e.g., M
m+xx+ and M
2m+x+y(x+y)+. Therefore, in aqueous solution, metal
nanoparticles become larger at a lower irradiation dose due to the formation of larger associated ions. In contrast, in the RTIL, the growth process of the metal
nanoparticles is in a similar situation as that in aqueous solution at low dose irradiation conditions, because the RTIL is quite stable toward γ-ray and accelerated electron beam irradiation compared to the aqueous solution system.
32 Under such a situation, larger associated ions, which make the resultant
nanoparticles larger, would be obtained at higher dose irradiation. In addition to this, we have to consider the thermal instability of the metal
nanoparticles yielded in the RTIL.
25,33,34 In fact, the mean particle size of metal
nanoparticles prepared in neat RTIL without a stabilizing agent can be controlled by heating the metal
nanoparticle-dispersed RTILs. We found that the sample vials are mildly heated by the collision energy derived from the accelerated electron beam during the irradiation experiments and the temperature increases with increasing irradiation dose. Therefore we believe that the slight temperature increase of the sample vials contributes to further
nanoparticle growth at 20 kGy irradiation, too.
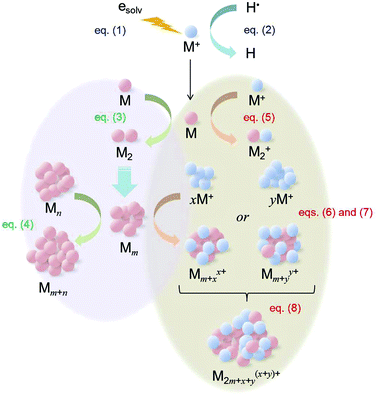 |
| | Fig. 3 Schematic illustration of the proposed metal nanoparticle formation process by accelerated electron beam irradiation. | |
Typical UV-vis spectra of [BuMeIm][Tf2N] with different NaAuCl4·2H2O concentrations after accelerated electron beam irradiation are shown in Fig. 4. Peculiar absorption associated with the surface plasmon absorption of the Au nanoparticles appeared and the peak wavelength was ca. 480 nm, independent of the experimental condition. Surface plasmon absorption of Au nanoparticles having a ca. 3–5 nm diameter produced by the magnetron sputtering method in [BuMeIm][Tf2N] without a stabilizing agent appears at 525 nm.35 There really is a difference between the reported value and our result. Considering that the dielectric constant of the solutions with Au nanoparticles affects the surface plasmon absorption,5 it may reflect the variation in the dielectric constant by the electron beam irradiation since the accelerated electron beam-irradiated [BuMeIm][Tf2N] is damaged.25 The stronger peak intensity observed at 20 kGy irradiation implies that more Au nanoparticles are produced.
![UV-vis spectra of [BuMeIm][Tf2N] with various NaAuCl4·2H2O concentrations after accelerated electron beam irradiation at () 6 or () 20 kGy. The concentrations were (a) 0.5, (b) 2.5, (c) 5.0 and (d) 20 mmol L−1. The background sample was neat [BuMeIm][Tf2N] without electron beam irradiation.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f4.gif) |
| | Fig. 4 UV-vis spectra of [BuMeIm][Tf2N] with various NaAuCl4·2H2O concentrations after accelerated electron beam irradiation at (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) 6 or ( ) 6 or (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) 20 kGy. The concentrations were (a) 0.5, (b) 2.5, (c) 5.0 and (d) 20 mmol L−1. The background sample was neat [BuMeIm][Tf2N] without electron beam irradiation. ) 20 kGy. The concentrations were (a) 0.5, (b) 2.5, (c) 5.0 and (d) 20 mmol L−1. The background sample was neat [BuMeIm][Tf2N] without electron beam irradiation. | |
Effect of side-chain length on cationic species in RTIL
In order to clarify the effect of side-chain length on the 1-alkyl-3-methylimidazolium cation ([R1MeIm]+) in the RTILs, we produced Au nanoparticles in five types of [R1MeIm][Tf2N] with 1.0 mmol L−1 NaAuCl4·2H2O. Fig. 5 and Fig. S1d (ESI†) show TEM images of the resulting Au nanoparticles after accelerated electron beam irradiation at 20 kGy. In cases where [EtMeIm][Tf2N] (Fig. 5a) and [BuMeIm][Tf2N] (Fig. S1d, ESI†) are used, we could observe Au nanoparticles although in the former RTIL most particles seemed to coalesce. But, as depicted in Fig. 5b–d, measurable Au nanoparticles did not appear in other RTILs consisting of [R1MeIm]+ having a longer alkyl chain than a hexyl group. It attributes to gauche defects that decrease with increasing the alkyl chain length of the [R1MeIm]+ cation, as described in several papers on Au nanoparticles obtained in an analogous fashion to this study.29,36 This is most likely to be a common behaviour for Au nanoparticle formation in RTILs. A similar result was also obtained at 6 kGy irradiation as shown in Fig. 1d and Fig. S2 (ESI†).
![TEM images of Au nanoparticles prepared in [R1MeIm][Tf2N] RTILs with 1.0 mmol L−1 NaAuCl4·2H2O by accelerated electron beam irradiation. The RTILs were (a) [EtMeIm][Tf2N], (b) [HexMeIm][Tf2N], (c) [MeOctIm][Tf2N] and (d) [DecMeIm][Tf2N]. The irradiation dose was 20 kGy.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f5.gif) |
| | Fig. 5 TEM images of Au nanoparticles prepared in [R1MeIm][Tf2N] RTILs with 1.0 mmol L−1 NaAuCl4·2H2O by accelerated electron beam irradiation. The RTILs were (a) [EtMeIm][Tf2N], (b) [HexMeIm][Tf2N], (c) [MeOctIm][Tf2N] and (d) [DecMeIm][Tf2N]. The irradiation dose was 20 kGy. | |
It has already been shown that Au clusters ranging in size from 28 atoms to picoscale particles show photoluminescence (PL) behaviour. Only nanoclusters having a smaller size than the Fermi wavelength of the conduction electrons show such peculiar behavior.10–14 By utilizing this feature, we attempted to demonstrate the existence of Au clusters in [HexMeIm][Tf2N], [MeOctIm][Tf2N] and [DecMeIm][Tf2N] after the accelerated electron beam irradiation experiments because as previously noted we could not observe Au nanoparticles in these RTILs. Fig. 6 and 7 exhibit typical examples that employed [HexMeIm][Tf2N] RTIL containing 1.0 mmol L−1 NaAuCl4·2H2O. By visual examination and analytical chemistry we could confirm blue light emission from the RTIL under UV irradiation. It suggests the existence of the Au clusters, which cannot be observed by an ordinary TEM system, in the RTIL. Similar blue light emission was also observed in the NaAuCl4·2H2O-dissolved [MeOctIm][Tf2N] and [DecMeIm][Tf2N] after the accelerated electron beam irradiation.
![Photographs of [HexMeIm][Tf2N] RTIL containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation. The irradiation doses were (a) 6 and (b) 20 kGy: (left) before and (right) under UV irradiation at 365 nm.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f6.gif) |
| | Fig. 6 Photographs of [HexMeIm][Tf2N] RTIL containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation. The irradiation doses were (a) 6 and (b) 20 kGy: (left) before and (right) under UV irradiation at 365 nm. | |
![Photoluminescence intensities of () neat [HexMeIm][Tf2N] RTIL and () [HexMeIm][Tf2N] containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation. The irradiation doses were (a) 6 and (b) 20 kGy. The UV irradiation was conducted at 370 nm.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f7.gif) |
| | Fig. 7 Photoluminescence intensities of () neat [HexMeIm][Tf2N] RTIL and () [HexMeIm][Tf2N] containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation. The irradiation doses were (a) 6 and (b) 20 kGy. The UV irradiation was conducted at 370 nm. | |
Effect of anionic species in RTIL
Fig. 8a–c depict TEM images of Au nanoparticles prepared in different [BuMeIm]+-based RTILs with 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation at 20 kGy. The mean particle size of the Au nanoparticles prepared in [BuMeIm][Tf2N], [BuMeIm][(FSO2)2N] and [BuMeIm][BF4] were 7.32 nm (standard deviation (SD): 1.89 nm), 20.4 nm (SD: 11.8 nm) and 5.18 nm (SD: 6.12 nm), respectively. The particle size and shape of the Au nanoparticles more or less depended on the RTIL species. UV-vis spectra of their RTIL solutions with Au nanoparticles are shown in Fig. 8d. Distinctive absorptions related to the surface plasmon absorption of the Au nanoparticle appeared and the peak wavelength was ca. 480 nm in the [BuMeIm][Tf2N] and [BuMeIm][BF4]. However, in the case of the [BuMeIm][(FSO2)2N] it shifted toward longer wavelength because of the larger mean nanoparticle size.7 Further investigation was conducted by using eight types of [EtMeIm]+-based RTILs with 1.0 mmol L−1 NaAuCl4·2H2O. In each case, measurable Au nanoparticles were yielded by the accelerated electron beam irradiation, but they formed spherical secondary nanoparticles or irregularly-shaped aggregated nanoparticles, if the irradiation dose was 20 kGy. On the other hand, as shown in Fig. 9, a variety of shapes of nanoparticles were produced at 6 kGy irradiation, as in the case of Au nanoparticle fabrication from aqueous solutions.4,37 Those are spherical secondary nanoparticles ([EtMeIm][Tf2N] (Fig. 9a), [EtMeIm][(FSO2)2N] (Fig. 9b)), star-shaped nanoparticles ([EtMeIm][BF4] (Fig. 9c)), sea urchin-shaped nanoparticles ([EtMeIm][BF3(C2F5)] (Fig. 9d)), rock sugar-shaped nanoparticles (1-ethyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate ([EtMeIm][PF3(C2F5)3]) (Fig. 9e), [EtMeIm][(FH)2.3F] (Fig. 9f)), oval-shaped nanoparticles (1-ethyl-3-methylimidazolium acetate ([EtMeIm][CH3COO]) (Fig. 9g)) and branched structures (1-ethyl-3-methylimidazolium L-(+)-lactate ([EtMeIm][L-(+)-CH3CH(OH)COO]) (Fig. 9h)). These results show that the anionic species in the RTILs have a significant influence on the morphology of the resulting Au nanoparticles. However, at this moment we do not understand the reason fully because there are many parameters influencing the Au nanoparticle formation process.
![TEM images of Au nanoparticles prepared by accelerated electron beam irradiation in [BuMeIm]+-based RTILs containing 1.0 mmol L−1 NaAuCl4·2H2O. The irradiation dose was 20 kGy. The RTILs were (a) [BuMeIm][Tf2N], (b) [BuMeIm][(FSO2)2N] and (c) [BuMeIm][BF4]. (d) UV-vis spectra of [BuMeIm]+-based RTILs containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation at 20 kGy. The RTILs were () [BuMeIm][Tf2N], (, blue) [BuMeIm][(FSO2)2N] and (, green) [BuMeIm][BF4]. The background samples for UV-vis spectroscopy were neat RTILs without electron beam irradiation.](/image/article/2012/RA/c2ra21353b/c2ra21353b-f8.gif) |
| | Fig. 8 TEM images of Au nanoparticles prepared by accelerated electron beam irradiation in [BuMeIm]+-based RTILs containing 1.0 mmol L−1 NaAuCl4·2H2O. The irradiation dose was 20 kGy. The RTILs were (a) [BuMeIm][Tf2N], (b) [BuMeIm][(FSO2)2N] and (c) [BuMeIm][BF4]. (d) UV-vis spectra of [BuMeIm]+-based RTILs containing 1.0 mmol L−1 NaAuCl4·2H2O after accelerated electron beam irradiation at 20 kGy. The RTILs were () [BuMeIm][Tf2N], (, blue) [BuMeIm][(FSO2)2N] and (, green) [BuMeIm][BF4]. The background samples for UV-vis spectroscopy were neat RTILs without electron beam irradiation. | |
![TEM images of Au nanoparticles prepared by accelerator electron beam irradiation in various [EtMeIm]+-based RTILs containing 1.0 mmol L−1. NaAuCl4·2H2O. The irradiation dose was 6 kGy. The RTILs were (a) [EtMeIm][Tf2N], (b) [EtMeIm][(FSO2)2N], (c) [EtMeIm][BF4], (d) [EtMeIm][BF3(C2F5)], (e) [EtMeIm][PF3(C2F5)3], (f) [EtMeIm][(FH)2.3F], (g) [EtMeIm][CH3COO] and (h) [EtMeIm][l-(+)-CH3CH(OH)COO].](/image/article/2012/RA/c2ra21353b/c2ra21353b-f9.gif) |
| | Fig. 9 TEM images of Au nanoparticles prepared by accelerator electron beam irradiation in various [EtMeIm]+-based RTILs containing 1.0 mmol L−1. NaAuCl4·2H2O. The irradiation dose was 6 kGy. The RTILs were (a) [EtMeIm][Tf2N], (b) [EtMeIm][(FSO2)2N], (c) [EtMeIm][BF4], (d) [EtMeIm][BF3(C2F5)], (e) [EtMeIm][PF3(C2F5)3], (f) [EtMeIm][(FH)2.3F], (g) [EtMeIm][CH3COO] and (h) [EtMeIm][L-(+)-CH3CH(OH)COO]. | |
The particle size of the Au nanoparticles prepared in [EtMeIm][Tf2N] (Fig. 9a), [EtMeIm][(FSO2)2N] (Fig. 9b) and [EtMeIm][BF4] (Fig. 9c) undoubtedly became larger in comparison with those in [BuMeIm]+-based RTILs depicted in Fig. 8a–c. This behaviour is well accorded with the results discussed in the preceding section, viz., the particle size increases with decreasing the alkyl chain length of the [R1MeIm]+ if the RTIL has the same anion species.
Conclusions
Au nanoparticles were produced with brief accelerated electron beam irradiation in various [R1MeIm]+-based RTILs. The resulting Au nanoparticles were able to remain in the nearly-monodispersed state in the RTILs for several weeks at least while the surface was not covered with a stabilizing agent, which is usually exploited in order to prevent aggregation behaviour. The particle size increased with increasing the irradiation dose and with decreasing the alkyl chain length of the [R1MeIm]+ if the anion species in the RTIL is the same. Interestingly, the shapes of the Au nanoparticle varied significantly with the anionic species of the RTILs.
Acknowledgements
Part of this research was supported by Grant-in-Aid for Scientific Research B, Grant No. 24350071, and for Scientific Research on Innovative Areas (Area No. 2206), Grant No. 23107518, from Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT), Kansai Research Foundation for Technology Promotion in Japan, The Noguchi Institute in Japan and General Sekiyu R&D Encouragement Assistance Foundation in Japan. We thank Mr. Kazuki Yoshii (Osaka University) for his help on TEM observation.
References
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- M. Grzelczak, J. P.-Juste, P. Mulvaney and L. M. L.-Marzán, Chem. Soc. Rev., 2008, 37, 1783 RSC.
-
Nanoparticles: From Theory to Application, ed. G. SchmidWiley-VCH, Weinheim, Germany, Second, Completely Revised and Updated Edition, 2010 Search PubMed.
- T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410 CrossRef CAS.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415 RSC.
- K. Yu, K. L. Kelly, N. Sakai and T. Tatsuma, Langmuir, 2008, 24, 5849 CrossRef CAS.
- T. Huang and R. W. Murray, J. Phys. Chem. B, 2001, 105, 12498 CrossRef CAS.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780 CrossRef CAS.
-
J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402/1 Search PubMed.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 2412 CrossRef CAS.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395 CrossRef CAS.
- E. Hutter, S. Boridy, S. Labrecque, M. L.-Hébert, J. Kriz, F. M. Winnik and D. Maysinger, ACS Nano, 2010, 4, 2595 CrossRef CAS.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647 RSC.
-
(a) J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965 RSC;
(b) E. I. Cooper and E. J. M. O'Sullivan, in: Proceedings of the Eighth International Symposium on Molten Salts, p. 386, ed. R. J. Gale, G. Blomgren, H. Kojima, PV92-16, The Electrochemical Society, Inc., Pennington, NJ, 1992 Search PubMed.
-
Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, Germany, Second Edition, 2008; M. Freemantle, An Introduction to Ionic Liquids, The Royal Society of Chemistry, Cambridge, UK, 2010; Electrodeposition from Ionic Liquids, ed. F. Endres, A. P. Abbott and D. R. MacFarlane, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
-
(a) R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792 CrossRef;
(b) M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS;
(c) K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H.-P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158 CrossRef CAS;
(d) S. Kuwabata, T. Tsuda and T. Torimoto, J. Phys. Chem. Lett., 2010, 1, 3177 CrossRef CAS.
-
(a) L. Zhao, C. Y. Zhang, L. Zhuo, Y. G. Zhang and J. Y. Ying, J. Am. Chem. Soc., 2008, 130, 12586 CrossRef CAS;
(b) K. S. Kim, D. Demberelnyamba and H. Lee, Langmuir, 2004, 20, 556 CrossRef CAS;
(c) H. Itoh, K. Naka and Y. Chujo, J. Am. Chem. Soc., 2004, 126, 3026 CrossRef CAS;
(d) R. Tatumi and H. Fujihara, Chem. Commun., 2005, 83 RSC.
-
(a) T. Torimoto, K. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka and S. Kuwabata, Appl. Phys. Lett., 2006, 89, 243117 CrossRef;
(b) K. Okazaki, T. Kiyama, T. Suzuki, S. Kuwabata and T. Torimoto, Chem. Lett., 2009, 38, 330 CrossRef CAS;
(c) T. Tsuda, K. Yoshii, T. Torimoto and S. Kuwabata, J. Power Sources, 2010, 195, 5980 CrossRef CAS;
(d) T. Suzuki, K. Okazaki, S. Suzuki, T. Shibayama, S. Kuwabata and T. Torimoto, Chem. Mater., 2010, 22, 5209 CrossRef CAS;
(e) C. W. Scheeren, G. Machado, S. R. Teixeira, J. Morais, J. B. Domingos and J. Dupont, J. Phys. Chem. B, 2006, 110, 13011 CrossRef CAS;
(f) K. Yoshii, T. Tsuda, T. Arimura, A. Imanishi, T. Torimoto and S. Kuwabata, RSC Adv., 2012, 2, 8262 RSC.
-
(a) S. A. Meiss, M. Rohnke, L. Kienle, S. Z. El Abedin, F. Endres and J. Janek, ChemPhysChem, 2007, 8, 50 CrossRef CAS;
(b) K. Baba, T. Kaneko and R. Hatakeyama, Appl. Phys. Lett., 2007, 90, 201501 CrossRef.
-
(a) T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196 CrossRef CAS;
(b) J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780 RSC;
(c) M.-A. Neouze, J. Mater. Chem., 2010, 20, 9593 RSC.
- T. Tsuda, S. Seino and S. Kuwabata, Chem. Commun., 2009, 6792 RSC.
- T. Tsuda, T. Sakamoto, Y. Nishimura, S. Seino, A. Imanishi and S. Kuwabata, Chem. Commun., 2012, 48, 1925 RSC.
-
(a) I. A. Shkrob and J. F. Wishart, J. Phys. Chem. B, 2009, 113, 5582 CrossRef CAS;
(b) D. Allen, G. Baston, A. E. Bradley, T. Gorman, A. Haile, I. Hamblett, J. E. Hatter, M. J. F. Healey, B. Hodgson, R. Lewin, K. V. Lovell, B. Newton, W. R. Pitner, D. W. Rooney, D. Sanders, K. R. Seddon, H. E. Sims and R. C. Thied, Green Chem., 2002, 4, 152 RSC;
(c) M. Qi, G. Wu, Q. Li and Y. Luo, Radiat. Phys. Chem., 2008, 77, 877 CrossRef CAS.
-
(a)
T. Tsuda and C. L. Hussey, in Modern Aspects of Electrochemistry, ed. R. E. White, Springer, New York, 2009, vol. 45, ch. 2, pp. 63–174 Search PubMed;
(b) T. Tsuda, K. Kondo, T. Tomioka, Y. Takahashi, H. Matsumoto, S. Kuwabata and C. L. Hussey, Angew. Chem., Int. Ed., 2011, 50, 1310 CrossRef CAS.
-
(a) R. Hagiwara, T. Hirashige, T. Tsuda and Y. Ito, J. Electrochem. Soc., 2002, 149 Search PubMed;
(b) R. Hagiwara D1;, K. Matsumoto, Y. Nakamori, T. Tsuda, Y. Ito, H. Matsumoto and K. Momota, J. Electrochem. Soc., 2003, 150, D195 CrossRef.
- A. Imanishi, S. Gonsui, T. Tsuda, S. Kuwabata and K. Fukui, Phys. Chem. Chem. Phys., 2011, 13, 14823 RSC.
- J. Belloni, Catal. Today, 2006, 113, 141 CrossRef CAS.
- K. Naghavi, E. Saion, K. Rezaee, W. Mahmood and M. Yunus, Radiat. Phys. Chem., 2010, 79, 1203 CrossRef CAS.
- J. F. Wishart, J. Phys. Chem. Lett., 2010, 1, 3225 CrossRef CAS.
- T. Tsuda, K. Yoshii, T. Torimoto and S. Kuwabata, J. Power Sources, 2010, 195, 5980 CrossRef CAS.
- T. Kameyama, Y. Ohno, T. Kurimoto, K. Okazaki, T. Uematsu, S. Kuwabata and T. Torimoto, Phys. Chem. Chem. Phys., 2010, 12, 1804 RSC.
- H. Wender, L. F. de Oliveira, P. Migowski, A. F. Feil, E. Lissner, M. H. G. Prechtl, S. R. Teixeira and J. Dupont, J. Phys. Chem. C, 2010, 114, 11764 CAS.
- Y. Hatakeyama, M. Okamoto, T. Torimoto, S. Kuwabata and K. Nishikawa, J. Phys. Chem. C, 2009, 113, 3917 CAS.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images. See DOI: 10.1039/c2ra21353b |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.