Synthesis of self-assembling glycerotriazolophanes†
Mohit
Tyagi
,
Nikhil
Taxak
,
Prasad V.
Bharatam
and
K. P. Ravindranathan
Kartha
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, SAS Nagar, Punjab, 160062, India. E-mail: rkartha@niper.ac.in
First published on 20th September 2012
Abstract
Twenty 16-membered triazole-based macrocycles possessing different pseudo-symmetry elements have been synthesized using the copper-catalyzed azide-alkynecycloaddition (CuAAC) reaction. The formation of the macrocycles was accompanied by their spontaneous self-assembly leading to solids with distinct layered structures as confirmed by scanning electron microscopy. The self-assembling properties of the product and the “on water” reaction media seemed to form the driving force for the reaction in a t-butanol–water system. The self-assembling properties of these macrocycles was supported by quantum chemical studies using Density Functional Theory.
Introduction
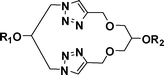
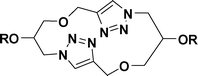
The Huisgen 1,3-dipolar cycloaddition is a widely applicable reaction for the synthesis of triazole-containing compounds.1 Although it is a relatively new addition to the synthetic organic chemistry arsenal, it has already found wide applications in the areas of medicinal chemistry,2 supramolecular chemistry,3 materials science,4polymer science,5etc., and click chemistry has proven to be quite versatile in accessing macrocyclic frameworks.6 Click macrocycles possess interesting attributes like anion-binding properties,7 self-assembling properties,8 the ability to act as artificial molecular receptors,9catenanes and rotaxanes,10 chemical sensors,11peptidomimetics,12drug carrier systems,13 expanded crown ethers,14enzyme inhibitors,15 chemosensors16 and artificial receptors,17etc. Whitesides et al. have described a wide range of application possibilities for materials possessing self-assembling properties.18 Besides these applications, the important attributes of the 1,4-substituted triazole ring that attracted our attention are its metabolic stability as well as the ability to show π-stacking, unlike the trans-amide bond.19 A series of glycerol-linked triazole-based macrocycles possessing interesting self-assembling properties are described herein.Results and discussion
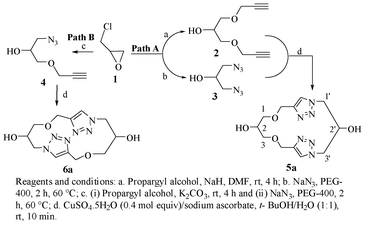
The three-carbon glycerol skeleton was sourced from the readily available epichlorohydrin (ECH, 1; Scheme 1). ECH, when reacted with propargyl alcohol in the presence of sodium hydride, gave the 1,3-dipropargyl ether of glycerol (2) in high yield.20 The diazide 3 was readily prepared by the reaction of 1 with sodium azide in PEG 400 as shown in Scheme 1.21 Treatment of 3 with 2 in the presence of CuSO4·5H2O and sodium ascorbate in t-BuOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave the triazolemacrocycle5 (Scheme 1; Path A). The cyclization was tried in several organic solvents and under different dilution conditions6 (Table 1); and the t-BuOH–H2O system (1:1) was found to be best suited for the purpose, in agreement with the observation by Sharpless et al. in which the “on water” reactions were seen to be distinctly superior.22
:1) gave the triazolemacrocycle5 (Scheme 1; Path A). The cyclization was tried in several organic solvents and under different dilution conditions6 (Table 1); and the t-BuOH–H2O system (1:1) was found to be best suited for the purpose, in agreement with the observation by Sharpless et al. in which the “on water” reactions were seen to be distinctly superior.22
 | ||
| Scheme 1 Synthesis of glycerotriazolophanes. | ||
| Solvent system | Reaction conditions | Yield (%) | ||
|---|---|---|---|---|
| Time | Reagents | Temp | ||
| a Reaction was carried out at high dilution, no macrocyclisation was found. b Product was not isolated in the form of self-assembled solid (in contrast to the case in which water was used as a co-solvent); no increment in the yields were found on prolongation of the reaction time. | ||||
| Toluenea | 20 d | CuI, DBU | rt | None |
| Acetonitrilea | 20 d | CuI, DIPEA | 40 °C | None |
| DMFb | 30 min | CuSO4/Na ascorbate | rt | 25 |
| t-BuOHb | 30 min | CuSO4/Na ascorbate | rt | 20 |
| THF–water | 15 min | CuSO4/Na ascorbate | rt | 84 |
| CH2Cl2–water | 15 min | CuSO4/Na ascorbate | rt | 80 |
| t-BuOH–water | 10 min | CuSO4/Na ascorbate | rt | 92 |
In t-BuOH–H2O it was found that the product 5a as formed was precipitated out as a near-colourless solid. The solids on separation by filtration and evaluation for their solubility after drying were found to possess characteristically very poor solubility in most protic/aprotic as well as polar/non-polar solvents. The product was seen to be sparingly soluble in DMSO as well as neat TFA. The 1H NMR spectrum of the compound exhibited a singlet of two protons at δ 8.05 ppm, corresponding to the hydrogen on each of the two triazole units in the structure.
Although the overall spectrum appeared to be relatively simple because of the symmetry present in the molecule, the OH-2 and the OH-2' were found to be distinctly different and were located at δ 5.69 and δ 4.92 ppm. On the other hand, H-1a, H-1b, H-1'a, H-3a, H-3b and H-3'a were observed at δ 4.50 ppm and H-1'b, H-2 and H-3'b at δ 4.17–4.16 ppm under two broad singlets. Likewise while the singlet at δ 3.55 ppm corresponded to H-2', 1-O-CH2 and 3-O-CH2 were located at δ 3.45–3.41 ppm, again as singlets. The assignments were based on the 1H–1H and 1H–13C correlation spectra as well as the deuterium exchange reactions. HRMS of the compound showed the only peak at 333.1290 Da, which corresponded to the value calculated for [M + Na]+. Some of these observations led us to consider the possibility of the formation of organized structures in the system. Thus, compound 5a as formed was subjected to SEM analysis. The micrographs were indeed confirmatory of ordered structures for the system. Layered structures with jigsaw-like individual blocks organized into a contiguous form in each of the layers were clearly evidenced. This of course indicated a possible arrangement of the basic structural units through secondary valence bonds, which was therefore verified by molecular modelling (to be described below). Various triazolemacrocycles (5b-5p) synthesized starting from their corresponding starting materials (corresponding derivatives of 2 and 3) in this manner are listed in Table 2. The pseudo-C2 symmetric macrocycles on the other hand were prepared by installing an azide residue on one end and a propargyl ether moiety on the other end of the three-carbon chain (Scheme 1; Path B). The reaction of propargyl alcohol with ECH in the presence of K2CO3 gave glycidyl propargyl ether which on further reaction with sodium azide in PEG 400 yielded 1-azido-1-deoxy-3-O-propargyl-glycerol (4, Scheme 1).21 The latter (4) on subjection to click reaction conditions led to the desired pseudo-C2 symmetric triazole-macrocycles6. The NMR and HRMS data for this pseudo-C2 symmetric macrocycle (6a) were also in accordance with the structure shown (Scheme 1 and Table 3).
|
|
|
||||||
|---|---|---|---|---|---|---|---|
| Entry | R 1 | R 2 | Product No. | Entry | R 1 | R 2 | Product No |
| 1 | H | H | 5a | 9 | Ts | OAc | 5i |
| 2 | H | Ac | 5b | 10 | Ts | Piv | 5j |
| 3 | H | Bz | 5c | 11 | Ts | Ts | 5k |
| 4 | H | Bn | 5d | 12 | Piv | H | 5l |
| 5 | Ac | H | 5e | 13 | Piv | Ac | 5m |
| 6 | Ac | Ac | 5f | 14 | Piv | Piv | 5n |
| 7 | Ac | Ts | 5g | 15 | Piv | Ts | 5o |
| 8 | Ts | H | 5h | 16 | Bz | Piv | 5p |

 6
6
|
 A typical scanning electron micrograph of the freshly prepared glycerotriazolophane 6a A typical scanning electron micrograph of the freshly prepared glycerotriazolophane 6a |
||
| Entry | R | Product | |
| 1 | H | 6a | |
| 2 | Piv | 6b | |
| 3 | Ts | 6c | |
| 4 | Bz | 6d | |
Electronic structure and interaction-analysis
In order to understand the structural details and the various intramolecular interactions, the structures of the macromolecule5a and 6a were optimized using a Gaussian03 package employing a density functional B3LYP/6-31+G(d) method, the 3D structures of which are given in Fig. 1 (for methodology details, see supporting information†).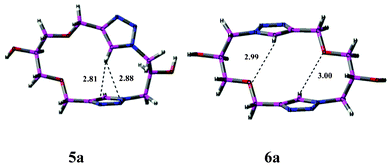 | ||
| Fig. 1 The monomeric arrangement of representative macrocycles, 5a and 6a. The intramolecular hydrogen bond distances are given in Å units. | ||
5a is characterized by pseudo-Cs symmetric properties. Further, a T-shaped interaction was revealed between the C–Htriazole and the nitrogen atoms on the other triazole ring in the molecule, with the former acting as the hydrogen bond donor, and involved a weak non-classical bifurcated C–H⋯N1N2 bonding interaction with the electronegative nitrogen atoms of the other triazole ring. The observed H-bond distance of 2.81 Å (CH⋯N1) and 2.88 Å (CH⋯N2) respectively were in accordance with this inference. AIM analysis23a (Fig. S2†) shows bond critical points (BCP) corresponding to both CH⋯N1 and CH⋯N2 intramolecular H-bonding interactions. There is also a ring critical point (RCP) corresponding to a seven-membered ring involving Ctriazole–N–C1′–C2′–C3′–N2–H formed due to these intramolecular hydrogen bond interactions. The images for AIM analysis showing BCP and RCP for several interactions are shown in the supporting information (Fig. S2†). In structure 6a, the T-shaped interaction is absent, instead, two weak H-bonding interactions between C–Htriazole and the ethereal oxygen were observed with H-bond distances of 2.99 and 3.00 Å.23b
Since the macromolecule was observed to show a self-assembling property, an analysis of its ‘dimeric’ structural form that could give an indication of the possible intermolecular interactions determining their self-assembling character was undertaken. The dimeric structures were constructed from the optimized geometry of the monomer as discussed above. The optimized 3D geometries of the dimer D-5a and D-6a are shown in Fig. 2.
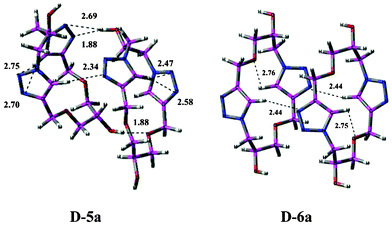 | ||
| Fig. 2 The self-assembly of representative macrocycles in the form of dimeric structures (D-5a and D-6a) showing different interactions. The intramolecular and intermolecular hydrogen bond distances are given in Å units. | ||
As shown in Fig. 2, there are three different intermolecular interactions in structure D-5a, namely H-bond interactions between (i) OH⋯Ntriazole, (ii) CHtriazole⋯Ntriazole and (iii) OH⋯Oether. The H-bond distances for these interactions were found to be 2.69, 2.34 and 1.88 Å respectively. These H-bond interactions stabilize the dimeric structure D-5a, and suggest the possibility of a self-assembling character. The intramolecular T-shaped interactions found in 5a continued to be present in the dimeric structure D-5a, with bond distances of 2.47–2.75 Å. The intermolecular CH⋯N interactions were confirmed by AIM analysis23c (Fig. S2†), which shows electron density at several bond critical points.
The structure D-6a represents the dimeric form of 6a, wherein it adopts a step-like (ladder) conformation due to the presence of intermolecular H-bonding interactions between the CHtriazole and the Ntriazole ring. These H-bonding interactions showed characteristic bond critical point parameter values for the intramolecular hydrogen bond as observed through AIM analysis.23d These H-bonding interactions give stabilization and a self-assembling property to this macromolecule. The intramolecular weak H-bonding CHtriazole⋯Oether interactions continued to be present in D-6a, similar to those in 6a. The dimer D-6a with a step-like or ladder conformation can be correlated to its SEM showing a diffused pattern. Since the SEM shows the self assembly of monomers and a higher connectivity between the monomers, the pattern can be extrapolated from the dimeric arrangement, where there is a vast amount of intermolecular CH⋯N hydrogen bond interactions.
The energy details of the monomers, 5a and 6a, and the energy required for dimerization are listed in Table 4. It can be observed that the monomer 6a, which is pseudo-C2 symmetric, is marginally more stable by 1.35 kcal mol−1 as compared to the pseudo-Cs symmetric monomer 5a. However, in contrast, the dimer D-5a was found to be comparatively more stable by 6.13 kcal mol−1 than D-6a due to the presence of additional H-bonding interactions of OH⋯Ntriazole and OH⋯Oether in addition to the T-shaped interactions.
| Macrocycle | Absolute energy (Hartree) | Relative energy (kcal mol−1) | Dimerization energya (kcal mol−1) |
|---|---|---|---|
| a Dimerization energy calculated by subtracting the summation of energies of monomer from the energy of the dimer. | |||
| Monomer 5a | −1097.431212 | 1.35 | — |
| Monomer 6a | −1097.433475 | 0.00 | — |
| Dimer D-5a | −2194.896536 | 0.00 | −19.10 |
| DimerD-6a | −2194.885194 | 6.13 | −10.27 |
The dimerization energies were calculated for both the monomers with respect to the summation of two monomers, and it was observed that the formation of D-6a releases ∼9 kcal mol−1 energy less than the dimer D-5a. The dimer formation in both cases was found to be exothermic. This also provides support for the higher stability of dimer D-5a as compared to D-6a.
The NBO charges calculated for the monomeric structures 5a and 6a were found to be similar in the dimeric structures D-5a and D-6a, except for differences in the NBO charge on the CHtriazole involved in several interactions with Ntriazole. It was observed that in 5a, the NBO charge on CHtriazole (0.261) increased to 0.268 for the T-shaped intramolecular interaction, and to 0.279 for the intermolecular H-bonding interaction. The charge on the Ntriazole involved in the intermolecular H-bonding interaction was found to be higher (−0.287) as compared to that involved in the T-shaped interaction (−0.263).
In the structure D-6a, the two intermolecular CH–NH-bonding interactions result in an increased charge on Ntriazole from −0.258 to −0.294. Moreover, the charge on the CHtriazole involved in intramolecular CH–Oether interaction increases from 0.259 to 0.274 and the charge on Oether from −0.606 to −0.611, indicating the strength of the interactions.
The relative and actual stabilization energies, different intra- and intermolecular interactions, NBO charges and AIM analysis confirm the self-assembling properties of these molecules. The SEM analysis of the macromolecules synthesised can be correlated to their dimeric structures, wherein the step-like arrangement of D-6a gives the impression of a flat structure akin to its diffused and flat SEM. This can be attributed to the close contact between two monomeric units via two CHtriazole–Ntriazole H-bonding interactions. Similarly, the stabilization of D-5a due to the presence of several additional H-bonding interactions gives this macromolecule a distinct shape and regularity that is translated to a characteristically defined pattern in its SEM (micrographs). Thus, a relation between the polymeric arrangement of these macromolecules and their corresponding SEM patterns could be identified using Density Functional Theory calculations (Fig. S1†). The self-assembling nature of these macrocycles can be due to thermodynamic factors leading to the release of several kcal mol−1 of energy, along with electrostatic factors.
Characterization by other physical methods
With a view to finding further support for molecular organization in the triazolophanes prepared, their characterization was also attempted by other physical methods. Compounds 5a and 6a possessed very low solubility in even powerful solvents such as DMF, DMSO, TFA etc. and our repeated attempts to get crystals suitable for crystallography proved unsuccessful. Efforts to generate diffractograms suitable enough for solving the structure by powder X-ray diffraction (PXRD) was also unsuccessful. Hence we turned our attention to analysis of the compound by high-resolution transmission electron microscopy (HRTEM) (Fig. 3). The micrographs (Fig. 3) clearly illustrate the ordered arrangement of the monomeric units in the macromolecular organization of compounds 5a and 6a. The small grain size observed possibly also explains the poor signal to noise ratio obtained while generating the PXRD data. Further, the endotherm at 65.6 °C for 5a and 72.2 °C for 6a observed in the thermograms (Supplementary Information 1†) obtained on subjecting compounds 5a and 6a to differential scanning calorimetry (DSC) analysis must correspond to the glass transition temperature (TG) of the respective organic compound. The Tm (187 °C for 5a and 260.5 °C for 6a) obtained by DSC was in agreement with the value (185–187 °C for 5a and 260.5 °C for 6a) obtained on determining the melting point on a laboratory melting point apparatus.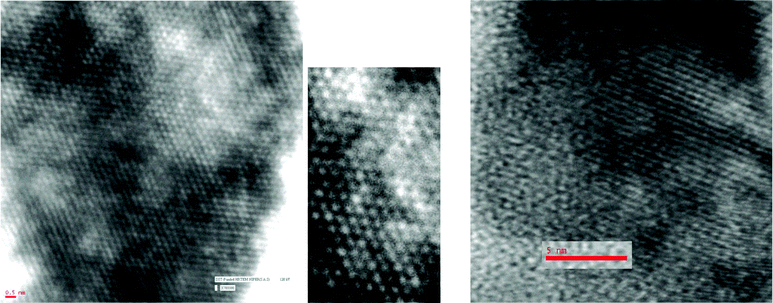 | ||
| Fig. 3 HRTEM imaging of glycerotriazolophanes. Left: compound 5a and middle: a portion of it magnified; right: compound 5b. | ||
Experimental methodology and characterization data
Differential scanning calorimetric thermograms were obtained on a Perkin Elmer Diamond DSC instrument equipped with a thermal analysis data station (Pyris software) at a heating rate of 5 °C min−1. Measurements were carried out on samples after drying under vacuum. An aluminium sample pan after loading (sample weight, 2–3 mg) was closed by placing the lid over the sample in the pan and sealing. An empty pan with a lid served as a reference. The instrument was calibrated with an indium standard before starting the experiment. The area under the endotherm was converted to energy units with the help of thermal analysis software. The heat of transition has been expressed as J g−1 dry wt.
Conclusions
Synthesis and characterization of a library of glycerotriazolophane-macrocycles with interesting self-assembling properties are described. The structure and properties of the compounds were rationalized by molecular modelling and quantum chemical analysis.Acknowledgements
Financial support by the Council of Scientific and Industrial Research (CSIR), New Delhi (to MT) and the Department of Science and Technology (DST), New Delhi (to NT and PVB) is gratefully acknowledged.References
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- (a) F. Reck, F. Zhou, M. Girardot, G. Kern, C. J. Eyermann, N. J. Hales, R. R. Ramsay and M. B. Gravestock, J. Med. Chem., 2005, 48, 499 CrossRef CAS; (b) W. S. Horne, C. A. Olsen, J. M. Beierle, A. Montero and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4718 CrossRef CAS; (c) Y. Naito, F. Akahoshi, S. Takeda, T. Okada, M. Kajii, H. Nishimura, M. Sugiura, C. Fukaya and Y. Kagitani, J. Med. Chem., 1996, 39, 3019 Search PubMed; (d) M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. D. Reischer, D. Stper and B. H. Yagi, J. Med. Chem., 2000, 43, 953 CrossRef CAS; (e) R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4194 Search PubMed; (f) D. R. Buckle, C. J. M. Rockell, H. Smith and B. A. Spicer, J. Med. Chem., 1986, 29, 2262 CrossRef CAS; (g) R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185 CrossRef CAS; (h) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961 CrossRef CAS.
- (a) J. F. Billing and U. J. Nilsson, J. Org. Chem., 2005, 70, 4847 CrossRef CAS; (b) J. L. Sessler, J. Cai, H. -Y. Gong, X. Yang, J. F. Arambula and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 14058 CrossRef CAS; (c) K. D. Bodine, D. Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638 CrossRef CAS; (d) S. Muthana, H. Yu, H. Cao, J. Cheng and X. Chen, J. Org. Chem., 2009, 74, 2928 CrossRef CAS.
- D. D. Diaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392 CrossRef CAS.
- (a) D. D. Diaz, J. J. M. Tellado, D. G. Vel_azquez and A. G. Ravelo, Tetrahedron Lett., 2008, 49, 1340 CrossRef; (b) V. A. -Leonetia, V. L. Campo, A. S. Gomes, R. A. Field and I. Carvalho, Tetrahedron, 2010, 66, 9475 CrossRef CAS.
- (a) K. D. Bodine, D. Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638 CrossRef CAS; (b) K. D. Bodine, D. Y. Gin and M. S. Gin, Org. Lett., 2005, 7, 4479 CrossRef CAS; (c) H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 5091 CrossRef CAS; (d) R. Rajesh, G. Periyasami and R. Raghunathan, Tetrahedron Lett., 2010, 51, 1896 CrossRef CAS; (e) A. Ray, K. Manoj, M. M. Bhadbhade, R. Mukhopadhay and A. Bhattacharjya, Tetrahedron Lett., 2006, 47, 2775 CrossRef CAS; (f) S. Dorner and B. Westermann, Chem. Commun., 2005, 2852 RSC; (g) N. Pietrzik, D. Schmollinger and T. Ziegler, Beilstein J Org. Chem., DOI:10.3762/bjoc.4.30, 2008, 4 Search PubMed; (h) G. V. Latyshew, M. S. Baranov, A. D. Averin, N. V. Lukashev and I. P. Beletskaya, Synthesis, 2009, 15, 2605 Search PubMed.
- (a) H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740 CrossRef CAS; (b) Y. Hua, R. O. Ramabhadran, E. O. Uduehi, J. A. Karty, K. Raghavachari and A. H. Flood, Chem.–Eur. J., 2011, 17, 312 CrossRef CAS; (c) Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630 CrossRef CAS; (d) Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838 CrossRef CAS; (e) S. Lee, Y. Hua, H. Park and A. H. Flood, Org. Lett., 2010, 12, 2100 CrossRef CAS; (f) Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649 CrossRef CAS.
- (a) F. García, M. R. Torres, E. Matesanz and L. Sánchez, Chem. Commun., 2011, 47, 5016 RSC; (b) T.-B. Yu, J. Z. Bai and Z. Guan, Angew. Chem., Int. Ed., 2009, 48, 1097 CrossRef CAS; (c) V. Haridas, K. Lal, Y. K. Sharma and S. Upreti, Org. Lett., 2008, 10, 1645 CrossRef CAS; (d) V. Haridas, S. Sahu and P. Venugopalan, Tetrahedron, 2011, 67, 727 CrossRef CAS.
- A. Kumar and P. S. Pandey, Org. Lett., 2008, 10, 165 CrossRef CAS.
- (a) K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240 RSC; (b) O. Š. Miljanić, W. R. Dichtel, I. Aprahamian, R. D. Rohde, H. D. Agnew, J. R. Heath and J. F. Stoddart;, QSAR Comb. Sci., 2007, 26, 1165 CrossRef CAS.
- Y. Hsieh, J. Chir, H. Wua, P. Chang and A. Wua, Carbohydr. Res., 2009, 344, 2236 CrossRef CAS.
- (a) N. G. Angelo and P. S. Arora, J. Am. Chem. Soc., 2005, 127, 17134 CrossRef CAS; (b) V. D. Bock, D. Speijer, H. Hiemstra and J. H. van Maarseveen, Org. Biomol. Chem., 2007, 5, 971 RSC; (c) Y. Liu, L. Zhang, J. Wan, Y. Li, Y. Xu and Y. Pan, Tetrahedron, 2008, 64, 10728 CrossRef CAS; (d) M. Nahrwold, T. Bogner, S. Eissler, S. Verma and N. Sewald, Org. Lett., 2010, 12(5), 1064 Search PubMed; (e) A. Isidro-Llobeta, T. Murilloa, P. Belloa, A. Cilibrizzia, J. T. Hodgkinsona, W. R. J. D. Gallowaya, A. Benderb, M. Welchc and D. R. Spring, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6793 CrossRef CAS.
- C. D. Hein, X. Liu and D. Wang, Pharm. Res., 2008, 25, 2216 CrossRef CAS.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654 CrossRef CAS.
- V. L. Campo, I. Carvalho, C. H. T. P. Da Silva, S. Schenkman, L. Hill, S. A. Nepogodiev and R. A. Field, Chem. Sci., 2010, 1, 507 RSC.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848 RSC.
- J. M. Beierle, W. S. Horne, J. H. V. Maarseveen, B. Waser, J. C. Reubi and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4725 CrossRef CAS.
- (a) N. Bowden, A. Terfort, J. Carbeck and G. M. Whitesides, Science, 1997, 276, 233 CrossRef CAS; (b) M. Boncheva and G. M. Whitesides, MRS Bull., 2005, 30, 736 CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS.
- A. Matijoska, Liet. TSR Mokslu, Akad. Darb. Ser. B, 1988, 4 Search PubMed 52-7 (Russ) Chemical Abstract 1989, 111, 684.
- A. R. Kiasat and M. Fallah-Mehrjardi, J. Iran. Chem. Soc., 2009, 6, 542 Search PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS.
- (a) AIM analysis (5a): BCPs were shown by a bond path connecting C–H⋯N1; bond critical point (BCP) is characterized by parameters typical to an intramolecular hydrogen bond, ρH–N = 0.051 × 10−1, ∇2 ρH—N = 0.047 × 10−1. A similar C–H⋯N2 intramolecular hydrogen bond interaction is found characterized by similar values of ρH–N = 0.049 × 10−1, ∇2 ρH–N = 0.082 × 10−1. There is also a ring critical point (RCP) (ρ = 0.038 × 10−1) corresponding to a seven-membered ring formed due to these intramolecular hydrogen bond interactions.; (b) Though the distances indicate intramolecular hydrogen bond, AIM analysis does not indicate the presence of a bond critical point. Hence, this intramolecular interaction may have only electrostatic component; (c) AIM analysis (D-5a): The electron density at several bond critical points was observed, with the values of ρH–N = 0.038 × 10−1, ∇2 ρH–N = 0.032 × 10−1. An intramolecular OH—Ntriazole interaction was characterized by values of ρH–N = 0.34 × 10−1 and ∇2 ρH–N = 0.24 × 10−1. The intramolecular hydrogen bond interaction between OH—Oether was characterized by ρH–O = 0.30 × 10−1 and ∇2 ρH–O = 0.015 × 10−1; (d) AIM analysis: (D-6a) The CHtriazole and Ntriazole interactions showed characteristic bond critical point parameter values; ρH–N = 0.115 × 10−1, ∇2 ρH–N = 0.089 × 10−1. Similar values were obtained for other CHtriazole⋯Ntriazole intramolecular H-bond interaction, ρH–N = 0.115 × 10−1, ∇2 ρH–N = 0.089 × 10−1.
- See Supplementary Information 2 for more details.
Footnote |
| † Electronic Supplementary Information (ESI) available: Images of polymeric structures of macromolecules and their SEM analysis; images of AIM analysis for 5a, 6a, D-5a, D-6a; Archive entries of all the optimized geometries; 1H and 13C spectra of parent compounds synthesized. See DOI: 10.1039/c2ra21341a |
| This journal is © The Royal Society of Chemistry 2012 |