Oxidation of sulfides to sulfoxides mediated by ionic liquids†
Bo
Zhang
a,
Ming-Dong
Zhou
b,
Mirza
Cokoja
a,
János
Mink
c,
Shu-Liang
Zang
bd and
Fritz E.
Kühn
*a
aMolecular Catalysis / Chair of Inorganic Chemistry, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straβe 1, D-85747, Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 289 13473; Tel: +49 89 289 13081
bSchool of Chemical and Material Science, Liaoning Shihua University, Dandong Road No. 1, 113001, Fushun, P. R. China
cChemical Research Centre of the Hungarian Academy of Sciences and Pusztaszeri út 59-67, 1025 Budapest, Hungary and Analytical Chemistry Research of the Hungarian Academy of Sciences and Research Institute of Chemical and Process Engeering University of Veszprém, Egyetem u. 10 8200, Veszprém, Hungary
dInstitute of Rare and Scattered Elements Chemistry, Liaoning University, Chongshan Middle Road No. 66, 110036, Shenyang, P. R. China
First published on 12th July 2012
Abstract
A highly selective and efficient oxidation of sulfides to sulfoxides is presented. The reactions were carried out at room temperature in the absence of a catalyst in the ionic liquid [Bmim][BF4] (Bmim = 1-butyl-3-methylimidazolium) using aqueous H2O2 (35%) as oxidant. The products were obtained in high yields. Compared to the analogous reactions in organic solvents, this system can be recycled without significant loss of activity and selectivity. Additionally, the reaction mechanism was examined by IR, Raman and NMR spectroscopy. Based on these examinations, it appears that the crucial step during the oxidation procedure is the formation of a hydrogen bond between the ionic liquid and the oxidant.
Introduction
The oxidation of sulfides is of considerable significance for industrial chemistry. The removal of sulfur-containing compounds is an important process in fuel industry.1 Therefore, research efforts focus on cost-efficient liquid phase processes, such as the oxidation of sulfides, which are corrosive agents for car engines and problematic for the environment. Furthermore, the selective transformation of organic sulfides to sulfoxides is of high interest in the synthesis of fine chemicals. Sulfoxides are valuable intermediates for the construction of various fine chemicals as useful building blocks in asymmetric synthesis.2 Thus, there is high demand for a low cost, efficient and highly selective method for the oxidation of sulfides to sulfoxides. There are numerous reports on the oxidation of sulfides to sulfoxides using molecular V-,3 Re-,4 Fe-,5 Mn-,6 Ti-,7 Mo-8 and W-9 based catalysts in organic solvents. However, these catalysts bear several disadvantages: firstly, the syntheses of the catalysts are often quite demanding, which renders them expensive. Secondly, over-oxidation to sulfones can often not be prevented. Some catalyst-free oxidations of sulfides were reported before.10 However, these reactions are usually quite time-consuming and accompanied by destruction of functional groups.Room temperature ionic liquids (RTILs) have received much attention due to their unique properties, such as negligible volatility, broad liquid temperature ranges and the ability to dissolve a wide range of inorganic and organic compounds.11 In the last 10 years, RTILs have been applied as alternative reaction media for a plethora of catalyzed reactions,12 including the oxidation of sulfides to sulfones.13 Previously, we and others have shown that ILs are very convenient reaction media for the epoxidation of olefins catalyzed by organometallic Re-14 and Mo-compounds.15 The catalytic activities in ILs surpass those achieved in organic solvents.16 More recently, we discovered that ionic liquids may have a beneficial effect on the activation of H2O2 in olefin epoxidation.17 In this work, a novel, efficient, highly selective and catalyst-free approach for the oxidation of sulfides to sulfoxides in ionic liquids is reported.
Results and discussion
Thioanisole was treated in a preliminary study with various oxidants in different reaction media to elucidate optimal reaction conditions (Scheme 1). It is known that the oxidant selection is very important for the oxidation of sulfide to sulfoxide and a variety of oxidants such as KMnO4,18 NaClO4,19 molecular halogens,20 NBS21 and tert-butyl hydroperoxide (TBHP).22 However, these oxidants are disadvantageous due to their low oxygen content, which not only makes product isolation difficult, but also leads to the formation of harmful byproducts. H2O2 is a good alternative oxidant for such a process not only due to its low cost and high oxygen content but also for being an environmental benign reagent, forming only water as byproduct. In this study, the activity of H2O2 for the oxidation of various sulfides using several ILs such as [Bmim][BF4] (Bmim = 1-butyl-3-methylimidazolium), [Emi][SE] (1-ethyl-3-methylimidazolium ethyl sulfate), [Bmim][HSO4] and [Dbmim][BF4] (1,2-dimethyl-3-butylimidazolium) both as solvent and promoter was examined. For sake of comparison, several other reactions using organic solvents, or different oxidants such as UHP and TBHP were also examined. All these results are summarized in Table 1. | ||
| Scheme 1 Oxidation of sulfides to sulfoxides. | ||
| Entry | Solvent | Oxidant | Time/h | Conv. (%)b | Yield (%)c |
|---|---|---|---|---|---|
| a Reaction conditions: 4 mL of solvent, 10 mmol of thioanisole, 20 mmol of oxidant. b Determined by GC-MS on the crude reaction mixture. c Isolated yield after column chromatography. | |||||
| 1 | n-Hexane | H2O2 | 24 | 10 | 7 |
| 2 | Toluene | H2O2 | 24 | 24 | 21 |
| 3 | CH2Cl2 | H2O2 | 24 | 29 | 28 |
| 4 | CH3CN | H2O2 | 24 | 72 | 69 |
| 5 | Methanol | H2O2 | 18 | >99 | 9910b |
| 6 | [Bmim][BF4] | — | 24 | — | — |
| 7 | [Bmim][BF4] | H2O2 | 4 | 98 | 95 |
| 8 | [Bmim][BF4] | UHP | 24 | 13 | 12 |
| 9 | [Bmim][BF4] | TBHP | 24 | 6 | 5 |
| 10 | [Dbmim][BF4] | H2O2 | 4 | 89 | 86 |
| 11 | [Bmim][HSO4] | H2O2 | 4 | 91 | 83 |
| 12 | [Emi][SE] | H2O2 | 4 | 92 | 80 |
It is seen in Table 1 that solvent selection is very important for sulfide oxidation. A high yield can be obtained only if the H2O2 solution can be dissolved in the selected solvent forming a homogeneous reaction system. In the case of using n-hexane, toluene or CH2Cl2 as solvent (Entries 1–3), a clear biphasic solution is observed, leading to a low sulfoxide yield (<30%). Conversely, a high yield (>65%) can be obtained when using CH3CN, methanol and ILs as solvent since all of them form a homogeneous phase with the H2O2 containing phase (Entries 5, 7, 10–12). Additionally, the polarity of the reaction medium may also be an important factor for the oxidation of sulfides. Methanol, despite very good conversion and yield, allows only quite long reaction time (18 h) and functional group decomposition occurs.10b The reaction becomes much faster when using ILs as reaction media (4 h) (Entries 7, 10–12). A high conversion (98%) and good yield (95%) within 4 h reaction time can be achieved in [Bmim]BF4 as solvent (Entry 7). This is mainly because that the applied IL acts not only as a solvent but also an activator in the reaction system. A more detailed discussion of the interaction between IL and H2O2, will be given in the spectroscopic part. Although most ILs in Table 1 represent high conversions (>89%) and yields (>80%), [Bmim][BF4] exhibits better stability than other ILs (such as [Bmim][HSO4], [Emi][SE]). The oxidation of thioanisole is completed within 4 h and less than 3% of sulfone is formed, even if the reaction time of 24 h is allowed. Therefore, [Bmim][BF4] was selected as solvent. The oxidation behavior in the IL system was also investigated (Entries 6–9). A blank experiment without oxidant was carried out and no significant oxidation was observed within 24 h (Entry 6), indicating that the oxygen source for the synthesis of sulfoxide is not air. For different oxidants such as TBHP and urea hydrogen peroxide adduct (UHP), only low conversions (13% for UHP, 6% for TBHP) and yields (12% for UHP, 5% for TBHP) were obtained (Entries 8, 9).
To generalize the developed protocol, a series of sulfides with different functional groups were selected (see Table 2) and all oxidation reactions were performed under the same conditions as Entry 7 in Table 1. Among all investigated substrates, dimethyl sulfide (Entry 6) was more easily oxidized (within 2 h) than other substrates with bulkier substituents. This indicates that the steric hindrance is an important factor for this reaction. In general, sulfides bearing strong electron-donating alkyl and alkene groups (Entries 1, 2, 5, 6, 9 and 11) display very good yields (>80%). On the other hand, the oxidation of sulfides containing electron-withdrawing groups such as alcoholate (Entry 7), ester (Entry 8), acetonyl (Entry 10) or less electron-donating groups (Entries 3, 4 and 12–14) lead to lower yields. Presumably, electron-donating groups increase the nucleophilicity of the sulfur atom and hence the reactivity of the substrates. Notably, neither epoxidation of the double bond of allyl phenyl sulfide (Entry 9) nor oxidation of the hydroxy group (Entry 7) was observed. Although the oxidation of diphenyl sulfide (Entry 3) requires a higher temperature compared to other aryl sulfides, the presented method still exhibits a relatively easy way to obtain diphenyl sulfoxides in a one-pot reaction. The reactivity of thioethers is affected by the nucleophilicity of the sulfur atom and the steric hindrance of the substituted groups. Higher reactivity is always obtained in the case of strong electron-donating groups and with smaller substituents.
| Entry | R1 | R2 | Time/h | Conv. (%)b | Yield (%)c |
|---|---|---|---|---|---|
| a Reaction conditions: 4 mL [Bmim][BF4], 10 mmol substrate, 20 mmol H2O2 (35%) at 25 °C. b Determined by GC-MS or 1H NMR on the crude reaction mixture. c Isolated yield after column chromatography. d Reaction conditions: 4 mL [Bmim][BF4], 10 mmol substrate, 20 mmol H2O2 (35%) at 50 °C. e iPr = isopropyl. f Bz = benzyl. | |||||
| 1 | Ph | Me | 4 | 98 | 95 |
| 2 | Ph | Et | 6 | 98 | 90 |
| 3 | Ph | Ph | 8d | 79 | 65 |
| 4 | Ph | Ph | 24 | 81 | 72 |
| 5 | nBu | nBu | 6 | 90 | 86 |
| 6 | Me | Me | 2 | 98 | 96 |
| 7 | Ph | CH2CH2OH | 8 | 81 | 75 |
| 8 | Ph | CH2COOMe | 8 | 78 | 71 |
| 9 | Ph | CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
8 | 83 | 80 |
| 10 | Ph | CH2OCH3 | 8 | 85 | 78 |
| 11 | Ph | iPre | 8 | 91 | 87 |
| 12 | Ph | Bzf | 8 | 79 | 73 |
| 13 | Bz | Bz | 6 | 89 | 85 |
| 14 | Dibenzothiophene | 8[d] | 38 | 36 | |
Regarding the mechanism of this reaction, the interaction between the ionic liquid and H2O2 appears to be crucial. Organic sulfides oxidized by hydrogen peroxide involve an electrophilic attack of the oxygen atom on the sulfur.23 This also explains the faster oxidation rate of aliphatic sulfides, which are more nucleophilic than aromatic sulfides. It may be assumed that the BF4 anion of the ionic liquid forms a hydrogen bond with H2O2 and increases the electrophilic ability of a peroxide oxygen atom of H2O2, and at the same time assists the leaving group H2O in departing from the reaction intermediate.10c (Scheme 2). However, it is very important to exclude other possible interactions with hydrogen peroxide. A thinkable scenario would be the reaction of hydrogen peroxide with the BF4 anion, which would generate hydrofluoric acid, or protons, respectively, which are also known to catalyze the oxidation of sulfides.24 Further, hypothetically, an oxygen atom could interact with the imidazolium cation. The interaction of H2O2 would be strongest with the hydrogen atom in the C2 position of the imidazolium ring, since the two neighboring N atoms have a relatively strong electron withdrawing ability, rendering the C–H bond more acidic.
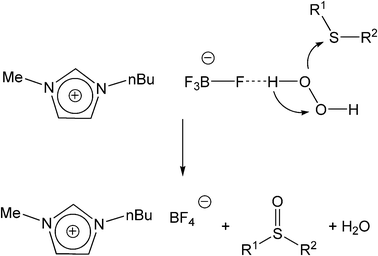 | ||
| Scheme 2 Suggested mechanism of the oxygen transfer from H2O2 to a sulfide in ionic liquids. | ||
A number of spectroscopic examinations of the interactions between water and ionic liquids were performed to date.25 Lendl et al. found that water interacts with BF4−via hydrogen bonding.26 A similar interaction between the anion of the IL and hydrogen peroxide could be expected. IR, Raman and NMR spectroscopy are convenient tools to support this hypothesis.
In order to answer the question, which type of interaction is most likely responsible for the activation of hydrogen peroxide, we performed vibration and nuclear magnetic resonance spectroscopic studies of the ionic liquid [Bmim][BF4] before and after treatment with hydrogen peroxide. Fig. 1 and 2 show the IR and Raman spectra of the B–F region of [Bmim][BF4] at different H2O2 concentrations. Pure [Bmim]BF4 has been examined in this range as well, bands were assigned as in previous reports.25i In this study, we concentrated on BF stretching vibrations regions of 900–1200 cm−1 for IR and 750–780 cm−1 for Raman as they are representative of structural changes in H2O2 (35%).
![IR spectra in the range of 900 to 1200 cm−1 for the treatment of [Bmim]BF4 with different H2O2 concentrations. The lines represent the spectra for 0.23, 0.53, 1.5, 3.8, mol·L−1 H2O2 concentrations from top to bottom (the topmost line represents pure [Bmim]BF4).](/image/article/2012/RA/c2ra21323k/c2ra21323k-f1.gif) | ||
| Fig. 1 IR spectra in the range of 900 to 1200 cm−1 for the treatment of [Bmim]BF4 with different H2O2 concentrations. The lines represent the spectra for 0.23, 0.53, 1.5, 3.8, mol·L−1 H2O2 concentrations from top to bottom (the topmost line represents pure [Bmim]BF4). | ||
![Raman spectra in the range of 750 to 780 cm−1 for the treatment of [Bmim]BF4 with different H2O2 concentrations. The lines represent spectra for 3.4 and 4.1 mol·L−1 H2O2 concentrations from top to bottom (the topmost line represents pure [Bmim]BF4).](/image/article/2012/RA/c2ra21323k/c2ra21323k-f2.gif) | ||
| Fig. 2 Raman spectra in the range of 750 to 780 cm−1 for the treatment of [Bmim]BF4 with different H2O2 concentrations. The lines represent spectra for 3.4 and 4.1 mol·L−1 H2O2 concentrations from top to bottom (the topmost line represents pure [Bmim]BF4). | ||
When increasing the H2O2 molar fraction, the symmetric BF4 stretching band only slightly shifts to a higher wavenumber (Fig. 1). Three bands have been observed at ∼1045 cm−1, ∼1033 cm−1, ∼1016 cm−1 for the pure ionic liquid.25i The BF4 anion displays Td symmetry resulting in four bands. Two of these bands are polarized (IR) and two are depolarized (Raman), and the two bands should be triplets in IR. However the three bands change into two (at ∼1068 cm−1, ∼1025 cm−1) after the addition of H2O2, due to the interaction between H2O2 and the BF4 anion. A B–F⋯H hydrogen bond may be assumed thus leading to a widening of the corresponding band (as compared to B-F). The original three bands have changed into two with the increasing concentration of H2O2, accordingly. In the Raman spectra, the symmetric stretching mode (B-F) of the ionic liquid is shifted from ∼764 cm−1 (pure form) to 767 cm−1 (Fig. 2). This finding is matched with the IR spectra, which also gives only a minor shift on the B–F vibration band after the addition of H2O2. The O–O vibration band of H2O2 was also studied with Raman techniques (Fig. 3). The shift difference of ν(O–O) is about 5 cm−1 between pure H2O2 (∼876 cm−1) and IL + H2O2 (∼871 cm−1), suggesting the H2O2 is weakly interacting. Furthermore, 11B NMR and 19F NMR were also performed to show hydrogen bonding in this system. The 11B NMR spectrum shows one resonance at −1.86 ppm (BF4−). According to the isotope effect of the boron atom (10B, 19.9% and 11B, 80.1%), two singlets are observed in the 19F NMR with an integral ratio of ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 at −148.66 and −148.71 ppm. When adding H2O2 to the IL, the 11B NMR resonance shows a slight shift to −2.16 ppm. The corresponding 19F NMR shows signals at −150.42 and −150.47 ppm (see ESI 2.1†). The spectroscopic investigations further show that the symmetry of the tetrafluoroborate anion has not changed and that no new species such as HF and BF3 are observed, which could in principle be formed by a reaction of BF4 with H2O2. The slight shift of the BF4 signals (see ESI 2.1†) points to a change in the coordination sphere of the anion, such as a weak H⋯F interaction, but not to the formation of BF3, which would lead to a significantly different peak position due to the symmetry change. On the other hand, the stretching modes of the imidazolium ring C–H groups (Raman and IR scattering contributions in the range of 3000–3500 cm−1) should be affected by this replacement. Yet, the measurements did not show appreciable changes in positions (see ESI 2.2–ESI 2.3†), maybe as a consequence of the difficulty of the spectral deconvolution due to the overlap between the C–H and the O–H stretching regions, especially for a high water content. To better address this issue, substitution of the C2 proton in the imidazolium ring with a CH3 group has been performed for oxidation of thioanisole under the same conditions, using [Dbmim][BF4] as solvent for oxidation of thioanisole (Table 1, Entry 10), yielding sulfoxides in 86%, which proves that an interaction of H2O2 with imidazolium protons does not take place.25e However, there is no possibility to exclude a situation in which they would interact with each other because of strong electrostatic forces, when the number of “free” cations and anions reaches a certain content.25a With the applied amount of aqueous H2O2, the interaction between H2O2 and the BF4 anion is much stronger and more stable than that between H2O2 and the cation.25e Based on the above described experimental and spectroscopic findings and in combination with published literature,25e a hydrogen bond formation between a peroxide oxygen and a BF4 anion appears to be the crucial step during the oxidation reaction in the [Bmim][BF4]+H2O2 (35%) system.
:4 at −148.66 and −148.71 ppm. When adding H2O2 to the IL, the 11B NMR resonance shows a slight shift to −2.16 ppm. The corresponding 19F NMR shows signals at −150.42 and −150.47 ppm (see ESI 2.1†). The spectroscopic investigations further show that the symmetry of the tetrafluoroborate anion has not changed and that no new species such as HF and BF3 are observed, which could in principle be formed by a reaction of BF4 with H2O2. The slight shift of the BF4 signals (see ESI 2.1†) points to a change in the coordination sphere of the anion, such as a weak H⋯F interaction, but not to the formation of BF3, which would lead to a significantly different peak position due to the symmetry change. On the other hand, the stretching modes of the imidazolium ring C–H groups (Raman and IR scattering contributions in the range of 3000–3500 cm−1) should be affected by this replacement. Yet, the measurements did not show appreciable changes in positions (see ESI 2.2–ESI 2.3†), maybe as a consequence of the difficulty of the spectral deconvolution due to the overlap between the C–H and the O–H stretching regions, especially for a high water content. To better address this issue, substitution of the C2 proton in the imidazolium ring with a CH3 group has been performed for oxidation of thioanisole under the same conditions, using [Dbmim][BF4] as solvent for oxidation of thioanisole (Table 1, Entry 10), yielding sulfoxides in 86%, which proves that an interaction of H2O2 with imidazolium protons does not take place.25e However, there is no possibility to exclude a situation in which they would interact with each other because of strong electrostatic forces, when the number of “free” cations and anions reaches a certain content.25a With the applied amount of aqueous H2O2, the interaction between H2O2 and the BF4 anion is much stronger and more stable than that between H2O2 and the cation.25e Based on the above described experimental and spectroscopic findings and in combination with published literature,25e a hydrogen bond formation between a peroxide oxygen and a BF4 anion appears to be the crucial step during the oxidation reaction in the [Bmim][BF4]+H2O2 (35%) system.
![Raman spectra of H2O2 (solid curve), [Bmim][BF4]+H2O2 (35%) (dashed curve) in the range of 840 to 950 cm−1.](/image/article/2012/RA/c2ra21323k/c2ra21323k-f3.gif) | ||
| Fig. 3 Raman spectra of H2O2 (solid curve), [Bmim][BF4]+H2O2 (35%) (dashed curve) in the range of 840 to 950 cm−1. | ||
Besides the activity and selectivity, the recyclability and stability of ILs for sulfide oxidations were also investigated.19F NMR (δ = −148.70 and −148.76 ppm) and 11B NMR (δ = −1.86 ppm) spectra of the [Bmim][BF4] were recorded after 4 recycling runs, confirming that the IL is stable and no structural change or decomposition took place. Furthermore, as shown in Fig. 4, no significant change of conversion, selectivity and yield are observed. The diphenyl sulfide has been oxidized using recycled [Bmim]BF4 as solvent under the same reaction conditions (Table 2, Entry 4). The obtained yield is 68% after 24 h, proving that this system has constant activity and stability during the recycling experiments.
![Oxidation of thioanisole with H2O2 in [Bmim][BF4] at 25 °C after four reaction runs.](/image/article/2012/RA/c2ra21323k/c2ra21323k-f4.gif) | ||
| Fig. 4 Oxidation of thioanisole with H2O2 in [Bmim][BF4] at 25 °C after four reaction runs. | ||
Experimental
General
All reactions were performed in oven-dried glassware under an argon atmosphere using standard Schlenk techniques. All solvents were collected from purification systems and kept over molecular sieves. 1H NMR, 13C NMR, 19F NMR and 11B NMR spectra were recorded on a Bruker Avance DPX-400 spectrometer. IR spectra were recorded on a Varian FTIR-670 spectrometer, using a GladiATR accessory with a diamond ATR element. Gas chromatography-mass spectroscopy (GC-MS) analysis was performed on an Agilent 6890 instrument using a capillary column (30 m × 0.25 mm × 0.25 μm) with MS detector. Raman spectra were recorded on a Bio-Rad FTS-60A. Melting points were measured by MPM-H2 melting point meters. TLC was performed on silica gel 60F254 plates procured from E. Merck. Silica gel (0.06–0.2 mm 60A) was used for column chromatography. All chemicals were purchased from Acros and ABCR and used without further purification. [Bmim][BF4], [Emi][SE] and [Bmim][HSO4] were synthesized according to literature procedures.27–29Typical procedure for the oxidation of the sulfides in [Bmim][BF4]
To the stirred solution of thioanisole (1.18 mL, 10 mmol) in 4 mL [Bmim][BF4], an aqueous solution of H2O2 (1.75 mL, 20 mmol, 35%) was added at room temperature forming a homogeneous reaction solution. The progress of the reaction was followed by TLC. The reaction mixture was extracted with diethyl ether (5 × 10 mL) and the extractant was dried over anhydrous MgSO4. The crude product was obtained by rolling evaporation and purified by column chromatography separation (silica gel using hexane–ethyl acetate 90:10 v/v). The RTIL phase was diluted with CH2Cl2 and treated with MnO2 to destroy the excess peroxide and dried over anhydrous MgSO4, filtered, and then dried in vacuo for 4 h at 50 °C to remove CH2Cl2. Fresh substrate and hydrogen peroxide were then added for a new reaction cycle. All products were characterized by 1H NMR, 13C NMR and IR spectroscopy (see ESI 1†).
Conclusions
A selective and efficient method for the oxidation of sulfides to the corresponding sulfoxides has been developed. The sulfoxidation can be performed under mild conditions without any catalyst or additional activation reagent when hydrogen peroxide is present. The good stability of this system is supported by 19F- and 11B-NMR spectroscopic results. No significant decrease of activity has been observed after four runs. According to IR, Raman and NMR examinations, the IL used in this system assists the nucleophilic attack of the sulfur atom by hydrogen bond formation between a OH group and a [BF4]− anion. High selectivity and seemingly good recyclability, as well as the absence of classical (and often quite expensive) molecular catalysts together with mild conditions make this method useful for the oxidation of sulfides to sulfoxides.Acknowledgements
B. Z. thanks the TUM Graduate School for financial support. M. D. Z. and S. L. Z. thank the National Natural Science Foundation of China (21071073 and 21101085) for financial support.References
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- (a) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (b) F. Brebion, B. Delouvrie, F. Najera, L. Fensterbank, M. Malacria and J. Vaissermann, Angew. Chem., Int. Ed., 2003, 42, 5342–5345 CrossRef CAS; (c) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 CrossRef CAS.
- (a) A. V. Anisimov, E. V. Fedorova, A. Z. Lesnugin, V. M. Senyavi, L. A. Aslanov, V. B. Rybakov and A. V. Tarakanova, Catal. Today, 2003, 78, 319–325 CrossRef CAS; (b) V. Conte, F. Fabbianesi, B. Floris, P. Galloni, D. Sordi, I. W. C. E. Arends, M. Bonchio, D. Rehder and D. Bogdal, Pure Appl. Chem., 2009, 81, 1265–1277 CrossRef CAS; (c) G. P. Romanelli, D. O. Bennardi, V. Palermo, P. G. Vázquez and P. Tundo, Lett. Org. Chem., 2007, 4, 544–549 CrossRef CAS; (d) R. Trivedi and P. Lalitha, Synth. Commun., 2006, 36, 3777–3782 CrossRef CAS.
- (a) J. H. Espenson, Chem. Commun., 1999, 479–488 RSC; (b) H. Q. N. Gunaratne, M. A. McKervey, S. Feutren, J. Finlay and J. J. Boyd, Tetrahedron Lett., 1998, 39, 5655–5658 CrossRef CAS; (c) K. J. Stanger, J. W. Wiench, M. Pruski, J. H. Espenson, G. A. Kraus and R. J. Angelici, J. Mol. Catal. A: Chem., 2006, 243, 158–169 CrossRef CAS.
- (a) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487–5489 CrossRef CAS; (b) M. Bagherzadeh and M. Amini, Inorg. Chem. Commun., 2009, 12, 21–25 CrossRef CAS; (c) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS; (d) A. Marques, M. Marin and M. F. Ruasse, J. Org. Chem., 2001, 66, 7588–7595 CrossRef CAS; (e) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 4225–4228 CrossRef CAS.
- (a) M. Bagherzadeh, R. Latifi, L. Tahsini and M. Amini, Catal. Commun., 2008, 10, 196–200 CrossRef CAS; (b) F. Xie, Z. Fu, H. S. Zhong, Z. P. Ye, X. P. Zhou, F. L. Liu, C. Y. Rong, L. Q. Mao and D. L. Yin, J. Mol. Catal. A: Chem., 2009, 307, 93–97 CrossRef CAS.
- M. Iwamoto, Y. Tanaka, J. Hirosumi, N. Kita and S. Triwahyono, Microporous Mesoporous Mater., 2001, 48, 271–277 CrossRef CAS.
- (a) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS; (b) N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Inorg. Chim. Acta, 2009, 362, 1089–1100 CrossRef CAS; (c) C. Yang, Q. P. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. G. Deng, Green Chem., 2009, 11, 1401–1405 RSC.
- (a) V. V. Thakur and A. Sudalai, Tetrahedron: Asymmetry, 2003, 14, 407–410 CrossRef CAS; (b) M. Ciclosi, C. Dinoi, L. Gonsalvi, M. Peruzzini, E. Manoury and R. Poli, Organometallics, 2008, 27, 2281–2286 CrossRef CAS.
- (a) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (b) J. Drabowicz and M. Mikolajczyk, Synth. Commun., 1981, 11, 1025–1030 CrossRef CAS; (c) W. L. Xu, Y. Z. Li, Q. S. Zhang and H. S. Zhu, Synthesis, 2004, 227–232 CrossRef CAS; (d) K. S. Ravikumar, F. Barbier, J-P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 1999, 95, 123–125 CrossRef CAS; (e) T. Noguchi, Y. Hirai and M. Kirihara, Chem. Commun., 2008, 3040–3042 RSC.
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS; (b) J. Tong, M. Hong, W. Guan, J. B. Li and J. Z. Yang, J. Chem. Thermodyn., 2006, 38, 1416–1421 CrossRef CAS.
- (a) S. L. Jain, S. Singhal and B. Sain, J. Organomet. Chem., 2007, 692, 2930–2935 CrossRef CAS; (b) I. P. Vasile and H. Christopher, Chem. Rev., 2007, 107, 2615–2665 CrossRef; (c) L. Lu, S. F. Cheng, J. B. Gao, G. H. Gao and M. Y. He, Energy Fuels, 2007, 21, 383–384 CrossRef CAS; (d) W. S. Zhu, H. M. Li, X. Jiang, Y. S. Yan, J. D. Lu, L. N. He and J. X. Xia, Green Chem., 2008, 10, 641–646 RSC; (e) Y. Chao, H. Li, W. Zhu, G. Zhu and Y. Yan, Pet. Sci. Technol., 2010, 28, 1242–1249 CrossRef CAS; (f) Y. Mochizuki and K. Sugawara, Energy Fuels, 2008, 22, 3303–3307 CrossRef CAS; (g) S. G. Zhang, Q. L. Zhang and Z. C. Zhang, Ind. Eng. Chem. Res., 2004, 43, 614–622 CrossRef CAS.
- (a) D. Betz, P. Altmann, M. Cokoja, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2011, 255, 1518–1540 CrossRef CAS; (b) A. A. Lindén, M. Johansson, N. Hermanns and J-E. Bäckvall, J. Org. Chem., 2006, 71, 3849–3853 CrossRef.
- D. Betz, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2009, 694, 3320–3324 CrossRef CAS.
- F. E. Kühn, J. Zhao, M. Abrantes, W. Sun, C. A. M. Afonso, L. C. Branco, I. S. Gonçalves, M. Pillinger and C. C. Romão, Tetrahedron Lett., 2005, 46, 47–52 CrossRef.
- D. Betz, A. Raith, M. Cokoja and F. E. Kühn, ChemSusChem, 2010, 3, 559–562 CrossRef CAS.
- I. I. E. Markovits, M. Cokoja, C. J. Münchmeyer, F. E. Kühn, S. Yue, M.-D. Zhou, B. Zhang, S.-L. Zang,W. A. Eger, A. Genest, N. Rösch and J. Mink, in preparation.
- G. W. Gokel, H. M. Gerdes and D. M. Dishong, J. Org. Chem., 1980, 45, 3634–3639 CrossRef CAS.
- J. M. Khurana, A. K. Panda, A. Ray and A. Gogia, Org. Prep. Proced. Int., 1996, 28, 234–237 CrossRef CAS.
- (a) G. Kar, A. K. Saikia, U. Bora, S. K. Dehury and M. K. Chaudhuri, Tetrahedron Lett., 2003, 44, 4503–4505 CrossRef CAS; (b) A. Shaabani, M. B. Teimouri and H. R. Safaei, Synth. Commun., 2000, 30, 265–271 CrossRef CAS; (c) K. Choudhary, D. Suri, S. Kothari and K. K. Banerji, J. Phys. Org. Chem., 2000, 13, 283–292 CrossRef CAS; (d) J. Drabowicz, W. Midura and M. Mikolajczyk, Synthesis, 1979, 39–40 CrossRef CAS.
- K. Surendra, N. S. Krishnaveni, V. P. Kumar, R. Sridhar and K. R. Rao, Tetrahedron Lett., 2005, 46, 4581–4583 CrossRef CAS.
- P. J. Kropp, G. W. Breton, J. D. Fields, J. C. Jung and B. R. Loomis, J. Am. Chem. Soc., 2000, 122, 4280–4285 CrossRef CAS.
- (a) V. Hulea, P. Moreau and F. D. Renzo, J. Mol. Catal. A: Chem., 1996, 111, 325–332 CrossRef CAS; (b) B. M. Choudary, B. Bharathi, C. V. Reddy and M. L. Kantam, J. Chem. Soc., Perkin Trans. 1, 2002, 2069–2074 RSC; (c) A. M. Al-Ajlouni, T. M. Daiafla and M. El-Khateeb, J. Mol. Catal. A: Chem., 2007, 275, 139–147 CrossRef CAS.
- G. Modena and L. Maioli, Gazz. Chim. Ital., 1957, 87, 1306–1316 CAS.
- (a) B. J. Sun, Q. Jin, L. S. Tan, P. Y. Wu and F. Yan, J. Phys. Chem. B, 2008, 112, 14251–14259 CrossRef CAS; (b) A. Mele, C. D. Tran and S. H. de Paoli Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364–4366 CrossRef CAS; (c) D. W. Kim, D. J. Hong, J. W. Seo, H. S. Kim, H. K. Kim, C. E. Song and D. Y. Chi, J. Org. Chem., 2004, 69, 3186–3189 CrossRef CAS; (d) Y. Jeon, J. Sung, D. Kim, C. Seo, H. Cheong, Y. Ouchi, R. Ozawa and H. Hamaguchi, J. Phys. Chem. B, 2008, 112, 923–928 CrossRef CAS; (e) L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC; (f) A. Engdahl, B. Nelander and G. Karlström, J. Phys. Chem. A, 2001, 105, 8393–8398 CrossRef CAS; (g) M. Pettersson, S. Tuominen and M. Räsänen, J. Phys. Chem. A, 1997, 101, 1166–1171 CrossRef CAS; (h) N. E. Heimer, R. E. D. Sesto, Z. Z. Meng, J. S. Wilkes and W. R. Carper, J. Mol. Liq., 2006, 124, 84–95 CrossRef CAS; (i) S. A. Katsyuba, E. E. Zvereva, A. Vikiš and P. J. Dyson, J. Phys. Chem. A, 2007, 111, 352–370 CrossRef CAS.
- M. López-Pastor, M. J. Ayora-Cañada, M. Valcáredl and B. Lendl, J. Phys. Chem. B, 2006, 110, 10896–10902 CrossRef.
- S. Park and R. J. Kazlauskas, J. Org. Chem., 2001, 66, 8395–8401 CrossRef CAS.
- J. Z Yang, X. M. Lu, J. S. Gui, W. G. Xu and H. W. Li, J. Chem. Thermodyn., 2005, 37, 1250–1255 CrossRef.
- V. Singh, S. Kaur, V. Sapehiyia, J. Singh and G. L. Kad, Catal. Commun., 2005, 6, 57–60 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21323k |
| This journal is © The Royal Society of Chemistry 2012 |