X-Ray absorption fine structure spectroscopy studies of thermal effects on ion-exchange equilibria†
Masami
Shibukawa
*a,
Makoto
Harada
b,
Tetsuo
Okada
b,
Yawara
Ogiyama
a,
Tomomi
Shimasaki
a,
Yoshiki
Kondo
a,
Asako
Inoue
a and
Shingo
Saito
a
aGraduate School of Science and Technology, Saitama University, 255 Shimo-Okubo, Sakura-ku, Saitama, 338-8570, Japan. E-mail: sibukawa@apc.saitama-u.ac.jp; Fax: +81-48-858-3520; Tel: +81-48-858-3520
bDepartment of Chemistry, Tokyo Institute of Technology, Meguro-ku, Tokyo, 152-8551, Japan
First published on 30th July 2012
Abstract
The effect of temperature on the first-shell coordination structures for Rb+, Sr2+, Br−, and I− in aqueous solution and those for Rb+ and Sr2+ in a sulfonated styrene-divinylbenzene copolymer cation-exchange resin have been evaluated by X-ray absorption fine structure (XAFS) spectroscopy in order to elucidate the origin of the effect of temperature on ion-exchange equilibria. XAFS measurements on aqueous solutions containing the inorganic ions in the temperature range from ambient temperature to 175 °C at 3 MPa indicate that the number of coordinated first shell water molecules decreases with an increase in temperature, while the first shell ion–oxygen (water) bond length remains almost unchanged (Sr2+ and I−) or slightly decreases (Rb+ and Br−). The spectral changes observed for Rb+ and Sr2+ in the hydrated cation-exchange resin were nearly the same as those for the ions in aqueous solution, though there is an indication for the contact ion pairs with the fixed sulfonate groups at high temperatures. These data represent the direct spectroscopic confirmation of structural change around inorganic ions in an ion-exchange resin as well as in aqueous solutions. The effect of temperature on separation selectivity in ion-exchange processes can be interpreted by the change in hydration of ions clarified in the present study.
1. Introduction
In recent years, it has been shown that increasing temperature causes the retention of some ions to increase, while decreasing the retention of others in ion-exchange chromatography and separation can be improved for particular analyte ions by careful choice of operating temperature.1–8 Consequently, there exists a great potential for substantial selectivity changes upon a change in temperature in ion-exchange processes, although the use of temperature as a way to alter ion-exchange selectivity is not common. However, the origin of thermal effects on the selectivity in ion-exchange separation has not been clarified yet.A number of theoretical and experimental works have been devoted to the study on ion-exchange processes and it has been revealed that the ion-exchange equilibrium is not determined solely by electrostatic forces between the exchangeable ions and the fixed charged groups but also by specific interaction of the ions with the matrix surfaces, which may include hydrophobic attraction, hydrogen bonding, etc.9–16 Since hydrophobic interactions as well as hydrogen bonding are strongly affected by temperature, the effect of temperature on ion-exchange selectivity, especially for the separation of organic ions, may result from a change in the specific interaction of the ions with the exchanger matrix. On the other hand, Coulomb interactions are independent of temperature. However, a change in the hydration structure of the entities that undergo the electrostatic interaction, i.e., hydrated ionic species in the system, may affect the strength of the force. Water molecules around ions are oriented in an ordered or disordered manner depending on the charge, size, and chemical structure of the ions, and the structure of the hydration shell may depend on temperature even for simple inorganic ions such as alkali metal ions and halide ions. Therefore the effect of temperature on ionic hydration structures should be elucidated in order to understand rigorously not only the thermal effects on the retention and separation selectivity of individual analyte ions in ion-exchange chromatography but also the molecular mechanism intrinsic to ion-exchange.
Shibukawa et al. have investigated the effect of temperature on the retention of alkali and alkaline-earth metal ions on a strongly acidic sulfonated ion exchanger over a wide temperature range, 40–175 °C, using superheated water, i.e., liquid water under pressure at temperatures above 100 °C, as the mobile phase and showed that the selectivity coefficient for a pair of alkali metal ions or that of alkaline-earth metal ions approaches unity as temperature increases.17 The retention of an analyte ion decreases with an increase in temperature when the eluent counterion in the mobile phase has stronger affinity than that of the analyte ion for the ion-exchanger, whereas it increases when eluted by a weaker counterion. Shibukawa et al. also demonstrated in a recent paper that temperature exerts a similar effect on anion-exchange selectivity.18 These results indicate that the charge on an ion becomes a predominant factor for determining the retention in ion-exchange chromatography at elevated temperatures. We presumed that the dependence of ion-exchange selectivity observed for simple inorganic ions on temperature can be attributed to the change in hydration of the ions in the solution phase and/or in the ion-exchange resin with temperature; hydration of ions can be assumed to become weaker as the temperature increases due to disruption of ion-dipole bonding by thermal motion of water molecules resulting in a reduction of the difference in hydration structure between different ions.
Among various experimental techniques, X-ray absorption and neutron diffraction spectroscopy enable us to give direct structural information of ion hydration.19–31 Particularly X-ray absorption fine structure (XAFS) is very useful for a precise measurement of the short-range structure of the hydration shells. Harada and Okada studied the local hydration structures of inorganic ions in some ion-exchange resins by XAFS and discussed the relation between the hydration structures of ions and ion-exchange selectivity.19–22 On the other hand, XAFS has also been used to explore the hydration of several inorganic ions in supercritical water or superheated water.23–27 Fulton et al. measured the degree of hydration of Sr2+ and Rb+ in ambient and supercritical water and reported the first observation of a significant reduction in hydration number around these ions under supercritical conditions.23–25 In subsequent papers, they investigated the structures of the hydration shells of Br− and Ca2+ ion in a temperature range from 25 to over 400 °C.26–28 A distinct dehydration was observed to occur even at 200 °C in superheated water. These observations suggest that the difference in hydration structures of ions may become smaller at elevated temperatures, leading to smaller differences in the affinity of ions having an identical charge to the ion-exchanger. This change in hydration of ions may explain the results we obtained by superheated water ion-exchange chromatography.
However, the data on ion hydration at elevated temperatures are scarce especially in the superheated or subcritical water temperature range. In addition, the effect of temperature on hydration structures of ions in ion-exchange resins has not been reported yet. In this paper we use XAFS to investigate the hydration of some alkali and alkaline-earth metal ions and halide ions not only in solution but also in ion-exchange resins in the temperature range from ambient temperature to 175 °C. This represents the most extensive examination of ion hydration in superheated water, exploring in detail the effects of temperature on the hydration structures of ions and the ion-exchange selectivity.
2. Experimental
2.1 Chemicals
All chemicals used in this study were obtained from commercial sources and were of reagent-grade unless otherwise stated. Water was purified subsequently with an ion-exchange cartridge PF-III H10 (Organo, Tokyo, Japan) and an Arium 611 DI (Sartorius, Tokyo, Japan). The ion-exchange resin used was DIAION MCI GEL CK10S (sulfonated polystyrene-divinylbenzene copolymer, particle diameter = 11 μm, Mitsubishi Chemical, Tokyo Japan). The sodium form of MCI GEL CK10S, a commercially available form, was converted to Sr2+ and Rb+ forms by treating it with 0.1 mol L−1 Sr(NO3)2 and RbNO3 aqueous solutions, respectively, and then washing them with pure water. The resins were dried in vacuum and were sealed in polyethylene pouches. The ion-exchange capacity of MCI GEL CK10S is 2.3 meq mL−1.2.2 XAFS experiments
A high temperature and high-pressure XAFS cell designed in our laboratory was composed of a block of stainless steel (SUS304) fitted with two opposing 5 mm diameter × 0.9 mm thick beryllium windows for transmission of X-ray beam. The structure of the cell is shown in Fig. S1 in the ESI.† These X-ray windows were mounted in such a way as to provide a path length (5–10 mm) suitable for a target element. The maximum operating pressure of this design is 10 MPa. Aqueous solutions of 0.1 mol L−1 Sr(NO3)2 and KBr, 0.05 mol L−1 Rb2SO4 and 0.5 mol L−1 KI were delivered into the cell with a JASCO (Tokyo, Japan) Model PU-2080 Plus HPLC pump, whereas water slurries of Sr2+ and Rb+ forms of MCI GEL CK10S were poured directly into the cell. To the KI solution, Na2SO3 was added at 0.05 mol L−1 in order to suppress oxidation of iodide ions.18 Then the pressure was applied at 3 MPa employing the HPLC pump and a JASCO Model SCF-Bpg/M back pressure regulator. The temperature of the cell was maintained to within ±2 °C using a PID temperature controller, CB103 (RKC Instruments, Tokyo, Japan).Strontium-K edge, bromine-K edge, and rubidium-K edge XAFS spectra were collected on beam-line BL-9C or BL-12C of the Photon Factory of High Energy Accelerator Research Organization (KEK) in Tsukuba, Japan. The XAFS measurements at iodine-K edge were carried out at the beam-line AR-NW10 of the Photon Factory of KEK. Typically, experiments were started at ambient temperature and subsequent spectra were collected at higher temperatures. The scattering amplitudes and phase shifts for the model systems were calculated with the FEFF8.20 program. The XAFS data were analyzed according to literature.32
3. Results and discussion
3.1 Effect of temperature on hydration structures of inorganic ions in solution
Fig. 1–4 show the k3-weighted XAFS oscillations, k3χ(k), acquired over temperatures for Rb+, Sr2+, Br−, and I− in aqueous solutions, respectively, where k is the wavenumber of the photoelectron ejected in the absorption process. As shown in these figures, increasing the temperature from ambient to the superheated region produces a definite change in the hydration structure. Particularly, the decrease in the amplitude of the oscillations shows a decrease in the hydration number of the first water solvation shell. On the other hand, locations of peaks are almost identically independent of temperature except for Rb+ and Br−, suggesting that the nearest neighbor distances approximately remain unchanged within the temperature range examined in this study for Sr2+ and I−. Coordinating atoms should be hydrogen for Br− and I−. However, the contributions of hydrogen atoms to overall XAFS oscillations can be neglected. Therefore X-ray scattering occurs at oxygen atoms in all the cases shown in Fig. 1–4.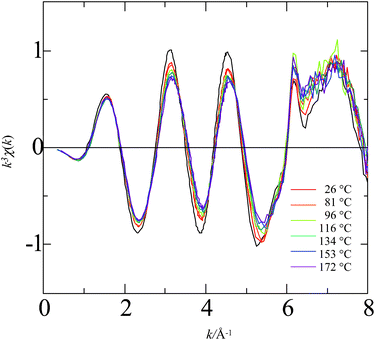 | ||
| Fig. 1 Plot of k3χ(k) for rubidium ion as a function of wavevector for a series of temperatures from 26 to 172 °C. Sample solution: 0.05 mol L−1 Rb2SO4. | ||
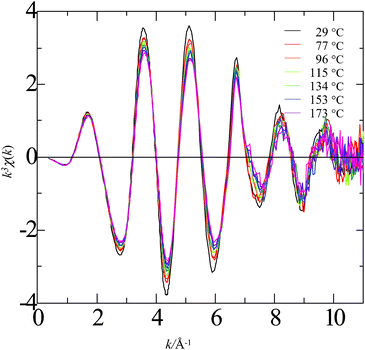 | ||
| Fig. 2 Plot of k3χ(k) for strontium ion as a function of wavevector for a series of temperatures from 29 to 173 °C. Sample solution: 0.1 mol L−1 Sr(NO3)2. | ||
 | ||
| Fig. 3 Plot of k3χ(k) for bromide ion as a function of wavevector for a series of temperatures from 29 to 137 °C. Sample solution: 0.1 mol L−1 KBr. | ||
 | ||
| Fig. 4 Plot of k3χ(k) for iodide ion as a function of wavevector for a series of temperatures from 27 to 160 °C. Sample solution: 0.5 mol L−1 KI + 0.05 mol L−1 Na2SO3. | ||
The spectra were thus analyzed in the usual way by assuming A+⋯O and A−⋯H–O as the scattering paths for the target cations (A+) and anions (A−), respectively. The number of scattering atoms, N, was determined by assuming N = 4 for Rb+, N = 8 for Sr2+, and N = 6 for Br− and I− at ambient temperature based on the results obtained by various studies.22,24,31,32 A typical example of the quality of the fits is given in Fig. 5 showing experimental and model fits for Sr2+ under ambient conditions. The range of curve-fitting was severely restricted for Rb+ by the multielectron excitation at k = ca. 6.1 Å−1,34 and thus the XAFS spectra were not analyzed past this k value for this ion.
 | ||
| Fig. 5 Comparison of k3χ(k) spectrum for strontium ions in aqueous solution at 29 °C with the corresponding k3χ(k) spectrum calculated by FEFF. The black curve represents experimental spectrum. The red curve shows the result of the calculation. | ||
Fig. 6 shows temperature dependence of the number of scattering atoms or the coordination number, the distance of the nearest-neighbors, r, and Debye–Waller coefficient, σ, for each ion on temperature (numerical values of XAFS parameters obtained by curve-fittings are listed in Table S1 in the ESI†). As expected from the XAFS spectra shown in Fig. 1–4, the N value decreases with an increase in temperature, indicating that all the ions studied experience dehydration in their first coordination shell at elevated temperatures. On the other hand, the distance of the nearest-neighbors for Sr2+ and I− remains almost unchanged, while Rb+ and Br− exhibit slight decreases in the distance as temperature increases. Debye–Waller coefficient exhibits a trend toward slightly increasing values at higher temperatures, which may be due to both static and thermal disorder.
 | ||
| Fig. 6 Dependence of XAFS structural parameters for Rb+, Sr2+, Br−, and I− in their aqueous solutions on temperature. Symbols: (●) Rb+, (▲) Sr2+, (□) Br−, (◇) I−. | ||
These observations are similar to the results obtained for the hydration of some inorganic ions in supercritical or subcritical water solutions.23–28 Seward et al. reported that there is a decrease in the Sr2+⋯O distance of ca. 0.05 Å with increasing temperature over the temperature range from 25 to 300 °C, which is associated with a decrease in the number of coordinated oxygens.31 Fulton et al. also observed the decrease in the Rb+⋯O distance in supercritical water.25 The results shown in Fig. 6 also indicate a decrease in the Rb+⋯O bond length with an increase in temperature. However, as shown in Fig. 6, any clear evidence of the contraction of the Sr2+⋯O distance was not obtained in this study. The hydration enthalpy for Sr2+ (−1445 kJ mol−1) is much larger than that for Rb+ (−296 kJ mol−1),35 which means that the Sr2+⋯O bond is much stronger than the Rb+⋯O bond. We have also shown that the selectivity coefficient for a pair of alkaline-earth metal ions in cation-exchange chromatography is much smaller than that for a pair of alkali metal ions.17 All these data and observations suggest that the difference in dependence of the bond length on temperature between Rb+ and Sr2+ reflects the difference in strength of hydration for these ions.
The contraction of the Br−⋯O distance by a rise of temperature indicates that one can assume the dehydration of Br− to proceed in a similar manner to that for Rb+. This assumption is supported by the facts that the hydration enthalpy for Br− (−336 kJ mol−1)35 is approximately the same as that of Rb+ and the temperature dependence of ion-exchange selectivity for a pair of halide ions and some other simple monovalent inorganic ions is larger compared to that for a pair of alkaline-earth metal ions.18 On the other hand, the distance of the nearest-neighbor water molecules for I− is nearly always independent of temperature in spite of its low hydration strength. This may be attributed to large ionic size of I− and weak interaction of the ion with water molecules. Tanida et al. investigated solvation structures of Br− and I− in various solvents by XAFS and demonstrated that the r value for I− is less dependent on the nature of the solvent compared with that for Br−.36,37 For example, the difference in distance between A−⋯O in water and A−⋯C in acetone for I− is 0.21 Å,37 while that for Br− is 0.38 Å.36 The I−⋯O distance may be determined solely by the ionic radius of I− and is not affected by small changes in the strength of the bond between the ion and a water molecule.
The decrease in the coordination number is consistent with a reduction in the number of ion–water bonds at elevated temperatures, which can be attributed to dehydration by thermal motion of water molecules around the ions or the change in coordination structure. This clearly indicates that disruption of the hydration structure of an ion occurs even in liquid water at temperatures lower than 100 °C. For concentrated solutions of some salts such as CaCl2 and SrCl2, the formation of contact ion-pairs was suggested to be another cause of the reduction of the coordination number,28,31 though it has been failed in some cases to obtain direct evidence of the ion-pairs from the XAFS experiments.26 We could not see any indication of the contact ion-pairs in the XAFS spectra obtained in this study. Seward et al. reported that the XAFS data for 1 mol kg−1 SrCl2 in 3 mol kg−1 HCl indicate the formation of strontium chloride ion pairs, while those for 0.1 mol kg−1 SrCl2 solution do not show any detectable ion paring.31 This suggests that the decrease in the coordination number of ions in aqueous solution observed in this study in the temperature range of 30–175 °C can be attributed to dehydration of ions by thermal effects.
3.2 Effect of temperature on hydration structures of inorganic ions in ion-exchange resins
Fig. 7 and 8 show the XAFS spectra obtained for Rb+ and Sr2+ present as the counterions of fixed sulfonate groups in the water-swollen cation-exchange resin as well as in the dried resin. We also tried to measure the XAFS spectra for some anion-exchange resin samples. However, reliable data could not be obtained at elevated temperatures because excess noise was produced even at 80 °C, which is probably due to partial decomposition of the resin.18,33 | ||
| Fig. 7 Plot of k3χ(k) for rubidium ion in the hydrated and dried cation-exchange resins as a function of wavevector for a series of temperatures from 28 to 176 °C. | ||
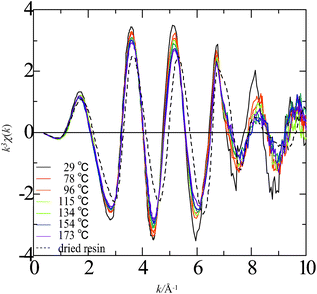 | ||
| Fig. 8 Plot of k3χ(k) for strontium ion in the hydrated and dried cation-exchange resins as a function of wavevector for a series of temperatures from 29 to 173 °C. | ||
In the resin soaked in water, the fixed sulfonate groups and counterions are hydrated. The spectral transitions observed for Rb+ and Sr2+ in the hydrated resin are similar to those for the ions in aqueous solution, suggesting that these cations have nearly the same structures of their first hydration shells in the interior of the cation-exchange resin as those in aqueous solution. Fig. 9 shows the effect of temperature on the N, r, and σ values for Rb+ and Sr2+ in the hydrated resin (full data are given in Table S2 in the ESI†). Similarly to the changes observed for aqueous solution samples, the coordination number decreases with an increase in temperature. The A+⋯O distance for Rb+ slightly decreases as temperature rises in a similar manner to that for the ion in a solution sample, whereas the distance for Sr2+ remains unchanged. These results suggest that the hydration structures of Rb+ and Sr2+ in the cation-exchange resin soaked in water can be assumed as approximately identical to those in aqueous solution even at elevated temperatures and thermal dehydration occurs not only in solution but also in the interior of the resin.
 | ||
| Fig. 9 Dependence of XAFS structural parameters for Rb+ and Sr2+ in water swollen MCI GEL CK10S resins on temperature. Symbols: (●) Rb+, (▲) Sr2+. | ||
If the counterions form contact ion-pairs with the sulfonate groups, the direct coordinating atom is the same as that of water, i.e., oxygen, and the A+⋯O distance for the contact is almost the same as that for the ion and water molecule. Consequently XAFS parameters do not provide clear information on the local structure of the countercations in the hydrated resin. However, there are slight phase shifts in oscillation in the high k region for both Rb+ and Sr2+ in the cation-exchange resin at elevated temperatures as shown in Fig. 7 and 8, which suggests a superposition of the spectra due to different coordination structures. The qualitative differences between water-swollen and dry conditions are seen in these figures; the phase of the oscillation for Rb+ and Sr2+ in the cation-exchange resin shifts to a higher k value when the resin is dried.
The estimated N values for the dry resin samples are 3.0 and 5.2 for Rb+ and Sr2+, respectively (full data of the XAFS parameters obtained for the dry resin samples are represented in Table S2 in the ESI†). If a cation is bound to one sulfonate group, the coordination number can be assumed to be four from the consideration of the contribution of sulfur atom to the oscillation amplitude.22 Although the N value is slightly smaller than 4, Rb+ can be considered to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 direct contact ion pair with sulfonate groups in the dried resin. On the other hand, Sr2+ may partly bind to two sulfonate groups.
:1 direct contact ion pair with sulfonate groups in the dried resin. On the other hand, Sr2+ may partly bind to two sulfonate groups.
The slight variation in oscillation of the XAFS spectra with temperature observed for the water-swollen cation-exchange resin samples implies that small amounts of Rb+ and Sr2+ may directly bind to the sulfonate groups in the resin at elevated temperatures. Since the ion-exchange capacity of the cation-exchange resin is 2.3 meq mL−1, the concentrations of these ions in the resin can be assumed to be more than 2 and 1 mol L−1 for Rb+ and Sr2+, respectively. In these concentrated solutions, the possibility of contact ion-pair formation could not be ruled out at elevated temperatures. Fig. 6 and 9 show that the change in the coordination number of Rb+ and Sr2+ with an increase in temperature for the resin samples is smaller than that observed for the corresponding aqueous solution samples. This can be ascribed to strong binding of Rb+ and Sr2+ to sulfonate ions in the resin phase by Coulomb force. The observed phase shifts in the XAFS spectra are too small to accurately evaluate the contribution from the contact ion-pairs. It is thus presumed that most of the ions may bind to the fixed ions with bridging water molecules under the conditions examined in this study. However, the results shown above suggest that the structure of the hydrated ion-exchange resin approximates to that of the dried one as temperature increases. The dehydration reaction of the ion-exchange resin may be schematically expressed as follows:
| –SO3−⋯H2O⋯An+ ⇆ –SO3−⋯An+ + H2O |
4. Conclusions
XAFS measurements have been carried out to establish the changes in the hydration of inorganic ions in an ion-exchange resin as well as in solution at elevated temperatures. The results obtained in this study demonstrate that there is a significant decrease in the number of coordinated water molecules in the first hydration shells of Rb+, Sr2+, Br−, and I− in aqueous solutions as temperature increases. It was also shown that the hydration numbers of Rb+ and Sr2+ present as counterions of fixed sulfonate groups in a water-swollen cation-exchange resin decrease with an increase in temperature. The data for 0.1 mol L−1 solutions of Sr(NO3)2 and KBr, 0.05 mol L−1 Rb2SO4 and 0.5 mol L−1 KI solutions provide no evidence for the formation of contact ion pairs, while evidence was observed for slight amounts of Rb+ and Sr2+ ions forming the ion pairs with fixed sulfonate groups at high temperatures in the cation-exchange resin, of which the ion-exchange capacity is 2.3 meq mL−1. However, it seems unlikely that the formation of contact ion pairs is primarily responsible for the dehydration of the inorganic ions. Instead, our results suggest that structural change of the hydration of the inorganic ions in the ion-exchange resin as well as in aqueous solutions are caused by thermal motion of water molecules at elevated temperatures. As we predicted in our previous studies on the effect of temperature on the separation selectivity in ion-exchange chromatography,17,18 the local structure around an ion can undergo a profound change from a fully hydrated species under ambient conditions to a species with partially disrupted hydration structure at high temperature where the hydrogen-bonded water network has broken down.The separation selectivity for a pair of alkali metal ions, alkaline-earth metal ions, and halide ions in ion-exchange processes decreases with increasing temperature as we have already demonstrated in previous papers.17,18 At elevated temperatures, electrostatic charge screening no longer effectively occurs, which may lead to smaller ion-exchange separation selectivity for different ions having an identical charge.
Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research No. 20350034 from Ministry of Education, Culture, Sports, Science and Technology, Japan and was performed under the approval of the Photon Factory Advisory Committee (Proposal Nos. 2008G092 and 2010G104)References
- N. E. Fortier and J. S. Fritz, Talanta, 1987, 34, 415–418 CrossRef CAS.
- P. K. Bokx and H. M. J. Boots, J. Phys. Chem., 1989, 93, 8243–8248 CrossRef.
- R. G. Smith, P. P. Drake and J. D. Lamb, J. Chromatogr., A, 1991, 546, 139–149 CrossRef CAS.
- M. G. Kolpachnikova, N. A. Penner and P. N. Nesterenko, J. Chromatogr., A, 1998, 826, 15–23 CrossRef CAS.
- P. Hatsis and C. A. Lucy, Analyst, 2001, 126, 2113–2118 RSC.
- P. Hatsis and C. A. Lucy, J. Chromatogr., A, 2001, 920, 3–11 CrossRef CAS.
- W. Bashir, E. Tyrrell, O. Feeney and B. Paull, J. Chromatogr., A, 2002, 964, 113–122 CrossRef CAS.
- H. Yu and R. Li, Chromatographia, 2008, 68, 611–616 CAS.
- R. W. Grimshaw and C. E. Harland, Ion-Exchange: Introduction to Theory and Practice, The Chemical Society, London, 1975 Search PubMed.
- S. Afrashtehfar and F. F. Cantwell, Anal. Chem., 1982, 54, 2422–2427 CrossRef CAS.
- R. A. Hux and F. F. Cantwell, Anal. Chem., 1984, 56, 1258–1263 CrossRef CAS.
- H. Liu and F. F. Cantwell, Anal. Chem., 1991, 63, 993–1000 CrossRef CAS.
- H. Liu and F. F. Cantwell, Anal. Chem., 1991, 63, 2032–2037 CrossRef CAS.
- G. Stahlberg, Anal. Chem., 1994, 66, 440–449 CrossRef.
- T. Okada, Anal. Chem., 1998, 70, 1692–1700 CrossRef CAS.
- J. S. Fritz, J. Chromatogr., A, 2005, 1085, 8–17 CrossRef CAS.
- M. Shibukawa, T. Shimasaki, S. Saito and T. Yarita, Anal. Chem., 2009, 81, 8025–8032 CrossRef CAS.
- M. Shibukawa, A. Taguchi, Y. Suzuki, K. Saitoh, T. Hiaki and T. Yarita, Analyst, 2012, 137, 3154–3159 RSC.
- M. Harada and T. Okada, J. Phys. Chem. B, 2002, 106, 34–40 CrossRef CAS.
- T. Okada and M. Harada, Anal. Chem., 2004, 76, 4564–4571 CrossRef CAS.
- M. Harada and T. Okada, J. Chromatogr., A, 2005, 1085, 3–7 CrossRef CAS.
- M. Harada and T. Okada, Chem. Commun., 2008, 5182–5184 RSC.
- D. M. Pfund, J. G. Darab, J. L. Fulton and Y. Ma, J. Phys. Chem., 1994, 98, 13102–13107 CrossRef CAS.
- B. J. Palmer, D. M. Pfund and J. L. Fulton, J. Phys. Chem., 1996, 100, 13393–13398 CrossRef CAS.
- J. L. Fulton, D. M. Pfund, S. L. Wallen, M. Newville, E. A. Stern and Y. Ma, J. Chem. Phys., 1996, 105, 2161–2166 CrossRef CAS.
- S. L. Wallen, B. J. Palmer, D. M. Pfund, J. L. Fulton, M. Newville, Y. Ma and E. A. Stern, J. Phys. Chem. A, 1997, 101, 9632–9640 CrossRef CAS.
- J. L. Fulton, S. M. Heald, Y. S. Badyal and J. M. Simonson, J. Phys. Chem. A, 2003, 107, 4688–4696 CrossRef CAS.
- J. L. Fulton, Y. Chen, S. M. Heald and M. Balasubramanian, J. Chem. Phys., 2006, 125, 094507 CrossRef.
- T. Yamaguchi and A. K. Soper, J. Chem. Phys., 1999, 110, 3529–3535 CrossRef CAS.
- S. Ansell and G. W. Neilson, J. Chem. Phys., 2000, 112, 3942–3944 CrossRef CAS.
- T. M. Seward, C. M. B. Henderson, J. M. Charnock and T. Driesner, Geochim. Cosmochim. Acta, 1999, 63, 2409–2418 CrossRef CAS.
- B. K. Teo, EXAFS: Basic Principles and Data Analysis, Springer-Verlag, Berlin, 1985 Search PubMed.
- H. F. Walton, Principles of Ion-Exchange, in Chromatography, ed. E. Heftmann,Van Nostrand, New York, 3rd edn, 1975 Search PubMed.
- A. Kodre, I. Arcon, J. P. Gomilsek, R. Preseren and R. Frahm, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 3497–3514 CrossRef CAS.
- J. Burgess, Ions in Solution, Basic Principles of Chemical Interactions, Horwood Publishing, Chichester, 1999 Search PubMed.
- H. Tanida, H. Sakane and I. Watanabe, J. Chem. Soc., Dalton Trans., 1994, 2321–2326 RSC.
- H. Tanida and I. Watanabe, Bull. Chem. Soc. Jpn., 2000, 73, 2747–2752 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available]. See DOI: 10.1039/c2ra21278a/ |
| This journal is © The Royal Society of Chemistry 2012 |