A theoretical study on the stereoconvergency of the intramolecular radical cation [2+2] cycloadditions of bis(styrenes)
Chenchen
Guo
,
Lijie
Cui
,
Bozhen
Chen
*,
Jianhua
Yuan
and
Zhiyuan
Tian
School of Chemistry and Chemical Engineering, Graduate University of Chinese Academy of Sciences, Yuquan Road 19A, Beijing, 100049, P. R. China. E-mail: bozhenchen@hotmail.com; Fax: +86 10 88256092; Tel: +86 10 88256129
First published on 23rd August 2012
Abstract
The intramolecular radical cation [2+2] cycloadditions of a series of bis(styrenes) have been explored by DFT (U)B3LYP method in conjunction with the 6-31G(d,p) and 6-311G(d,p) basis sets. According to our calculations, the pathway of the cycloaddition is stepwise via the formation of a 5-membered ring intermediate. The final cyclobutane products are formed by electron transfer between the long-bond radical cation product and the neutral reactant. The understanding of the mechanism gives a new insight into the stereoconvergency of the cycloaddition.
Introduction
Recently, there has been considerable interest in producing cyclobutane, which is the remarkable structural feature of many bioactive natural products,1 by [2+2] cycloaddition of olefins. Although the [2+2] cycloaddition is symmetry-forbidden by the Woodward–Hoffmann rule, the corresponding radical cation/anion reaction easily occurs under moderate experimental conditions. Since the first description of the radical cation cycloadditions of electron-rich olefins upon one-electron oxidation by Ledwith2 in 1969, the cycloadditions of styrene derivatives, with the characters of high cycloaddition rates and low activation barriers, have been well investigated by many researchers.3–20 Besides, in the past decade, research workers have investigated the inter- or intramolecular radical anion cycloaddition of various enones using photocatalysis,21 electrocatalysis22 and chemically induced23 methods.By now, the mechanism of the intermolecular radical cation cycloaddition of styrene derivatives has been investigated by both experimental5–8,11,20,24 and computational25 methods. The pathway of the cycloaddition was predicted to be stepwise with the formation of an acyclic radical cation intermediate5 or concerted to give a long-bond cyclobutane in one step.6,7 In 2008, Marquez et al.19 detected the existence of the distonic radical cation intermediate by extractive electrospray ionization spectrometry, giving a strong evidence for the stepwise mechanism.
It has been found that the radical cation cycloaddition shows excellent stereoselectivity. The trans cyclobutane was observed to be the major product of the intermolecular cycloaddition of the E-styrene experimentally.24 Based on DFT B3LYP calculations,25 the stereoselectivity was predicted to be caused by the greater thermodynamic stability of the trans isomer compared to the cis one. However, according to experimental studies26 on the intramolecular cycloaddition of bis(styrene) catalyzed by Ru(bpy)3Cl2 upon irradiation with visible light, the cis cyclobutane P 1 was the major product of (E,E) bis(styrene) R 1 (see the definition in Scheme 1), and it was also the major product of (E,Z) bis(styrenes) R 2 and R 3 (see Scheme 1) with the trans cyclobutanes P 2 and P 3 as minor products. The authors predicted that R 3 underwent isomerization to R 1 at a relatively fast rate compared to that of the cycloaddition from their GC results (see Supporting Information in ref. 26). They therefore concluded that the origin of the stereoconvergency was the isomerization from R 3 to R 1, and the [2+2] cycloaddition step was itself stereospecific. However, the GC results24 of the cycloaddition of R 1 upon irradiation with ultraviolet light showed that the rate of olefin isomerization from R 1 to R 3 was slower than that of the cycloaddition. According to the previous investigation,25,27 the overall energy barriers of the radical cation/anion cycloadditions were low (no more than 20 kcal mol−1), while the activation energies of the thermal isomerizations of olefins via rotation around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds are very high. Therefore, the rate of the thermal isomerization from R 3 to R 1 should be slower than that of the cycloaddition. Generally, the photoisomerizations of olefins are afforded by the light falling in the ultraviolet region. Given these precedents above, it is necessary to find an alternative pathway of the isomerization and the origin of the stereoconvergency.
C double bonds are very high. Therefore, the rate of the thermal isomerization from R 3 to R 1 should be slower than that of the cycloaddition. Generally, the photoisomerizations of olefins are afforded by the light falling in the ultraviolet region. Given these precedents above, it is necessary to find an alternative pathway of the isomerization and the origin of the stereoconvergency.
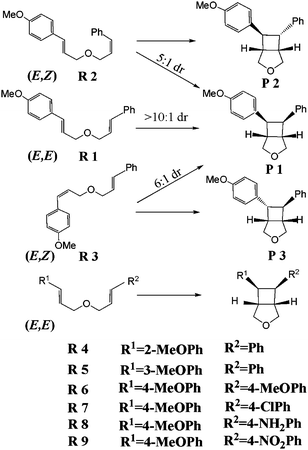 | ||
| Scheme 1 The definitions of the nine kinds of bis(styrenes) and their cyclobutane products. | ||
In this paper, we investigate the cycloaddition pathways of nine bis(styrene) derivatives (see Scheme 1) to find the origin of the stereoconvergency and to illuminate the effects of substituents and solvents on the reactions.
Computational methods
All calculations were performed using the Gaussian 09 Programs. The geometries in the gas-phase were fully optimized with the DFT (U)B3LYP (Becke, 3-parameter, Lee–Yang–Parr) method.28 Each structure of the stationary points along the reaction paths was classified as a minimum (no imaginary frequencies) or a transition-state structure (one imaginary frequency) by frequency analysis calculations at the B3LYP/6-31G(d,p) level. The energies reported in this paper were obtained by performing the B3LYP/6-311G(d,p) single-point energy calculations at the B3LYP/6-31G(d,p) geometries. Additionally, all energies were corrected with zero-point energies (ZPEs), which were evaluated under assumption of harmonic oscillator at the B3LYP/6-31G(d,p) level. Gibbs free energies (including the ZEPs) were calculated at the B3LYP/6-311G(d,p)//B3LYP/6-31G(d,p) level at 25 °C. The intrinsic reaction coordinate (IRC) calculations were also performed at the same level in order to check whether the transition states connect the reactants, intermediates, or products.The effect of CH3CN solvent on the reaction enthalpies was explored by the B3LYP/6-311G(d,p) single-point energy calculations at the optimized geometries from the B3LYP/6-31G(d,p) calculations in gas-phase using the polarized continuum model (PCM)29 in the Self-Consistent Reaction Field (SCRF) theory. It is known that PCM performs better than CPCM for polar solvents.
The <S2> values for all the reactants, transition states, intermediates, and products along the reaction pathways at the (U)B3LYP/6-31G(d,p) and (U)B3LYP/6-311G(d,p) levels are less than 0.78.
Results and discussion
To observe the alternative explanation of the stereoconvergency, we firstly investigate the mechanism of the [2+2] cycloaddition of bis(styrene) isomers. The possible mechanisms of this reaction are shown in Scheme 2. The first step is the formation of the radical cation R·+via one-electron oxidation. Then R·+ proceeds via a stepwise or a concerted mechanism, giving a cyclobutane radical cation P·+. Subsequently, P·+ is reduced by the neutral reactant R, producing R·+ and the neutral cyclobutane product P. These processes mentioned above form the complete electron transfer catalytic cycle. Considering that the electron transfer process is independent from the cycloaddition process, we investigated these two processes respectively.![The possible mechanism of intramolecular [2+2] cycloaddition reactions of bis(styrenes).](/image/article/2012/RA/c2ra21236f/c2ra21236f-s2.gif) | ||
| Scheme 2 The possible mechanism of intramolecular [2+2] cycloaddition reactions of bis(styrenes). | ||
Investigation of the electron transfer reaction
For the electron transfer catalysis [2+2] cycloaddition, the electron transfer is an indispensable process, not only for affording the radical cation reactant but also promoting the formation of cyclobutane. In order to confirm our prediction of the electron transfer catalytic cycle in Scheme 2, we calculated the adiabatic ionization potentials (AIPs) of all the reactants and products. The possibility of the electron transfer between the radical cation product and the corresponding neutral reactant was determined based on the law that the greater the AIP, the more difficult to remove an electron. The adiabatic ionization potentials are evaluated here according to the following definition:| AIP = |
| E |
| (optimized cation) − |
| E |
| (optimized neutral) |
Table 1 shows the AIPs of the reactants and cyclobutane products. The AIP values were obtained by performing the B3LYP/6-311G(d,p)//B3LYP/6-31G(d,p) calculations in the gas phase and CH3CN solvent. The calculated AIPs for each species in the gas phase are all larger than the corresponding values in CH3CN solvent, indicating that the radical cations could be stabilized in CH3CN solvent. Besides, the AIPs of the reactants are all smaller than those of their corresponding products in both the gas phase and CH3CN solvent. Therefore, the electron transfer from the neutral reactant to the radical cation product, which forms the electron transfer catalytic cycle, is energetically feasible. Considering that the AIP of the initiator Ru(bpy)32+ used in the oxidative visible light photocatalysis cycloaddition is about 132.6 kcal mol−1,30 which is lower than that of almost all cyclobutane products, the electron transfer to the radical cation products from Ru(bpy)32+ is also possible. However, this electron transfer is less feasible thermodynamically, since the AIP of Ru(bpy)32+ is higher than that of the reactants. Additionally, as the concentration of the initiator is much lower than the neutral reactant, it is reasonable to conclude that the electron transfer to the radical cation product from the neutral reactant is dominating. It is worth mentioning that the impossibility of the reverse electron transfer plays an important role in producing the neutral product (see below).
| Reactants | AIP | AIP-CH3CN | Products | AIP | AIP-CH3CN |
|---|---|---|---|---|---|
| R 1 | 158.9 | 126.7 | P 1 | 166.8 | 133.3 |
| R 2 | 160.4 | 129.1 | P 2 | 168.9 | 134.6 |
| R 3 | 160.3 | 128.7 | P 3 | 169.8 | 135.4 |
| R 4 | 160.4 | 129.5 | P 4 | 166.7 | 133.8 |
| R 5 | 164.2 | 133.1 | P 5 | 173.4 | 139.1 |
| R 6 | 153.0 | 124.5 | P 6 | 160.4 | 129.8 |
| R 7 | 160.5 | 127.7 | P 7 | 168.9 | 134.4 |
| R 8 | 147.8 | 117.7 | P 8 | 154.6 | 121.3 |
| R 9 | 165.1 | 128.7 | P 9 | 174.9 | 135.8 |
The initiator Ru(bpy)32+ is sufficient to oxidize the reactants listed in Table 1 except R 5. This fact may explain why R 5 couldn't undergo the cycloaddition, producing the cyclobutane, but other reactants could, under the experimental conditions.26
The mechanism of the radical cation [2+2] cycloaddition reaction of R 1·+
The [2+2] cycloaddition process from R·+ to P·+, involving the break of the C1C2 and C3C4 π bonds and the formation of the C1–C4 and C2–C3 σ bonds (see the definitions in Fig. 1), could proceed via two possible pathways. Take the cycloaddition of R 1·+ for instance, as with the previous investigation of the intramolecular cycloaddition of the bis(enone)27 in our laboratory, we chose the conformation with the shorter C2–C3 distance and lower energy as the structure of R 1·+. According to the earlier studies on radical cation/anion cycloaddition,25,27 we are more inclined to support the stepwise mechanism. To investigate the possibility of a stepwise mechanism, an intermediate structure with a 5- or 7-membered ring was first located by taking the distance between C2 and C3 or the distance between C1 and C4 as the reaction coordinate. The calculations show the formation of the intermediate with a 5-membered ring. However, we didn't get the structure with a 7-membered ring since the energy kept increasing rapidly with C1 gradually approaching to C4. This phenomenon indicates that the pathway via a 7-membered ring intermediate is unfavorable energetically. Therefore, we conclude that the mechanism of cycloaddition of R 1·+ is stepwise with the initial formation of a 5-membered ring intermediate.
![The key geometry changes in the intramolecular [2+2] cycloaddition of R 1·+ predicted by the B3LYP/6-31G(d,p) method. Bond lengths are given in angstroms.](/image/article/2012/RA/c2ra21236f/c2ra21236f-f1.gif) | ||
| Fig. 1 The key geometry changes in the intramolecular [2+2] cycloaddition of R 1·+ predicted by the B3LYP/6-31G(d,p) method. Bond lengths are given in angstroms. | ||
The major reaction pathway of R 1·+ and the geometry changes along the reaction pathway predicted by our calculations are shown in Fig. 1. In radical cation reactant R 1·+, the C2–C3 and C1–C4 distances are predicted to be 2.859 and 3.751 Å.; the C1–C2 and C3–C4 bonds are 1.369 and 1.359 Å, respectively. The moderately longer C1–C2 and C3–C4 bonds than those in the neutral reactant R 1 (1.342 and 1.341 Å) probably is caused by the decrease of the electron on the π bonding orbitals. In the first transition state TS1 1, the C2–C3 distance shortens more rapidly than the C1–C4 distance, which indicates the priority to form the C2–C3 bond in the cycloaddition reaction, giving a 5-membered ring intermediate IM 1. Just as we expected, in IM 1, the C2–C3 distance is predicted to be 1.568 Å, showing the formation of a single bond, whereas the C1–C4 distance to be 2.466 Å. The C1–C4 distance shortens to 1.885 Å in the second transition state TS2 1. However, this distance in the radical cation product P 1·+ is predicted to be 1.810 Å, only 0.075 Å shorter than that of TS2 1. The formation of the long-bond cyclobutane P 1·+ is probably attributed to the electronic effect on the cyclobutane.
Fig. 2 shows the schematic diagram of the potential energy curve (PEC) along the predicted reaction pathway of the cycloaddition of R 1·+. Along the reaction pathway, there is a moderate barrier of 8.9 kcal mol−1 for the formation of the 5-membered intermediate IM 1. The activation energy from IM 1 to TS2 1 is 2.6 kcal mol−1. It is noted that the energy of P 1·+ is 0.3 kcal mol−1 higher (0.1 kcal mol−1 lower without the ZPE correction) than that of TS2 1, which indicates that the TS2 1 may disappear at the higher level of theory. Although P 1·+ is 11.6 kcal mol−1 higher in energy than R 1·+, the electron transfer to the radical cation product from the neutral reactant reduces the concentration of P 1·+, which makes the reactions proceed forward and simultaneously produce P 1.
![Schematic diagram of the potential energy curves along the intramolecular cycloaddition reaction pathways of R 1·+, R 2·+ and R 3·+. Energies with ZEP corrections are given in kcal mol−1, with results from B3LYP/6-311G(d,p) calculations in normal texts, and Gibbs free energies at 25 °C in [brackets].](/image/article/2012/RA/c2ra21236f/c2ra21236f-f2.gif) | ||
| Fig. 2 Schematic diagram of the potential energy curves along the intramolecular cycloaddition reaction pathways of R 1·+, R 2·+ and R 3·+. Energies with ZEP corrections are given in kcal mol−1, with results from B3LYP/6-311G(d,p) calculations in normal texts, and Gibbs free energies at 25 °C in [brackets]. | ||
Investigation of the stereoconvergency of the cycloaddition reaction
The cycloaddition pathways of the (E,Z) bis(styrene) isomers R 2 and R 3, giving the trans cyclobutanes P 2 and P 3 respectively, are predicted to be analogous to the case of R 1. As shown in Fig. 2, the radical cations R 2·+ and R 3·+ are 4.0 and 3.9 kcal mol−1 higher in energy than R 1·+, respectively. The first barriers forming the trans intermediates IM 2 and IM 3 are both predicted to be 7.8 kcal mol−1, and the second barriers forming the trans radical cation products P 2·+ and P 3·+ to be 2.0 and 1.8 kcal mol−1. It is obvious that the trans radical cation cyclobutanes P 2·+ and P 3·+ could be formed via the cycloaddition of R 2·+ and R 3·+, respectively, in the same situation as P 1·+, which is in agreement with the experimental fact that the trans cyclobutanes are observed in the cycloadditions of R 2 and R 3. However, as mentioned above, experimentally observed major products of both R 2 and R 3 are cis cyclobutanes P 1. In the following paragraphs we will discuss the stereoconvergency in detail.Firstly, we investigated the possibility of the isomerization from R 3 to R 1 by calculating the energy barrier of thermal isomerization via rotating around the CC double bonds. Just as we expected, the calculated energy barrier (78.5 kcal mol−1) is much larger than that of the cycloaddition (see above), and it is also larger than the visible light energy (47.7–71.6 kcal mol−1). Therefore, the thermal isomerization is unfeasible under irradiation of visible light. For the photoisomerization, the HOMO–LUMO gap and the singlet–triplet excitation energy of R 3 are predicted to be 105.4 and 140.2 kcal mol−1, respectively. Therefore, the direct isomerization from R 3 to R 1 is unfeasible energetically.
As shown in Fig. 2, there is a branching point along the cycloaddition pathway of both R 2·+ and R 3·+, occurring at the trans intermediates, which subsequently lead to the trans and cis cyclobutanes, respectively. The structure parameters in Fig. 1 show that the C1–C2 and C3–C4 bonds have changed into C–C single bond in IM 1, indicating that the trans-cis intermediate conversion is very easy. Therefore the stereoconvergency of the cycloaddition may come from the trans-cis intermediate conversion. However, there must be competition between the trans-cis conversion and the cyclization at the intermediate. Considering that the energy barriers of the cyclizations of IM 2 and IM 3 are only 2.0 and 1.8 kcal mol−1, respectively, it is necessary to calculate the energy barrier of the trans-cis conversion. We therefore carried out the single point energy calculations scanning the corresponding dihedral angles, H1–C1–C2–C3 and H2–C4–C3–C2 (see the definitions in Fig. 3), with a step-size of 10°.
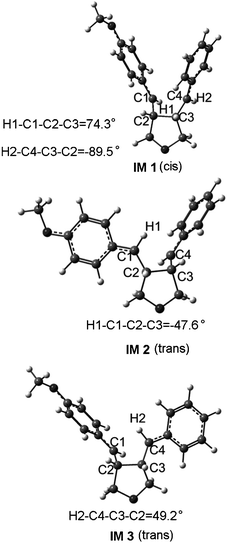
The barrier of the conversion from IM 2 to IM 1 is predicted to be 0.9 kcal mol−1, which is lower than that of the ring closure of IM 2. In addition, the energy of IM 1 is 1.8 kcal mol−1 lower than that of IM 2. Therefore, the trans-cis conversion is favorable both thermodynamically and kinetically. The barrier of the conversion from IM 3 to IM 1 is predicted to be 2.2 kcal mol−1. Although it is a little larger than that of the ring closure of IM 3, the conversion is still an advantaged pathway as the energy of IM 1 is much lower than that of IM 3 while P 3·+ is higher in energy than IM 3. After the trans-cis intermediate conversion, there are two competitive reaction pathways from IM 1. One is to form R 1·+ with a barrier of 0.2 kcal mol−1viaTS1 1, and the other is to form P 1·+ with a barrier of 2.6 kcal mol−1viaTS2 1. It is obvious that the first pathway is more favorable. This explains why the rate of the isomerization from R 3 to R 1 is faster than that of the cycloaddition predicted by Ischay et al.26 Meanwhile, the formative R 1·+ could also undergo the cycloaddition, forming P 1·+. The aforementioned facts could exactly explain why the cis cyclobutane is observed as the major product of R 2 and R 3. However, the energy barrier of the conversion from IM 1 to IM 3 is much higher than that of the ring closure reaction of IM 1, which also explains the GC result of Schepp et al.,24 that the rate of the isomerization from R 1 to R 3 is slower than that of the cycloaddition.
The effect of the position of methoxyl on phenyl ring
According to the experimental results, the position of methoxyl has an important effect on the cycloaddition reactions of the bis(styrenes).26 Therefore, we further investigated the cycloadditions of R 4 with methoxyl at the ortho position and R 5 with methoxyl at the meta position.Our calculations indicate that the energies of R 1, R 5, and R 4 increase in order. This result probably is caused by the increase of the steric hindrance with the approach of the two substituents on the phenyl ring. However, the energy order rearranges in their corresponding radical cations (R 5·+ being highest in energy). It is known that the methoxyl at para or ortho position is electron-donating by the conjugated effect, but the methoxyl at meta position is electron-withdrawing by the inductive effect. The electron-withdrawing effect would decrease the energy of the highest occupied molecular orbital, making R 5 need more energy to lose one electron. Therefore, R 5·+ is less stable than R 1·+ and R 4·+. The above explanation is also confirmed by the fact that the AIP of R 5 is larger than that of R 1 and R 4.
As shown in Fig. 4, along the reaction pathway of R 4·+, the barrier for the formation of IM 4 is 8.1 kcal mol−1, close to the situation of R 1·+. However, IM 4 couldn't undergo the ring closure reaction, as the energy increases monotonously with C1 approaching to C4. We tried to locate P 4·+ starting from the geometry of P 4, but got the structure of IM 4, suggesting that P 4 may be formed via the electron transfer between IM 4 and R 4. The mechanism and energy barriers of the cycloaddition of R 5·+ are similar to those of R 1·+. The energy of IM 5 is 0.7 kcal mol−1 higher (0.4 kcal mol−1 lower without the ZPE correction) than that of TS1 5, and similarly P 5·+ is 0.4 kcal mol−1 higher (0.2 kcal mol−1 lower without the ZPE correction) in energy than TS2 5. These results indicate that TS1 5, IM 5 and TS2 5 may disappear at the higher level of theory, which imply that the cycloaddition of R 5·+ may be concerted. On the basis of our calculations, we could conclude that the cycloaddition of R 5·+ should occur theoretically. However, R 5 couldn't be oxidized by the initiator, producing R 5·+ (see above). This may be the key reason why R 5 couldn't form the cyclobutane under the experimental conditions.26
![Schematic diagram of the potential energy curves along the intramolecular cycloaddition reaction pathways of R 1·+, R 4·+ and R 5·+. Energies with ZEP corrections are given in kcal mol−1, with results from B3LYP/6-311G(d,p) calculations in normal texts, and Gibbs free energies at 25 °C in [brackets].](/image/article/2012/RA/c2ra21236f/c2ra21236f-f4.gif) | ||
| Fig. 4 Schematic diagram of the potential energy curves along the intramolecular cycloaddition reaction pathways of R 1·+, R 4·+ and R 5·+. Energies with ZEP corrections are given in kcal mol−1, with results from B3LYP/6-311G(d,p) calculations in normal texts, and Gibbs free energies at 25 °C in [brackets]. | ||
The effect of the electron-donating and electron-withdrawing substituents
Only one methoxyl at the para position on one styrene moiety is necessary for the [2+2] cycloaddition of bis(styrenes) according to the experimental results.26 To investigate the effect of the electron-donating and electron-withdrawing substituents on the other styrene moiety on the reaction, we studied the cycloadditions of R 6, R 7, R 8 and R 9, in which the H atom of the para position on the phenyl group is substituted by methoxyl, chlorine, amino and nitryl, respectively (see Scheme 1).As shown in Table 2, the cycloaddition pathways of R 6·+, R 7·+, R 8·+ and R 9·+ and the energy barriers are all analogous to the situation of R 1·+, indicating that the electron-donating and electron-withdrawing substituents have no obvious effect on the cycloaddition reactions. These results are in agreement with the experimental facts that both R 6 and R 7 give good yields of the cyclobutane products. However, these substituents significantly influence the AIPs of the reactants. The order of the calculated AIPs is R 9 > R 7 > R 1 > R 6 > R 8 (see Table 1), which is opposite to the order of the electron-donating capacities of the substituents: –NO2 < –Cl < –H < –OCH3 < –NH2. Therefore, electron-donating substituents are beneficial for the formation of radical cation. However, too strong electron-donating substituent would stabilize the radical cation species. This stabilization gives the radical cation intermediate enough time to rearrange, leading to the side reaction. On the other hand, the AIP of R 9 is even higher than that of R 5, indicating that too strong an electron-withdrawing substituent with high AIP may be not able to afford the radical cation reactant to undergo the cycloaddition. Thus, too strong electron-donating and electron-withdrawing substituents are both unfavorable for cycloaddition.
| Bis(styrene) | R·+ | TS1 | IM | TS2 | P·+ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| a Energy of the trans cyclobutane product. b The second transition state and the radical cation product of R 4 wasn't obtained according to the calculations. | ||||||||||
| R 1 | 0 | [0] | 8.9 | [8.3] | 8.7 | [7.3] | 11.3 | [9.9] | 11.6 | [10.0] |
| (0) | (8.9) | (8.8) | (11.8) | (11.5) | ||||||
| R 2 a | 0 | [0] | 7.8 | [6.7] | 6.5 | [5.4] | 8.5 | [7.2] | 7.4 | [4.7] |
| (0) | (8.1) | (6.5) | (9.4) | (6.7) | ||||||
| R 3 a | 0 | [0] | 7.8 | [7.4] | 6.8 | [6.0] | 8.6 | [7.6] | 8.3 | [5.9] |
| (0) | (8.2) | (6.8) | (9.2) | (7.5) | ||||||
| R 4 | 0 | [0] | 8.1 | [7.4] | 8.0 | [6.5] | —b | —b | ||
| (0) | (8.4) | (8.6) | ||||||||
| R 5 | 0 | [0] | 9.1 | [7.7] | 9.8 | [7.7] | 12.0 | [10.4] | 12.4 | [9.6] |
| (0) | (8.9) | (9.9) | (12.3) | (12.3) | ||||||
| R 6 | 0 | [0] | 8.6 | [7.4] | 8.8 | [6.8] | 10.9 | [8.9] | 11.4 | [9.8] |
| (0) | (8.6) | (8.6) | (11.5) | (11.6) | ||||||
| R 7 | 0 | [0] | 9.2 | [8.5] | 9.1 | [7.5] | 11.1 | [9.6] | 12.2 | [10.5] |
| (0) | (9.7) | (9.7) | (12.5) | (12.9) | ||||||
| R 8 | 0 | [0] | 9.2 | [9.4] | 8.6 | [7.9] | 10.8 | [10.0] | 10.9 | [8.5] |
| (0) | (9.8) | (8.7) | (11.4) | (11.8) | ||||||
| R 9 | 0 | [0] | 10.2 | [9.5] | 9.1 | [6.2] | 13.3 | [12.0] | 13.4 | [10.9] |
| (0) | (10.8) | (9.5) | (14.0) | (13.3) | ||||||
The effect of the solvent on the reaction
We studied the effect of CH3CN solvent (ε = 36.6) on the cycloaddition reaction using the polarized continuum model (PCM), since the experimental investigation is usually carried out in solvents. As shown in Table 2, the energy barriers of the R 1·+–R 8·+ cycloadditions in CH3CN solvent decrease by 0.5–1.5 kcal mol−1, indicating that the polar solvent doesn't have much effect on the cycloaddition process, which is consistent with there being no large solvent effect in hydrocarbon cycloadditions and lack of significant electron delocalization.31–33 However, the solvent effect on the cycloaddition of R 9·+ is much stronger, probably due to the electron delocalization caused by the nitryl on the phenyl ring. As we discussed above, the polar solvent significantly stabilizes the radical cations, providing more time for the radical cation rearrangement, which may benefit the side reaction. Therefore, a polar surrounding is unfavorable for the [2+2] radical cation cycloadditions.Conclusions
We have systematically investigated the mechanisms of the intramolecular radical cation cycloadditions of variety of bis(styrenes) by performing DFT B3LYP calculations. The calculations show that the cycloaddition is stepwise via the formation of a 5-membered ring intermediate. The final cyclobutane products are formed via the electron transfer between the long-bond radical cation product and the neutral reactant. The origin of the stereoconvergency for the cycloaddition of the (E,E) and (E,Z) bis(styrenes) is attributed to the conversion from trans intermediate to cis intermediate. The properties and the position on the phenyl ring of the substituents have strong effect on the stability of the radical cation, which would lead the side reaction or the impossibility to form the radical cation. In addition, the polar solvent may be unfavorable for the radical cation [2+2] cycloaddition.Acknowledgements
We want to express our gratitude to the Professor Zhi-Xiang Wang for providing computing equipment. And we gratefully acknowledge the National Natural Science Foundation of China (No. 21173262) for supporting this work.References
- (a) T. V. Hansen and Y. Stenstrøm, in Naturally Occurring Cyclobutanes in Organic Synthesis: Theory and Applications, Elsevier, Oxford, U.K., 2001, pp. 1 Search PubMed; (b) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1 CrossRef CAS.
- (a) F. A. Bell, R. A. Crellin, N. Fugii and A. Ledwith, J. Chem. Soc., Chem. Commun., 1969, 251 RSC; (b) R. A. Carruthers, R. A. Crellin and A. Ledwith, J. Chem. Soc., Chem. Commun., 1969, 252–253 RSC.
- M. Kojima, H. Sakuragi and K. Toukumaru, Bull. Chem. Soc. Jpn., 1989, 62, 3863 CrossRef CAS.
- V. Nair, J. Mathew, P. P. Kanakamma, S. B. Paniekera, V. Sheeba, S. Zeena and G. K. Eigendorf, Tetrahedron Lett., 1997, 38, 2191 CrossRef CAS.
- (a) S. L. Mattes and S. Farid, J. Am. Chem. Soc., 1986, 108, 7356 CrossRef CAS; (b) S. L. Mattes and S. Farid, Org. Photochem., 1983, 6, 233 CAS; (c) S. L. Mattes and S. Farid, J. Am. Chem. Soc., 1983, 105, 1386 CrossRef CAS; (d) S. L Mattes and S. Farid, J. Am. Chem. Soc., 1982, 104, 1454 CrossRef CAS.
- (a) N. L. Bauld and R. Pabon, J. Am. Chem. Soc., 1983, 105, 633 CrossRef CAS; (b) N. L. Bauld, Tetrahedron, 1989, 45, 5307 CrossRef CAS.
- (a) N. P. Schepp, D. Shukla, H. Sarker and N. L. Bauld, J. Am. Chem. Soc., 1994, 116, 6985 CrossRef; (b) N. P. Schepp and L. J. Johnston, J. Am. Chem. Soc., 1996, 118, 2872 CrossRef CAS.
- F. D. Lewis and M. Kojima, J. Am. Chem. Soc., 1988, 110, 8664 CrossRef CAS.
- O. Wiest, J. Phys. Chem. A, 1999, 13, 7907 CrossRef.
- F. L. Cozens, R. Bogdanova, M. Régimbald, H. García, V. Martí and J. C. Scaiano, J. Phys. Chem. B, 1997, 101, 6921 CrossRef CAS.
- F. D. Lewis, A. M. Bedell, R. E. Dykstra, J. E. Elbert, I. R. Could and S. Farid, J. Am. Chem. Soc., 1990, 112, 8055 CrossRef CAS.
- R. M. Wilson, J. G. Dietz, T. A. Shepherd, D. M. Ho, K. A. Schnapp, R. C. Elder, J. W. Watkins, L. S. Geraci and C. F. Campanat, J. Am. Chem. Soc., 1989, 11, 1749 CrossRef.
- M. Kojima, A. Kakehi, A. Ishida and S. Takamuku, J. Am. Chem. Soc., 1996, 118, 2612 CrossRef CAS.
- Y. Roh, D. Gao and N. L. Bauld, Adv. Synth. Catal., 2001, 343, 481 CrossRef CAS.
- M. Arata, T. Miura and K. Chiba, Org. Lett., 2007, 9, 4347 CrossRef CAS.
- Y. Okada, R. Akaba and K. Chiba, Org. Lett., 2009, 11, 1033 CrossRef CAS.
- Y. Okada and K. Chiba, Electrochim. Acta, 2011, 56, 1037 CrossRef CAS.
- T. Miyashi, H. Ikeda and Y. Takahashi, Acc. Chem. Res., 1999, 32, 815 CrossRef CAS.
- C. A. Marquez, H. Wang, F. Frabbretti and J. O. Metzger, J. Am. Chem. Soc., 2008, 130, 17208 CrossRef CAS.
- G. A. Mirafzal, T. Kim, J. Liu and N. L. Bauld, J. Am. Chem. Soc., 1992, 114, 10968 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS.
- G. A. N. Felton, Tetrahedron Lett., 2008, 884 CrossRef CAS.
- J. Yang, G. A. N. Felton, N. L. Bauld and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 1634 CrossRef CAS.
- N. P. Schepp, D. Shukla, H. Sarker, N. L. Bauld and L. J. Johnston, J. Am. Chem. Soc., 1997, 119, 10325 CrossRef CAS.
- L. L. O'Neil and O. Wiest, J. Org. Chem., 2006, 71, 8926 CrossRef CAS.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef CAS.
- Q. Zhang, Z. Li and B. Chen, THEOCHEM, 2009, 901, 202 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frish, J. Phys. Chem., 1944, 98, 11623 CrossRef.
- (a) S. Miertus, E. Scrocco and E. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS; (b) V. Barone and M. Cossi, J. Phys. Chem. A, 1988, 102, 1995 CrossRef; (c) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS.
- R. Seidel, M. Faubel, B. Winter and J. Blumberger, J. Am. Chem. Soc., 2009, 131, 16127 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- C. Amovilli, V. Barone, R. Cammi, E. Cances, M. Cossi, B. Mennucci and C. S. Pomelli, Adv. Quantum Chem., 1998, 32, 227 CrossRef.
- C. J. Cramer and D. G. Truhlar, Chem. Rev., 1999, 99, 2161 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |