Photophysics and photostability of 9,10-bis(phenylethynyl)anthracene revealed by single-molecule spectroscopy
Masaaki
Mitsui
*,
Yuya
Kawano
,
Ryoya
Takahashi
and
Hiroki
Fukui
Department of Chemistry, Faculty of Science, Shizuoka University, 836 Ohya Suruga-ku, Shizuoka, 422-8529, Japan. E-mail: smmitsu@ipc.shizuoka.ac.jp; Fax: +81-54-238-4755
First published on 20th August 2012
Abstract
The photophysics and photostability of 9,10-bis(phenylethynyl)anthracene (BPEA) diluted in a 40-nm-thick Zeonex polymer film have been investigated by single-molecule spectroscopy (SMS). The single-molecule detection of BPEA was verified by recording fluorescence intensity trajectories, fluorescence lifetimes, and fluorescence spectra. The intensity trajectories showed frequent on/off blinking and one-step photobleaching behaviors. The observed blinking was attributed to the temporary occupation of the excited triplet state T1via intersystem crossing (ISC). Assuming a three-state model (e.g., S0, S1, and T1), the distributions of triplet lifetime and S1→T1 ISC quantum yield of BPEA were both derived from the analyses of the blinking statistics and the intensity autocorrelation. We found extremely low ISC yields (on the order of 10−5–10−4), which were theoretically rationalized by the large energy gap between 3B2u and S1(1B1u)/T1(3B1u) states. SMS measurements were also conducted under both air and Ar atmospheres in order to gain insight into the influence of oxygen on photobleaching. The results reveal that, although the presence of oxygen considerably degraded the photostability of BPEA, under deoxygenated conditions, BPEA delivers more than 107 photons before photobleaching and possesses an appreciably low photobleaching yield of 10−9–10−8. This study shows that BPEA has a relatively high degree of photostability at room temperature and can serve as a useful green fluorescent probe for SMS studies.
1. Introduction
9,10-Bis(phenylethynyl)anthracene (BPEA, Fig. 1) is well known as one of the most efficient green light-emitting aromatic hydrocarbons. In addition, it shows high solubility in a variety of solvents, and good chemical and thermal stabilities. Accordingly, BPEA has been widely used as a molecular probe in various spectroscopic studies,1–7 and its photophysical properties have been studied by a number of spectroscopic methods.8–12 BPEA and its derivatives have been recently demonstrated to possess attractive optical and electronic properties, thereby making them promising candidates for a wide variety of optoelectronic and photonic applications such as light-emitting diodes,13 photoswitches,14 chemiluminescent light sources,15 optical waveguides,16 and solar cells.17 Moreover, bis(arylethynyl)arene compounds have recently received considerable attention as molecular wires.18 These facts have thus led to renewed efforts to gain a more comprehensive understanding of the photophysical and photochemical properties of BPEA. | ||
| Fig. 1 Molecular structure of BPEA. | ||
Photophysical parameters of BPEA, obtained via the singlet manifold (e.g., fluorescent quantum yield, Φf, and fluorescence lifetime, τf), have been widely reported in the literature.8,12,19–21 In contrast, few experimental efforts have been hitherto invested in the study of photophysics involving the triplet manifold of BPEA9 whose Φf value (ca. 1) prevents information about the triplet state from being obtained by conventional spin resonance and thermal lensing techniques. The high Φf value indicates that the intersystem crossing (ISC) rate to the triplet state is very low in BPEA, and consequently, this parameter does not play a significant role in the photophysics of this compound. However, once the long-lived π,π* triplet state (T1) is formed through ISC from the lowest excited singlet state (S1), it can efficiently produce a singlet oxygen 1O2(1Δg) that may react with the ground-state molecule.22 Unfortunately, such photochemical oxidation reactions are known to provide a major route to the irreversible photobleaching of anthracene and its derivatives,22–25 thereby limiting the active time of these materials.
Single-molecule spectroscopy (SMS) has been widely employed in recent years to obtain more information about the photophysics and photochemistry of dye molecules. The detection of single-molecule emission allows the observation of so-called photoblinking as well as one-step photobleaching. Photoblinking is characterized by a reversible “on-off” emission as a result of the temporary occupation of non- or low-emissive states such as excited triplet states,26–35 photoisomers,34,36,37 or other reversible metastable species generated by charge transfer.35,38–42 Among them, the photoblinking that originates from the triplet-state excursion is often referred to as “triplet blinking.” Previous studies on the triplet blinking26–35 revealed that on- and off-times (i.e., the durations of high intensity “on” and background “off” levels, respectively) are exponentially distributed on the range of microsecond to milliseconds since the ISC process toward and from the triplet state is characterized by a single rate constant. The analysis of the triplet blinking can provide several parameters such as the triplet lifetime (τT), the ISC rate constant (kISC), and the ISC quantum yield (ΦISC) of single molecules. In contrast to photoblinking, photobleaching is an irreversible process caused by light-induced chemical reactions and is responsible for the permanent loss of photon emission in fluorophores.42–55 By evaluating the number of total emitted photons before photobleaching, one can estimate the photobleaching quantum yield (Φb) that can serve as a useful index of the molecular photostability.42,46,55 Thus, SMS of BPEA is expected to render new quantitative information on the triplet-state kinetics (relatively elusive by ensemble experiments owing to the high Φf value) and the molecular photostability of this compound. To the best of our knowledge, however, no SMS studies on BPEA and its derivatives have been reported so far, even though they are one of the promising classes of light-emitting organic materials, as mentioned above.
In this contribution, we report on an SMS study of the photophysics and photobleaching characteristics of BPEA in a Zeonex polymer matrix at room temperature. Photophysical parameters of BPEA such as τT and ΦISC were determined through the analysis of the on/off blinking as well as the intensity autocorrelation traces. In addition, the photostability of BPEA was examined by evaluating the total number of emitted photons before photobleaching and the photobleaching statistics for single BPEA molecules. Consequently, the present study demonstrates that BPEA meets the indispensable requirements for SMS studies such as high quantum fluorescence yields in a rigid matrix and appreciably low photobleaching yields, leading to a substantial amount of emitted photons (>107).
2. Experimental
BPEA (97%, Sigma-Aldrich), a nonpolar cyclo-olefin polymer Zeonex (330 R, Tg = 123 °C, Zeon Chemicals), cyclohexane, and toluene (spectroscopic grade, Wako) were used as received. Ensemble absorption spectra of BPEA in both cyclohexane and a Zeonex film were recorded on a Lambda 650 spectrometer (Perkin-Elmer), while fluorescence emission spectra were obtained with a RF-5300PC fluorometer (Shimadzu). BPEA-doped Zeonex film samples for ensemble absorption and fluorescence measurements were prepared after evaporation of the solvent from 10−7 to 10−5 M toluene solutions of BPEA containing 50 mg mL−1 of Zeonex.The samples for the SMS experiments were prepared by spin-coating (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) one drop of a toluene solution containing BPEA (∼10−10 M) and Zeonex (5 mg mL−1) onto thoroughly cleaned cover glasses (Matsunami). As a result, BPEA molecules were dispersed in a Zeonex thin film with a thickness of ca. 40 nm, as measured by atomic force microscopy (AFM, SPM-9700, Shimadzu). As shown in Fig. 2a, the sample-coated cover glass was set on an O-ring and acted as the top face of a small vacuum chamber. This setup allowed us to observe the sample from the upper surface of the coverglass with the help of an oil immersion objective. Before starting the SMS measurements, the sample was evacuated for 30 min under low vacuum conditions (<0.1 Pa) in order to remove residual solvents and oxygen from the polymer film as much as possible. In SMS measurements, the oxygen concentration in the polymer film was reduced by continuously flushing the sample with Ar gas.
000 rpm) one drop of a toluene solution containing BPEA (∼10−10 M) and Zeonex (5 mg mL−1) onto thoroughly cleaned cover glasses (Matsunami). As a result, BPEA molecules were dispersed in a Zeonex thin film with a thickness of ca. 40 nm, as measured by atomic force microscopy (AFM, SPM-9700, Shimadzu). As shown in Fig. 2a, the sample-coated cover glass was set on an O-ring and acted as the top face of a small vacuum chamber. This setup allowed us to observe the sample from the upper surface of the coverglass with the help of an oil immersion objective. Before starting the SMS measurements, the sample was evacuated for 30 min under low vacuum conditions (<0.1 Pa) in order to remove residual solvents and oxygen from the polymer film as much as possible. In SMS measurements, the oxygen concentration in the polymer film was reduced by continuously flushing the sample with Ar gas.
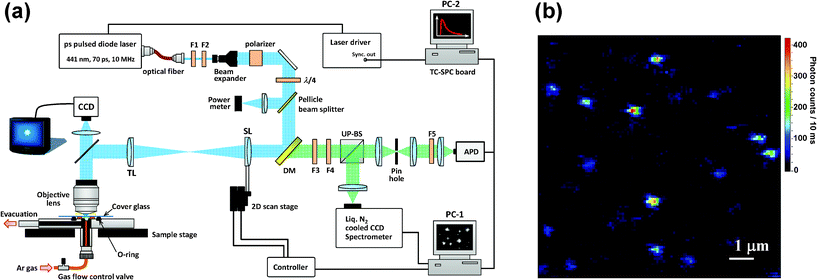 | ||
| Fig. 2 (a) Experimental setup used for single-molecule spectroscopy. F1: bandpass filter; F2: neutral density filter; F3: longpass filter; F4: notch filter; F5: shortpass filter; DM: dichroic mirror; SL: scan lens (f = 180 mm); TL: tube lens (f = 180 mm); UP-BS: unpolarized beam splitter; PC: personal computer. (b) Fluorescence image of BPEA in a Zeonex film. | ||
SMS experiments were performed in a home-built laser-scanning optical microscope, as depicted schematically in Fig. 2a. The excitation light source used was a 441 nm pulsed laser diode (PLP10-044C, Hamamatsu Photonics) with a pulse width of 70 ps FWHM (full-width at half-maximum), and a repetition rate of 10 MHz. The excitation light was spatially filtered by a polarization-maintaining single-mode fiber and subsequently collimated and attenuated to an average power of 0.2 to 1.5 μW at the sample by a neutral density filter. The resulting beam passed through a band-pass filter (F2, FF01-438/24-25, Semrock), a 2× beam expander (which increases the laser beam diameter to fill the back aperture of the microscope objective), and a Glan-Thompson polarizer (extinction ratio 106:1, Edmund Optics). A λ/4 plate (AQWP05-M-600, Thorlabs) was used to shift the beam polarization from linear to circular.
The laser-scanning method used in our microscope was developed on the basis of the laser-scanning scheme and algorithm reported by Yu and co-workers,56 by utilizing the magnification power of an objective. The setup included an oil immersion and infinity-corrected objective lens (100×, NA 1.4, Olympus), which focuses the excitation beam to a diffraction-limited spot size of ca. 210 nm FWHM. A dichroic mirror (DM, Di01-R442-24 × 36, Semrock) redirects the laser beam to the scanning lens (SL), which is moved two dimensionally by an x − y translational stage equipped with Nanomover actuators (Melles Griot). The distances from the tube lens (TL) to the SL and to the back aperture of the objective were both 2f, where f is the focal length of the TL. This setup assures that the laser beam always fills the back aperture of the objective.56 For both the TL and the SL, we used achromatic lenses (Melles Griot) with f = 180 mm to fully utilize the power of the objective magnification (i.e., 100×). The position repeatability of this actuator is higher than 200 nm, so the positioning repeatability of the laser spot is expected to be less than or equal to 2 nm (200 nm/100) on the sample plane. This repeatability ensures the location of a single molecule inside the focused laser spot. A fluorescence image (10 × 10 μm2) of the sample was acquired by raster scanning of the laser focal spot, a typical example of which is shown in Fig. 2b. The fluorescence imaging confirmed a density of coverage of approximately 0.1 molecules per μm2, which ensures single-molecule imaging with a diffraction-limited fluorescence spot on the sample plane. An electronic shutter was used to block the excitation beam before data acquisition. After the laser focus was moved to the position of the molecule of interest, the shutter was reopened and fluorescence photons from the excited molecule were collected through the same objective, and then, passed through the DM and long-pass (F3, LP02-442RS-25, Semrock) and notch (F4, NF01-442U-25, Semrock) filters to block the scattered laser light. The fluorescence was then split by a 50:50 unpolarized beam splitter (UP-BS). Half of the detected fluorescence signal was sent to a polychromator (SpectraPro 2300i) coupled to a liquid nitrogen-cooled charge coupled device (CCD) camera (Spec-10:100B/LN, Roper Scientific). The other half was focused onto a pinhole (75 μm diameter) for rejection of out-of-focus background. Finally, the signal passed through short-pass filters (F5, Edmund Optics) and directed onto an avalanche photodiode (APD, SPCM-AQR-14 Perkin-Elmer). The total detection efficiency at the APD in the present setup was estimated to be about 4% by considering the transmission of the totality of optical parts used in the detection path (i.e., objective lens, filters, etc.) and the quantum efficiency of the APD detector. The fluorescence signals detected by the APD were shared by two PC plug-in cards. A data-acquisition board (PCI 6602 Counter-Timer, National Instruments) in PC-1 was used for the continuous counting of photon detection events from the APD. The imaging and positioning process were controlled by a home-made LabView program. The signals from the APD were also sent to a time-correlated single photon counting (TCSPC) PC card (TimeHarp 200, PicoQuant) in PC-2, which computed the time-tagged and time-resolved (T3R) modes. This methodology allowed simultaneous registration of each detected photon on two independent time scales: (i) the arrival time after the beginning of the acquisition and (ii) the time lag between the excitation pulse and the fluorescence photon. Data acquisition and fluorescence decay and autocorrelation analyses were performed using the software SymPhoTime v5.2.4 (PicoQuant). Fluorescence intensity, spectrum (3 s per spectrum), and lifetime trajectories (∼1000 photons per decay) were acquired until a photobleaching event occurred. Note that the selected integration time (3 s) for the spectral trajectories was intentionally long in comparison to the timescale of spectral jumps observed in organic dyes34,57 with the purpose of verifying that the observed signals originate from BPEA. To determine the wavelength of the fluorescence maximum (λmax), each spectrum was fitted with the appropriate number of Gaussians. Monoexponentials were fitted to the fluorescence decay curves by maximum-likelihood estimation (MLE) to determine the fluorescence lifetime, as the MLE method provides reasonable results even at photon counts lower than 1000.58 All measurements were carried out at room temperature.
3. Results and discussion
3.1. Ensemble results
Fig. 3 shows the UV-vis absorption and fluorescence emission spectra of BPEA in both cyclohexane and a Zeonex film. When BPEA was dissolved in cyclohexane, the fluorescence spectral shape was considerably different than that of the absorption spectrum (i.e., they were not a mirror image). This has been previously attributed to the existence of a variety of conformations of BPEA in which the two phenyl groups can rotate almost freely in a solution at room temperature, whereas a planar conformation is mostly responsible for the fluorescence emission spectra.8,12 The absorption spectrum became somewhat broader, and λmax, the wavelength of absorption maximum, was slightly red-shifted (−5 nm) when BPEA was embedded in the Zeonex film. However, the overall spectral shape remained unchanged, thereby suggesting that various BPEA conformations were also present in the film. We note that this result differs from that of BPEA in polyethylene in which only the planar conformation was formed.8 The higher free volume of the amorphous Zeonex as compared to that of crystalline polyethylene can account for this difference. The emission spectrum of BPEA in Zeonex was also somewhat broader than in cyclohexane since the molecules embedded in the amorphous solid matrix can have a large variety of local surroundings. However, as in the case of cyclohexane, the emission spectral shape was noticeably different from the shape of the absorption spectrum. This result strongly suggests that excited-state structural planarization (i.e., the rotations or twists of the phenyl groups) also occurs in the Zeonex matrix. Such a conformational relaxation has been often observed for dye molecules embedded in glassy polymers.59–62 At temperatures well below the glass transition point, the polymer can still relax by local rearrangements of chain segments and exhibit local density fluctuations (or motion of holes).63 This local density fluctuation is known to cause characteristic fluctuations of the fluorescence lifetime and result in an asymmetric lifetime distribution.64,65 However, this distribution was not observed in the lifetime trajectories for BPEA in Zeonex, even though the integration time was down to 0.05 s. Thus, we believe that the amorphous Zeonex matrix contains sufficiently steadily microscopic free spaces that facilitate the structural relaxation of BPEA.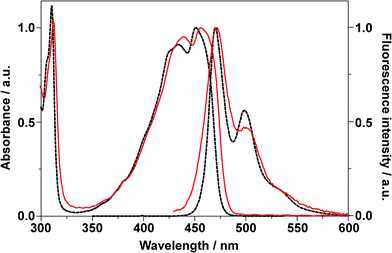 | ||
| Fig. 3 Normalized UV-vis absorption and emission (λex = 400 nm) spectra of BPEA in cyclohexane (dotted lines) and in a Zeonex film (solid lines). | ||
3.2. Identification of single BPEA molecules
To examine the photophysics of BPEA at the single-molecule level, time trajectories of fluorescence intensity, lifetime, and spectra were recorded for 85 individual BPEA molecules embedded in a Zeonex film. The exemplary trajectories are shown in Fig. 4. The fluorescence intensity trace (Fig. 4a) showed frequent discrete jumps between the stable (∼260 counts per 10 ms, refer to as “on” level) and the background (∼11 counts per 10 ms, “off” level) intensity levels before ending with an irreversible photobleaching at 211 s. As shown in Fig. 4b, the fluorescence lifetime was distributed around 3.5 ns for this molecule. The distribution mean, obtained from 64 molecules, was 3.4 ns, which was in good agreement with the reported lifetime of BPEA (e.g., 3.2 ns at room temperature and 3.8 ns at 77 K) in nonpolar solvents (methylcyclohexane).8 As indicated in Fig. 4c, the fluorescence spectra was much narrower than the corresponding ensemble spectrum (Fig. 3) owing to the elimination of the ensemble averaging effect, and they displayed a clear vibronic structure similar to the ensemble spectrum obtained in cyclohexane. This result further indicates that the fluorescence emission of BPEA in Zeonex does indeed originate from the planar conformation. λmax obtained from the fluorescence spectra (Fig. 4c) remained practically constant at 469 nm, and the average value of λmax obtained for 149 single molecules was found to be 471 nm, which is in excellent agreement with that of the corresponding ensemble spectrum (λmax = 471 nm). The on/off blinking, one-step photobleaching, and agreement of the fluorescence parameters strongly indicate that the fluorescence signal indeed originated from a single BPEA molecule. The photophysical parameters of BPEA in the Zeonex film are summarized in Table 1.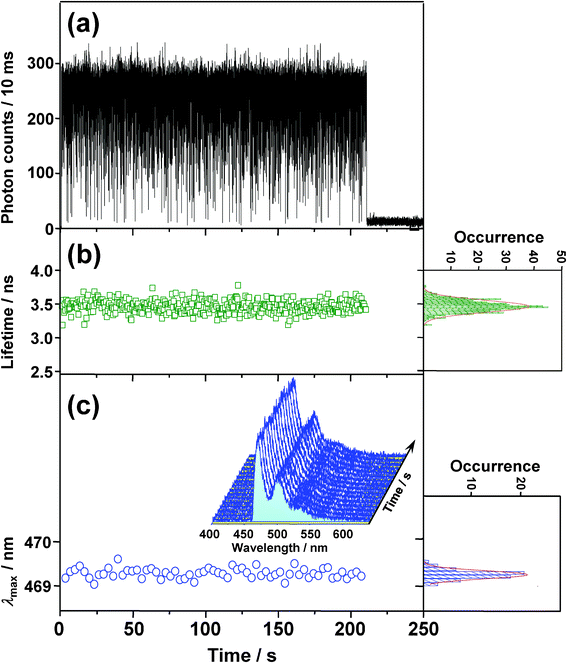 | ||
| Fig. 4 Typical time evolutions of (a) fluorescence intensity, (b) lifetime, and (c) wavelength of emission maximum (λmax) obtained from the fluorescence spectra (3 s integration) shown in (c) as an inset. The right-hand panels in b and c show frequency histograms of each trajectory, along with fitted Gaussian functions (lines). | ||
| Atmosphere | λ max/nm | τ f/ns | Φ ISC | k ISC/s−1 | τ T/ms | N tot | Φ b d | |
|---|---|---|---|---|---|---|---|---|
| histogram | ACF | |||||||
| a Values in parentheses indicate the number of molecules yielding a mean value. b 3 of 85 molecules yielded very narrow distributions of Non data points, so they were excluded from the analysis. c No blinking was observed. d Obtained by assuming a Poissonian distribution of photobleaching quantum yield.55 | ||||||||
| Ar | 471 (149) a | 3.4 (64) | 5.0 × 10−5 (82) b | 1.2 × 104 | 2.70 (85) | 2.82 (85) | 5.6 × 107 (149) | 1.5 × 10−8 (149) |
| Air | 473 (58) | 3.4 (73) | — c | — | — | — | 1.3 × 106 (73) | 1.0 × 10−6 (73) |
3.3 Triplet blinking
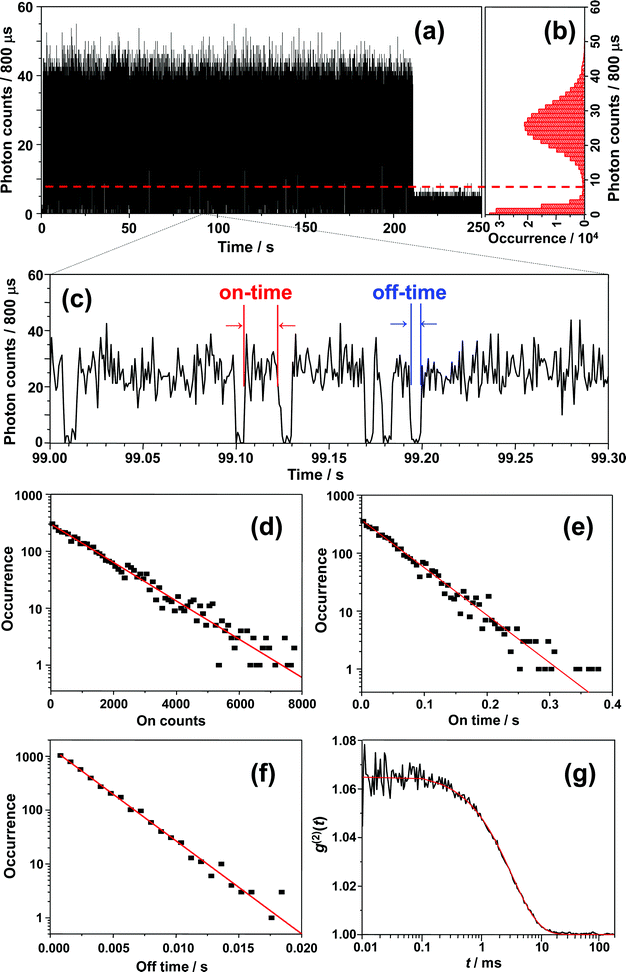
Before starting the analysis of the observed blinking, we systematically examined the effect of the selected bin-time on the resultant average on-time τon, off-time τoff, and on-counts 〈Non〉. The bin-time of one single-molecule data can be arbitrarily changed by means of data collection via T3R mode. With the aim to determine τoff, τon, and 〈Non〉 at each bin-time, a simple threshold analysis (the so-called histogram method) was used to differentiate between “on” and “off” levels and extract on-counts (Non, i.e., the total number of photons detected during on-time periods). As an example, the results for a bin-time of 800 μs are shown in Fig. 5. Fig. 5a shows the 800 μs binned trajectory of data represented in Fig. 4a, along with the corresponding histogram (Fig. 5b) and a 0.3 s zoom (Fig. 5c). As can be seen in Fig. 5b, the intensity histogram of the “on” level displayed a symmetric distribution, and it can be satisfactorily fitted with a Gaussian, which implies that the selected bin-time (800 μs) was likely enough to resolve the observed blinking kinetics.35 The unbiased threshold level (Ith) was determined using the equation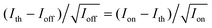 , where Ion and Ioff are the average intensity levels of “on” and “off” states, respectively.35,66 The intensity at a given time above and below Ith is assigned to “on” and “off” states, respectively. Fig. 5d, e and f present semi-log plots of the histograms for the on-counts, on-time, and off-time durations, respectively, obtained from the 800 μs binned trajectory (Fig. 5a). All the histograms approximated well to a straight line (i.e., a single-exponential distribution), yielding average 〈Non〉, τon, and τoff of 1289 ± 24, 51 ± 1 ms, and 2.51 ± 0.04 ms, respectively. The same analysis was performed for other bin-times, and the corresponding plots of 〈Non〉, τon, and τoff against the bin-time are shown in Fig. 6. Importantly, 〈Non〉 and τon exponentially decreased with reducing the bin-time in a range of 0.4–7 ms, although this decrease became more intense at bin-times below 0.6 ms. In the triplet blinking, the probability density of off-times increased exponentially with decreasing bin-times (Fig. 5f). These trends thus demonstrate that a considerable number of “off”-events, which are shorter than the selected bin-times, are still involved in on-time periods. Ideally, bin-time must be reduced until 〈Non〉 (or τon) becomes independent of this parameter. This, however, is practically unachievable in the histogram method because the influence of shot-noise becomes more remarkable at shorter bin-times, thereby causing artificial decreases in 〈Non〉 and τon. This effect is indeed visible below 600 μs bin-time (Fig. 6a and b). With the aim to avoid an arbitrary choice of bin-time and thus estimate a more reliable value for 〈Non〉 and τon, we herein adopted the extrapolated value (i.e., bin-time→0) obtained from the exponential fit of the 〈Non〉 or τonversus bin-time (Fig. 6a or b). For the single molecule of Fig. 5, the values of 〈Non〉 and τon were finally determined to be 874 ± 61 and 35.5 ms, respectively, which will be used in the estimation of ΦISC described below. In contrast to 〈Non〉 and τon, the value of τoff was bin-time independent and nearly constant within the experimental error over a bin-time range of 0.4 to 7 ms (Fig. 6c). For power-law blinking of single semiconductor quantum dots, Kuno and co-workers reported that the experimentally determined average off-time shows an apparent bin-time dependence because “averages” are not well defined for power-law kinetics.67 In triplet blinking, however, single exponential kinetics are dominated by a well-defined average value. Consequently, bin-time independent values are obtained for τoff in a wide range of bin-times. From now on, we will use τoff values obtained at 800 μs bin-time.
, where Ion and Ioff are the average intensity levels of “on” and “off” states, respectively.35,66 The intensity at a given time above and below Ith is assigned to “on” and “off” states, respectively. Fig. 5d, e and f present semi-log plots of the histograms for the on-counts, on-time, and off-time durations, respectively, obtained from the 800 μs binned trajectory (Fig. 5a). All the histograms approximated well to a straight line (i.e., a single-exponential distribution), yielding average 〈Non〉, τon, and τoff of 1289 ± 24, 51 ± 1 ms, and 2.51 ± 0.04 ms, respectively. The same analysis was performed for other bin-times, and the corresponding plots of 〈Non〉, τon, and τoff against the bin-time are shown in Fig. 6. Importantly, 〈Non〉 and τon exponentially decreased with reducing the bin-time in a range of 0.4–7 ms, although this decrease became more intense at bin-times below 0.6 ms. In the triplet blinking, the probability density of off-times increased exponentially with decreasing bin-times (Fig. 5f). These trends thus demonstrate that a considerable number of “off”-events, which are shorter than the selected bin-times, are still involved in on-time periods. Ideally, bin-time must be reduced until 〈Non〉 (or τon) becomes independent of this parameter. This, however, is practically unachievable in the histogram method because the influence of shot-noise becomes more remarkable at shorter bin-times, thereby causing artificial decreases in 〈Non〉 and τon. This effect is indeed visible below 600 μs bin-time (Fig. 6a and b). With the aim to avoid an arbitrary choice of bin-time and thus estimate a more reliable value for 〈Non〉 and τon, we herein adopted the extrapolated value (i.e., bin-time→0) obtained from the exponential fit of the 〈Non〉 or τonversus bin-time (Fig. 6a or b). For the single molecule of Fig. 5, the values of 〈Non〉 and τon were finally determined to be 874 ± 61 and 35.5 ms, respectively, which will be used in the estimation of ΦISC described below. In contrast to 〈Non〉 and τon, the value of τoff was bin-time independent and nearly constant within the experimental error over a bin-time range of 0.4 to 7 ms (Fig. 6c). For power-law blinking of single semiconductor quantum dots, Kuno and co-workers reported that the experimentally determined average off-time shows an apparent bin-time dependence because “averages” are not well defined for power-law kinetics.67 In triplet blinking, however, single exponential kinetics are dominated by a well-defined average value. Consequently, bin-time independent values are obtained for τoff in a wide range of bin-times. From now on, we will use τoff values obtained at 800 μs bin-time.
 | ||
| Fig. 5 (a) 800 μs binned fluorescence intensity trajectory of Fig. 4a, along with (b) the corresponding intensity histogram. (c) Fluorescence intensity trajectory of (a) from 99.0 to 99.3 s magnified to clearly display an on-time and off-time. Histograms of (d) on-counts, (e) on-time duration, and (f) off-time duration. The lines are single-exponential fits. (g) Intensity autocorrelation curve obtained from data of Fig.4a, together with corresponding single-exponential fit. | ||
 | ||
| Fig. 6 Bin-time dependence of (a) 〈Non〉, (b) τon, and (c) τoff. The lines in (a) and (b) represent single-exponential fits. The extrapolated values for 〈Non〉 and τon were 874 and 35.5 ms, respectively. | ||
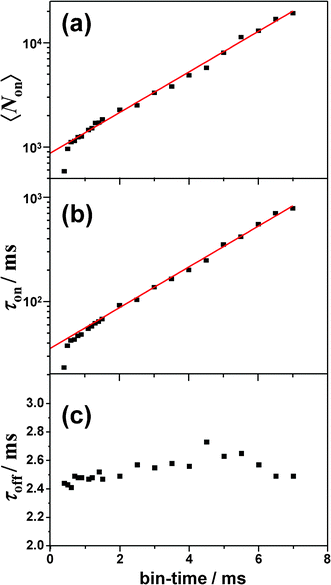
The exponential distribution of on-counts and on-/off-time indicated that ISC toward and from the triplet state was responsible for the photoblinking of BPEA.26–35 Based on the three-electronic-states (S0, S1, and T1) model to account for the triplet blinking of single molecules, the quantum yield of S1-T1 ISC can be estimated by the expression ΦISC = ξdet/〈Non〉, where ξdet (∼4%) refers to the total detection efficiency. We note that in this estimation, the internal conversion process (which does not create a photon) was not taken into consideration, and consequently, the number of S0→S1 transition events may be underestimated. Hence, the value thus obtained should be considered as an upper limit of the true ΦISC value. The triplet lifetime (τT) was directly obtained by an average off-time (τoff). The ISC yield and triplet lifetime for the single-molecule in Fig. 4 were found to be 4.6 × 10−5 and 2.5 ms, respectively. As shown in Fig. 5g, the analysis of the second-order correlation function g(2)(t) of the fluorescence intensity trajectory gave a comparable result of τon = 38.8 ms and τT = (τoff =) 2.4 ms, which were also obtained on the basis of the three-state model.26–28 It is worth noting that the value of τon obtained by the autocorrelation function (ACF) analysis was in good agreement with that determined by the extrapolation to zero bin-time in the histogram method (35.5 ms, see Fig. 6b). Similarly, ISC yields and triplet lifetimes of the single BPEA molecules in Zeonex were determined by both the histogram method and the ACF analysis, and their distributions are displayed in Fig. 7a and b. The triplet lifetime histograms obtained by both methods exhibited symmetric distributions, and the mean values of τT determined from Gaussian fits of the histograms are summarized in Table 1. The mean value obtained by the ACF analysis (2.8 ms) was found to be almost comparable to that obtained by the histogram method (2.7 ms). As can be seen in Fig. 7c, the ISC yields of single BPEA molecules exhibited a Poissonian-type distribution, and they were distributed in the 10−5–10−4 range, thereby quantitatively demonstrating, for the first time, very low ISC yields of BPEA. In the case of Φf ∼ 1 (ΦISC ≪ 1), the radiative rate constant (kr) can be approximated as the inverse of fluorescence lifetime (1/τf), and the rate constant of S1–T1 ISC (kISC) was estimated to be about 104 s−1 by using the relation of ΦISC/τf.
 | ||
| Fig. 7 Histograms of triplet lifetimes of 85 molecules obtained by using (a) histogram method and (b) autocorrelation analysis. (c) Histogram of ISC yields of 82 molecules. Note that 3 of 85 molecules yielded only few data points of on-counts, and they were thus excluded from the analysis. | ||
According to the three-state model,26–28τon is related to ΦISC through the expression 1/τon = kexcΦISC, where kexc represents the excitation rate. By using the extrapolated values of τon and ΦISC for a single-molecule in Fig. 5 (i.e., τon = 35.5 ms, ΦISC = 4.6 × 10−5) in this equation, kexc was calculated to be 6.1 × 105 s−1. We note that kexc was almost bin-time independent, i.e., (6.2 ± 0.5) × 105 s−1, although τon and ΦISC (or 〈Non〉) were found to exponentially depend on bin-time (Fig. 6). Since the absorption cross section of BPEA at 441 nm wavelength was 1.18 × 10−16 cm2, the excitation probability of BPEA, provided by the diffraction-limited focal spot (ca. 3.5 × 10−10 cm2) with a 0.2 μW laser power and a 10 MHz repetition rate, was calculated to be 0.015 per pulse. This indicates that the estimated excitation rate (6 × 105 s−1) is reasonable at 107 excitation pulses per second.
To gain an insight into the origin of the extremely low ΦISC value of BPEA, we performed density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. In this study, the hybrid B3PW91 functional68,69 and 6-31G(d) basis set were used for ground-state (S0) geometry optimization and predictions of the vertical transition energies from the optimized ground-state geometry to excited singlet and triplet states. This theoretical approach has been previously reported to reproduce well the experimental electronic excitation energies of BPEA.9 The geometry optimization for the lowest singlet excited state (S1) was also conducted at a time-dependent Hatree-Fock (TD-HF)/6-31G(d) level with no symmetry constraint.70 Harmonic vibrational frequencies of the optimized geometries for S0 and S1 states were also calculated to ensure that they represented true potential minima. All calculations were carried out using the GAUSSIAN 09 package.71 The optimized geometries for S0 and S1 were both found to conform to planar structures with a D2h symmetry. As mentioned above, the ensemble- and single-molecule fluorescence spectra of BPEA in Zeonex suggest that the planar D2h structure is always formed through conformational relaxation in S1, even though BPEA molecules in S0 take multiple conformations when embedded in this polymer. Therefore, the symmetry restriction of D2h regarding spin–orbit coupling (SOC) is operative in the following discussion. The vertical transition energies to excited singlet and triplet states, calculated at this optimized geometry, are shown in Fig. 8. The symmetries of S1 and T1 of BPEA were both B1u, thereby revealing no direct SOC between these two states. Consequently, the vibronically induced SOC via Herzberg–Teller vibronic coupling should play a predominant role in BPEA. Direct SOC of 1B1u is possible with 3B2u, 3B3u, or 3Au in D2h symmetry. Among them, only 3B2u (T5 and T8) exists in the vicinity of S1(1B1u), although their energy gaps are relatively large (0.8–1 eV). In addition, vibronic coupling is possible between 3B1u and 3B2u through b3g vibrational modes. Thus, the relative energetic position of 3B2u with respect to S1(1B1u)/T1(3B1u) becomes significant. In the case of anthracene, the S1(1B1u) and 3B2u states are nearly isoenergetic (less than 200 cm−1)72 so that anthracene has a moderate ISC yield of 0.3 (kISC ∼108 s−1).22 However, the present theoretical result suggests that 3B2u level of BPEA is located at ∼6000 cm−1 above S1(1B1u) [or ∼16000 cm−1 above T1(3B1u)], which is too high for effective mixing with S1, resulting in a considerably smaller SOC compared to anthracene. It is therefore likely that this large energy gap is responsible for the appreciably low ISC yield of BPEA.
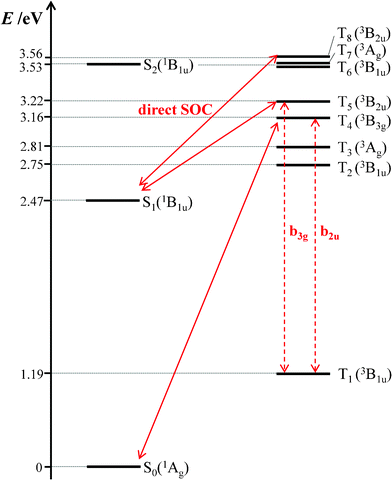 | ||
| Fig. 8 Energy level diagram of BPEA obtained by TD-DFT calculations with the B3PW91/6-31G(d) level of theory. In this diagram, the energy scale is not linear. Key singlet and triplet levels in S1→T1 and T1→S0 ISC processes, which can be coupled through direct spin–orbit coupling (SOC) and vibronic interactions, are connected by solid and dashed arrows, respectively. | ||
In contrast to the S1→T1 ISC process, the triplet lifetime of BPEA (2.7–2.8 ms) was shorter than that of anthracene (13 ms) measured in polymethylmethacrylate (PMMA) under degassed conditions.73 This result suggests that the T1→S0 ISC rate of BPEA is faster than that of anthracene. In the case of the T1→S0 ISC process, the 3B3g state should play a major role since it can couple with S0(1Ag) via direct SOC and with T1(3B1u) via vibronic coupling. As can be seen in Fig. 8, the energy gap between 3B3g and T1(3B1u) states was ca. 2 eV for BPEA, which is considerably larger than that of anthracene (∼0.5 eV),72 thereby suggesting a longer triplet lifetime for the former. With regard to this point, two remarks can be made. First, oxygen is known to dissolve better in less polar matrixes such as Zeonex films. Moreover, Zeonex has a relatively high permeability for oxygen.74 Consequently, it is possible that the triplet lifetime obtained under Ar atmosphere was still shortened due to the quenching effect produced by a trace amount of residual oxygen in the Zeonex film. Second, a shortening of the triplet lifetime with increasing laser power has been reported for several organic dyes, and this effect has been attributed to a reverse ISC process via higher excited triplet states (Tn→Sn).31,75,77 Then, we have examined the laser power dependence of the triplet lifetime of BPEA by changing the excitation intensity from 0.2 to 1.4 MW cm−2. However, we found that the triplet lifetime hardly decreased with the excitation intensity, thereby suggesting that the T1 state of BPEA does not appreciably absorb a 441 nm photon. In other words, the T1→Tn absorptions for BPEA would not largely overlap the S0→S1 absorptions at this excitation wavelength, which is compatible with the T1→Tn absorption spectrum recorded for a BPEA derivative in which no strong absorption band was observed around 440 nm.78
3.4 Photobleaching
With the aim of obtaining information on the effect of oxygen on photobleaching of BPEA, intensity trajectories were recorded both under air (i.e., high O2 concentration) and Ar gas flow conditions (i.e., low O2 concentration), and results are shown in Fig. 9a and b. Note that since the survival time until the molecules photobleach was remarkably shorter in air than in Ar, the excitation intensity was reduced to less than half of that used under an inert atmosphere. Irrespective of the lower excitation intensity applied, the survival time was still considerably shorter in air (mean value: 20 s) with respect to Ar atmosphere (135 s). Furthermore, as can be seen in Fig. 9b, the triplet blinking observed under pure Ar atmosphere was completely unresolved in the 10 ms-trajectory measured in air. This was also the case in trajectories with shorter bin-times (<1 ms). Since the occurrence of on/off blinking was discernible in intensity trajectories at bin-times up to ∼30τoff, the triplet lifetime in air was roughly estimated as less than 100 μs owing to fast quenching by O2. In this quenching process, the T1 state energy of BPEA (1.3–1.8 eV),9,76 can transfer to a neighbouring oxygen molecule, thus producing a highly reactive single oxygen O2(1Δg) whose transition energy is about 1 eV.22 As is well known, anthracene and its derivatives efficiently react with O2(1Δg) to form the corresponding endoperoxides.22–25 It is therefore very likely that the singlet oxygen formed through self-sensitization of BPEA oxidizes the same molecule and affords the endoperoxides, leading to terminal photochemical destruction of the molecule.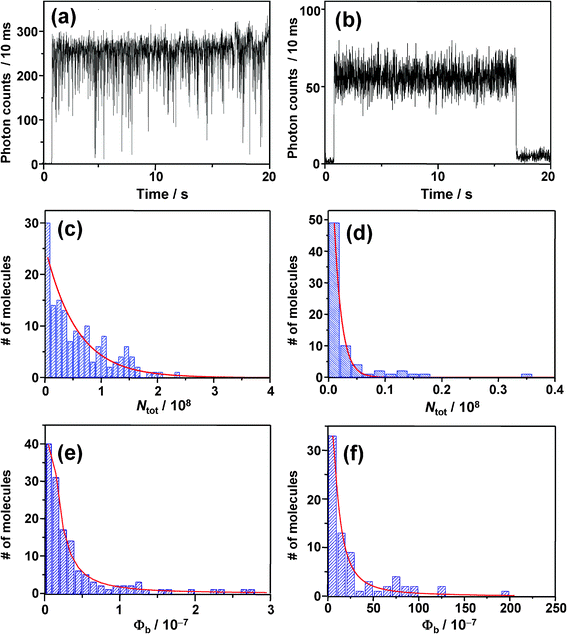 | ||
| Fig. 9 (a) and (b) Fluorescence intensity trajectories; (c) and (d) total number of photons emitted by single molecules and; (e) and (f) photobleaching quantum yields, obtained under Ar atmosphere (a, c, e) on left side and in air on right side (b, d, f). | ||
In order to gain quantitative insights into the photostability of BPEA, the photobleaching quantum yield Φb, defined as Φb = Φf/Ntot, was evaluated.79 In this expression, Φf represents the fluorescence quantum yield while Ntot refers to the total number of photons emitted by each single-molecule before photobleaching. Histograms of Ntot obtained under Ar and air atmospheres are shown in Fig. 9c and d, respectively. Both histograms displayed single exponential distributions with mean values 〈Ntot〉 of 5.6 × 107 and 1.3 × 106 photons under Ar and air atmospheres, respectively. Considering that other highly fluorescent organic dyes typically emit 104–107 photons before permanent photobleaching occurs,46 the photostability of BPEA is quite good. The photobleaching quantum yields of each single molecule can be subsequently calculated using the relationship mentioned above. Since the Φf values reported for BPEA in nonpolar solvents range within 0.85–1.0,8,12,19–21 a mean value of 0.93 was used in the calculation of Φb. The histograms of Φb under Ar and air atmospheres are displayed in Fig. 9e and f. The photostability markedly dropped after switching from Ar to ambient air (i.e., an increase in Φb of almost two orders of magnitude), indicating that photobleaching is indeed dominated by the surrounding oxygen concentration. Comparison of the present data with previous ones is problematic since the photobleaching yield is quite sensitive to the system itself and to experimental conditions such as excitation intensity and oxygen concentration. In particular, rhodamine dyes show Φb values on the order of 10−7–10−6 for a laser intensity below 1 kW cm−2, where the two-photon processes (S1→Sn and T1→Tn) are negligible.46 Even though a rather high laser intensity of ∼1 MW cm−2 was used in our case provided by ps-pulsed excitation, most of the single molecules of BPEA in the deoxygenated environment exhibited very low Φb values of 10−9–10−8 (Fig. 9e), thereby demonstrating that BPEA possesses a high degree of photostability. As mentioned above, photobleaching from excited states higher than the first excited singlet and triplet states are not likely to occur for BPEA. Because of such an “inert” photophysical property of BPEA, unexpected and complex photophysical phenomena can be avoided even under high excitation intensities, thereby making this molecule an excellent green fluorescent probe for SMS studies.
4. Conclusions
We used SMS to investigate the fluorescence blinking and photobleaching of BPEA embedded in a Zeonex polymer film. The triplet lifetimes and ISC yields for single BPEA molecules were determined using the histogram method and the autocorrelation analysis. The histogram method, in which the effect of the bin-time on the resultant average τon, τoff, and 〈Non〉 are examined, revealed a more reliable estimate of ISC yields for single molecules. We found very small ΦISC values (10−5–10−4) of BPEA, which were attributed to the large energy gap between 3B2u and S1(1B1u)/T1(3B1u) states based on the results of the TD-DFT calculation. The results of the present study also showed that BPEA combines a moderate fluorescence photon count rate with a high photostability (i.e., very low Φb values of 10−9–10−8) at room temperature. All these properties of BPEA make this molecule a promising fluorescent probe for SMS studies and a single-photon source. It is expected that a superior long-term photostability of BPEA can be achieved by further reduction in the oxygen concentration in the polymer matrix. Such a strategy was recently reported for single molecules of dibenzoterrylene in rigorously oxygen-free polymers.55 Alternatively, photostability can be further enhanced by providing physical insulation to the molecules by means of crystalline host matrices50,54 and molecular capsules. We are currently exploring the photostability of BPEA derivatives protected by a self-assembled molecular capsule.Acknowledgements
The authors are grateful to Prof. Hisao Murai from Shizuoka University for loan of a fluorescence spectrometer. Takeo Saito from Shimadzu Corporation is acknowledged grateful for experimental work with AFM. Theoretical calculations were partly performed using Research Center for Computational Science, Okazaki, Japan. This work is partly supported by Grant-in-Aids for Young Scientists (A), No. 20685001, Scientific Research on Priority Areas “Molecular Science for Supra Functional Systems”, No. 20050011, and Challenging Exploratory Research, No.23655007.References
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171 CrossRef CAS.
- A. Ponnu and E. V. Anslyn, Supramol. Chem., 2010, 22, 65 CrossRef CAS.
- M. P. Heitz and M. Maroncelli, J. Phys. Chem. A, 1997, 101, 5852 CrossRef CAS.
- M. T. Cicerone and M. D. Ediger, J. Chem. Phys., 1996, 104, 7210 CrossRef CAS.
- F. Luschtinetz and C. Dosche, J. Colloid Interface Sci., 2009, 338, 312 CrossRef CAS.
- C. Wang and M. D. Ediger, Macromolecules, 1997, 30, 4770 CrossRef CAS.
- M. V. Suneesh, C. V. Vinayak and K. R. Gopidas, J. Phys. Chem. C, 2010, 114, 18735 Search PubMed.
- M. Levitus and M. A. Garcia-Garibay, J. Phys. Chem. A, 2000, 104, 8632 CrossRef CAS.
- A. V. Kukhta, I. N. Kukhta, S. M. Kazakov, O. V. Khristophorov and O. L. Neyra, J. Chem. Phys., 2007, 127, 084316 CrossRef CAS.
- A. Zhu, J. O. White and H. G. Drickamer, J. Phys. Chem. A, 2002, 106, 9209 CrossRef CAS.
- J. C. Ribierre, A. Ruseckas, H. Cavaye, H. S. Barcena, P. L. Burn and I. D. W. Samuel, J. Phys. Chem. A, 2011, 115, 7401 CrossRef CAS.
- A. Beeby, K. S. Findlay, A. E. Goeta, L. Porrès, S. R. Rutter and A. L. Thompson, Photochem. Photobiol. Sci., 2007, 6, 982 CAS.
- T. M. Swager, D. J. Gil and M. S. Wrighton, J. Phys. Chem., 1995, 99, 4886 CrossRef CAS.
- C. Wang, Y. Liu, Z. Wei, H. Li, W. Xu and W. Hu, Appl. Phys. Lett., 2010, 96, 143302 CrossRef.
- B. Li, W. Miao and L. Cheng, Dyes Pigm., 2000, 46, 81 CrossRef CAS.
- Y. S. Zhao, J. Xu, A. Peng, H. Fu, Y. Ma, L. Jiang and J. Yao, Angew. Chem., Int. Ed., 2008, 47, 7301 CrossRef CAS.
- L. Valentini, D. Bagnis, A. Marrocchi, M. Seri, A. Taticchi and J. M. Kenney, Chem. Mater., 2008, 20, 32 CrossRef CAS.
- J. Reichert, R. Ochs, D. Beckmann, H. B. Weber, M. Mayor and H. v. Löhneysen, Phys. Rev. Lett., 2002, 88, 176804 CrossRef CAS.
- P. J. Hanhela and D. B. Paul, Aust. J. Chem., 1984, 37, 553 CrossRef CAS.
- C. A. Heller, R. A. Henry, B. A. McLaughlin and D. E. Bliss, J. Chem. Eng. Data, 1974, 19, 214 CrossRef CAS.
- D. R. Maulding and B. G. Roberts, J. Org. Chem., 1969, 34, 1734 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, California, 2010 Search PubMed.
- W. Fudickar and T. Linker, Chem. Commun., 2008, 1771 RSC.
- R. C. Olivares, G. Günther, A. L. Zanocco and E. Lemp, J. Photochem. Photobiol., A, 2009, 207, 160 CrossRef.
- J. -M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668 CrossRef CAS.
- T. Ha, Th. Enderle, D. S. Chemla, P. R. Selvin and S. Weiss, Chem. Phys. Lett., 1997, 271, 1 CrossRef CAS.
- W.-T. Yip, D. Hu, J. Yu, D. A. V. Bout and P. F. Barbara, J. Phys. Chem. A, 1998, 102, 7564 CrossRef CAS.
- K. D. Weston, P. J. Carson, J. A. DeAro and S. K. Buratto, Chem. Phys. Lett., 1999, 308, 58 CrossRef CAS.
- J. A. Veerman, M. F. G. Garcia-Parajo, L. Kuipers and N. F. v. Hulst, Phys. Rev. Lett., 1999, 83, 2155 CrossRef CAS.
- D. S. English, A. Furube and P. F. Barbara, Chem. Phys. Lett., 2000, 324, 15 CrossRef CAS.
- D. S. English, E. J. Harbron and P. F. Barbara, J. Phys. Chem. A, 2000, 104, 9057 CrossRef CAS.
- T. Vosch, J. Hofkens, M. Cotlet, F. Köhn, H. Fujiwara, R. Gronheid, K. V. D. Biest, T. Weil, A. Herrmann, K. Müllen, S. Mukamel, M. V. d. Auweraer and F. C. De Schryver, Angew. Chem., Int. Ed., 2001, 40, 4643 CrossRef CAS.
- C. G. Hübner, A. Renn, I. Renge and U. P. Wild, J. Chem. Phys., 2001, 115, 9619 CrossRef.
- Y. Hou and D. A. Higgins, J. Phys. Chem. B, 2002, 106, 10306 CrossRef CAS.
- M. Haase, C. G. Hübner, E. Reuther, A. Herrmann, K. Müllen and T. Baschè, J. Phys. Chem. B, 2004, 108, 10445 CrossRef CAS.
- F. Köhn, J. Hofkens, R. Gronheid, M. V. d. Auweraer and F. C. De Schryver, J. Phys. Chem. A, 2002, 106, 4808 CrossRef.
- E. J. G. Peterman, S. Brasselet and W. E. Moerner, J. Phys. Chem. A, 1999, 103, 10553 CrossRef CAS.
- R. Liu, M. W. Holman, L. Zang and D. M. Adams, J. Phys. Chem. A, 2003, 107, 6522 CrossRef CAS.
- J. N. Clifford, T. D. M. Bell, P. Tinnefeld, M. Heilemann, S. M. Melnikov, J. Hotta, M. Sliwa, P. Dedecker, M. Sauer, J. Hofkens and E. K. L. Yeow, J. Phys. Chem. B, 2007, 111, 6987 CrossRef CAS.
- E. K. L. Yeow, S. M. Melnikov, T. D. M. Bell, F. C. D. Schryver and J. Hofkens, J. Phys. Chem. A, 2006, 110, 1726 CrossRef CAS.
- J. P. Hoogenboom, J. Hernando, E. van Dijk, N. F. van Hulst and M. F. Garcia-Parajo, ChemPhysChem, 2007, 8, 823 CrossRef CAS.
- M. Haase, C. G. Hübner, F. Nolde, K. Müllen and T. Baschè, Phys. Chem. Chem. Phys., 2011, 13, 1776 RSC.
- T. Christ, F. Kulzer, P. Bordat and T. Baschè, Angew. Chem., Int. Ed., 2001, 40, 4192 CrossRef CAS.
- T. Gensch, M. Böhmer and P. F. Aramendía, J. Phys. Chem. A, 2005, 109, 6652 CrossRef CAS.
- W. Göhde Jr., U. C. Fischer, H. Fuchs, J. Tittel, T. Baschè, Ch. Bräuchle, A. Herrmann and K. Müllen, J. Phys. Chem. A, 1998, 102, 9109 CrossRef.
- C. Eggeling, J. Widengren, R. Rigler and C. A. M. Seidel, Anal. Chem., 1998, 70, 2651 CrossRef CAS.
- J. P. Hoogenboom, E. M. H. P. van Dijk, J. Hernando, N. F. van Hulst and M. F. Garcia-Parajo, Phys. Rev. Lett., 2005, 95, 097401 CrossRef.
- C. R. Viteri, J. W. Gilliland and W. T. Yip, J. Am. Chem. Soc., 2003, 125, 1980 CrossRef CAS.
- M. Vacha, Y. Koide, M. Kotani and H. Sato, Chem. Phys. Lett., 2004, 388, 263 CrossRef CAS.
- R. J. Pfab, J. Zimmermann, C. Hettich, I. Gerhardt, A. Renn and V. Sandoghdar, Chem. Phys. Lett., 2004, 387, 490 CrossRef CAS.
- H. Piwoński, R. Kołos, A. Meixner and J. Sepioł, Chem. Phys. Lett., 2005, 405, 352 CrossRef.
- C. Julien, A. Débarre, D. Nutarelli, A. Richard and P. Tchénio, J. Phys. Chem. B, 2005, 109, 23145 CrossRef CAS.
- M. Banasiewicz, D. Wiącek and B. Kozankiewicz, Chem. Phys. Lett., 2006, 425, 289 CrossRef CAS.
- C. Toninelli, K. Early, J. Bremi, A. Renn, S. Goetzinger and V. Sandoghdar, Opt. Express, 2010, 18, 6577 CrossRef CAS.
- M. Nothaft, S. Höhla, F. Jelezko, J. Pflaum and J. Wrachtrup, Phys. Status Solidi B, 2012, 249, 661 CrossRef CAS.
- L. Zhang, S. Aite and Z. Yu, Rev. Sci. Instrum., 2007, 78, 083701 CrossRef.
- H. P. Lu and X. S. Xie, Nature, 1997, 385, 143 CrossRef CAS.
- M. Maus, M. Cotlet, J. Hofkens, T. Gensch and F. C. De Schryver, Anal. Chem., 2001, 73, 2078 CrossRef CAS.
- F. Stracke, C. Blum, S. Becker, K. Müllen and A. J. Meixner, Chem. Phys. Lett., 2000, 325, 196 CrossRef CAS.
- M. Lippitz, C. G. Hübner, T. Christ, H. Eichner, P. Bordat, A. Herrmann, K. Müllen and T. Baschè, Phys. Rev. Lett., 2004, 92, 103001 CrossRef.
- M. Cotlet, S. Masuo, G. Luo, J. Hofkens, M. van der Auweraer, J. Verhoeven, K. Müllen, X. S. Xie and F. D. Schryver, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14343 CrossRef CAS.
- H. Yoo, J. Yang, A. Yousef, M. R. Wasielewski and D. Kim, J. Am. Chem. Soc., 2010, 132, 3939 CrossRef CAS.
- D. Wöll, E. Braeken, A. Deres, F. C. D. Schryver, H. Uji-I and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313 RSC.
- R. A. L. Vallée, N. Tomczak, L. Kuipers, G. J. Vancso and N. F. van Hulst, Phys. Rev. Lett., 2003, 91, 038301 CrossRef.
- R. A. L. Vallée, M. van der Auweraer, F. C. D. Schryver, D. Beljonne and M. Orrit, ChemPhysChem, 2005, 6, 81 CrossRef.
- M. Lippitz, F. Kulzer and M. Orrit, ChemPhysChem, 2005, 6, 770 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter, 1996, 54, 16533 CrossRef CAS.
- C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., , J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- N. Nijegorodov, V. Ramachandran and D. P. Winkoun, Spectrochim. Acta, Part A, 1997, 53, 1813 CrossRef.
- W. H. Melhuish and R. Hardwick, Trans. Faraday Soc., 1962, 58, 1908 RSC.
- T. Vosch, M. Cotlet, J. Hofkens, K. V. D. Biest, M. Lor, K. Weston, P. Tinnefeld, M. Sauer, L. Latterini, K. Müllen and F. C. De Schryver, J. Phys. Chem. A, 2003, 107, 6920 CrossRef CAS.
- L. Fleury, J. M. Segura, G. Zumofen, B. Hecht and U. P. Wild, Phys. Rev. Lett., 2000, 84, 1148 CrossRef CAS.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171 CrossRef CAS.
- P. Tinnefeld, V. Buschmann, K. D. Weston and M. Sauer, J. Phys. Chem. A, 2003, 107, 323 CrossRef CAS.
- C. V. Suneesh and K. R. Gopidas, J. Phys. Chem. C, 2009, 113, 1606 CAS.
- W. P. Ambrose, P. M. Goodwin, J. C. Martin and R. A. Keller, Phys. Rev. Lett., 1994, 72, 160 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |