An expedient approach to highly enantioenriched cyclic nitrones mediated by robust and recoverable C3-symmetric cinchonine-squaramide catalysts†
Xin
Han
a,
Xiangfei
Wu
a,
Chang
Min
a,
Hai-Bing
Zhou
ab and
Chune
Dong
*ab
aLaboratory of Combinatorial Biosynthesis and Drug Discovery, Ministry of Education, Wuhan University School of Pharmaceutical Sciences, Wuhan, 430071, China
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China. E-mail: cdong@whu.edu.cn; Tel: 86-2768759586
First published on 13th June 2012
Abstract
The C3-symmetric cinchonine-squaramide catalyzed asymmetric Michael addition of β-ketosulfones to nitroalkenes is presented. Subsequent transformation leads to chiral cyclic nitrones with excellent results (up to 85% yield and >99% ee). The catalyst can be recovered and reused for six cycles without losing activity and selectivity.
Introduction
The catalytic asymmetric Michael addition of sulfones to nitroalkenes represents a prime approach toward chiral nitroalkanes or cyclic nitrones bearing sulfone functionality in organic synthesis,1,2 which are extremely versatile building blocks for the construction of numerous natural products and pharmaceuticals as well.3–6 As a consequence of this, many different catalytic enantioselective versions of this fundamental transformation have been reported.7,8 Among them, chiral cinchona alkaloids have provided an alternative way to reach this goal. Recently, Mancheño and co-workers reported thiourea cinchona alkaloid catalyzed Michael addition of ketosulfones to nitroalkenes followed by reductive cyclization to form the chiral cyclic nitrones in good yields and high enantioselectivity.9 However, the existing catalytic system is still limited and the enantioselectivity decreased dramatically when substituted nitrostryrenes were employed, in many cases, further crystallization was required in order to achieve high enantioselectivity. Due to the difficulty in catalyst recycling, the application of the catalytic system in pharmaceutical production was precluded. Accordingly, the development of a practical, highly efficient and recyclable chiral catalyst for this type of Michael addition reaction is particularly attractive.On the other hand, C3-symmetric chiral molecules have recently emerged as powerful organocatalysts in several asymmetric transformations,10–13 as well as its application in the areas of molecular recognition and material sciences.14,15 Due to its unique chiral environment in asymmetric transformations, the C3-symmetric chiral molecule has great potential to explore. In consideration of the excellent performance of squaramide organocatalysts, pioneered by Rawal's group and later exploited by other groups,16,17 we recently developed a novel C3-symmetric cinchonine-squaramide for asymmetric Michael addition of 1,3-dicarbonyl compounds to nitroalkenes, high yields with excellent enantioselectivities and diastereoselectivities were achived.18a During our ongoing study on novel C3-symmetric squaramide catalyzed asymmetric transformations,18 we envisioned the possibility of applying these chiral C3-symmetric squaramide catalysts to develop a highly enantioselective conjugate addition of β-ketosulfones to nitroalkenes. Also, the poor solubility of the C3-squaramide in organic solvents enable its easy recovery by a simple precipitation method, allowing its operational recycling. Moreover, there is no report on the C3-symmetric squaramide catalyzed conjugate addition of ketosulfones so far. In our work, we discovered that the application of readily available C3-symmetric cinchonine-squaramide results in a drastic improvement in enantioselectivity, providing a straightforward protocol for a direct synthesis of optical pure nitrones from enantioselective Michael addition of β-ketosulfones to nitroalkenes.
Herein, we describe the first C3-symmetric cinchonine-squaramides as a highly efficient, recyclable robust catalyst for the enantioselective addition of β-ketosulfones to nitroalkenes, which afforded valuable chiral nitrones in satisfactory yields and excellent enantioselectivities (up to >99% ee).
Results and discussion
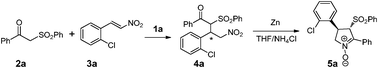
First, we initiated the conjugate addition of nitroalkene 2a to β-ketosulfone 3a by employing 1 as the catalyst. Some of the key findings are illustrated in Table 1. Gratifyingly, in all cases, the Michael addition reaction proceeded smoothly to generate adduct 4a in almost quantitative conversion and a moderate diastereomeric ratio was obtained (d.r. = 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :37), which was selectively transformed into chiral nitrone by Zn/NH4Cl in THF to provide the desired trans nitrone 5a as single diastereoisomer (d.r. > 99:1).9 The high diastereomeric ratio of nitrones 5 observed might be attributable to the easy enolization of the α-position of the ketone and the obtainment of the most stable trans compound. The data matched well with previous literature.9 With 5 mol% of 1a as the catalyst at room temperature, various solvents have been examined for this reaction (Table 1, entries 4–9). Except for DMF, the asymmetric Michael addition could be carried out smoothly in several conventional solvents such as methylene chloride (98% ee), benzene (97% ee) and toluene (95% ee). The use of THF resulted in the highest enantioselectivity of >99% ee (Table 1, entry 6). Hence, THF was determined to be the most appropriate solvent for this study. Lowering the reaction temperature to −20 °C resulted in an obvious erosion of the enantioselectivity of the reaction (Table 1, entry 5). Also, adjusting the catalyst loading demonstrated a great influence on the enantioselectivity of the reaction. The use of 1 mol% of catalyst gave the desired product 5a in only 92% ee (entry 3). For comparison, bissquaramide 1b and monosquaramide 1c were also employed under the same reaction condition. Both catalysts 1b and 1c led to the same enantiomer as 1a gave, however, lower ees were noted (entries 10–11). With respect to enantioselectivity, further screening indicated that the reaction conducted in THF at room temperature in the presence of 5 mol% 1a offered the best result. Furthermore, the C3-symmetric cinchonidine-squaramide 1d, prepared from cinchonidine, induced high enantioselectivity, giving the product with opposite configuration in 60% yield and 96% ee (entry 12).
:37), which was selectively transformed into chiral nitrone by Zn/NH4Cl in THF to provide the desired trans nitrone 5a as single diastereoisomer (d.r. > 99:1).9 The high diastereomeric ratio of nitrones 5 observed might be attributable to the easy enolization of the α-position of the ketone and the obtainment of the most stable trans compound. The data matched well with previous literature.9 With 5 mol% of 1a as the catalyst at room temperature, various solvents have been examined for this reaction (Table 1, entries 4–9). Except for DMF, the asymmetric Michael addition could be carried out smoothly in several conventional solvents such as methylene chloride (98% ee), benzene (97% ee) and toluene (95% ee). The use of THF resulted in the highest enantioselectivity of >99% ee (Table 1, entry 6). Hence, THF was determined to be the most appropriate solvent for this study. Lowering the reaction temperature to −20 °C resulted in an obvious erosion of the enantioselectivity of the reaction (Table 1, entry 5). Also, adjusting the catalyst loading demonstrated a great influence on the enantioselectivity of the reaction. The use of 1 mol% of catalyst gave the desired product 5a in only 92% ee (entry 3). For comparison, bissquaramide 1b and monosquaramide 1c were also employed under the same reaction condition. Both catalysts 1b and 1c led to the same enantiomer as 1a gave, however, lower ees were noted (entries 10–11). With respect to enantioselectivity, further screening indicated that the reaction conducted in THF at room temperature in the presence of 5 mol% 1a offered the best result. Furthermore, the C3-symmetric cinchonidine-squaramide 1d, prepared from cinchonidine, induced high enantioselectivity, giving the product with opposite configuration in 60% yield and 96% ee (entry 12).

| Entry | Catalyst (mol%) | Solvent | T (°C) | Yield 5a (%)b | ee (%)c |
|---|---|---|---|---|---|
|
a Reaction conditions: 0.2 mmol of 2a, 0.4 mmol of 3a, indicated amount of catalyst in 1.0 mL of appropriate solvent for 24 h. The adduct 4a was reduced by zinc (70 equiv.) in THF/NH4Cl (1:1) at rt for 1.5 h.
b Isolated yield of 5a after two steps.
c ee values were determined by HPLC analysis on nitrone compound 5a (3R, 4S) using a chiralpac AD column.
d Opposite configuration was observed.
|
|||||
| 1 | 5 (1a) | CH2Cl2 | rt | 66 | 98 |
| 2 | 2.5 (1a) | CH2Cl2 | rt | 67 | 96 |
| 3 | 1 (1a) | CH2Cl2 | rt | 73 | 92 |
| 4 | 5 (1a) | CH2Cl2 | 0 | 65 | 98 |
| 5 | 5 (1a) | CH2Cl2 | −20 | 68 | 91 |
| 6 | 5 (1a) | THF | rt | 76 | >99 |
| 7 | 5 (1a) | C6H6 | rt | 71 | 97 |
| 8 | 5 (1a) | Toluene | rt | 61 | 95 |
| 9 | 5 (1a) | DMF | rt | 65 | 81 |
| 10 | 5 (1b) | THF | rt | 63 | 88 |
| 11 | 5 (1c) | THF | rt | 61 | 90 |
| 12 | 5 (1d) | THF | rt | 60 | 96d |
Encouraged by these results, we next probed the scope of the reaction with a variety of nitro olefins and different β-ketosulfones with 5 mol% of C3 catalyst 1a in THF at room temperature. The results are summarized in Table 2. Generally, the Michael addition reaction catalyzed by 1a underwent cleanly to give adduct 4 in full conversion, followed by reduction, the desired cyclic nitrones 5 were furnished in up to 85% overall yield and >99% ee. As illustrated in Table 2, nitroalkenes bearing electron-withdrawing groups on the aryl rings reacted smoothly with ketosulfone 2a, exclusively affording the corresponding products in excellent ee values (>99%) (Table 2, entries 1–4). In addition, alkylketosulfone 2b was also evaluated, giving chiral nitrones 5e and 5h in 98% ee and 86% ee, respectively (entries 5 and 8). Under the same reduction conditions, nitrone 5l was afforded in 83% ee (entry 14). With respect to alkyl-substituted ketosulfone, somewhat higher enantionselectivity was achieved when xylene was used as a solvent. For example, in the case of 2b, changing the solvent from THF to xylene resulted in an obvious improved enantioselectivity (entry 9, 80% ee vs. entry 8, 86% ee). Furthermore, the reaction of 2a with nitrostyrene in xylene gave the desired product in >99% ee (entry 2). Interestingly, heteroaromatic nitroalkene was also tolerated, leading to the corresponding product 5i in 70% yield and 82% ee (entry 10). Alkyl nitroalkenes generally were less active in this kind of asymmetric transformation. We found that in our case, the alkyl nitroalkene was also tolerated compared to its aryl congeners. For example, when 1-nitropent-1-ene was used in this kind of asymmetric transformation, the desired product was obtained in good enantioselectivities (91% ee) (entry 15).

| Entry | R1 | R2 | R3 | Yield of 5 (%)b | ee (%)ce |
|---|---|---|---|---|---|
|
a Unless otherwise noted, the reactions were carried out with 0.2 mmol of ketosulfone 2, 0.4 mmol of nitroalkene 3 in 1.0 mL of THF in the presence of 0.01 mmol of 1a at rt for 24 h. The adduct 4 was reduced by zinc (70 equiv.) in THF/NH4Cl (1:1) at rt, 1.5 h. The nitrones 5 (3R, 4S) were formed as single diastereoisomers.
b Isolated yield of 5 after two steps.
c ee values were determined by HPLC analysis on nitrone compounds 5 using a chiralpac IB-H or AD-H column.
d Xylene was used as solvent.
e Absolute configuration was assigned by the comparison of their CD spectrum with that of analogues (5f, 5h, 5k) or optical rotation with that reported in the literature9 (5a–e, 5i–m).
f The absolute configuration was not determined.
g Catalyzed by 1d, the product was obtained with opposite configuration.
|
|||||
| 1 | Ph | Ph | o-ClC6H4 | 5a 66 | >99 |
| 2d | Ph | Ph | Ph | 5b 65 | >99 |
| 3 | Ph | Ph | p-ClC6H4 | 5c 65 | >99 |
| 4 | Ph | Ph | p-FC6H4 | 5d 70 | >99 |
| 5 | Me | p-MeC6H4 | 2,4-(Cl)2C6H3 | 5e f 69 | 98 |
| 6 | Ph | Ph | 2,4-(Cl)2C6H3 | 5f 70 | 90 |
| 7 | Me | Ph | 2,4-(Cl)2C6H3 | 5g f 64 | 90 |
| 8d | Me | p-MeC6H4 | o-ClC6H4 | 5h 65 | 86 |
| 9 | Me | p-MeC6H4 | o-ClC6H4 | 5h 65 | 80 |
| 10 | Ph | Ph | 2-Thienyl | 5i 70 | 82 |
| 11 | Ph | Ph | 3,5-(CF3)2C6H3 | 5j 85 | 80 |
| 12d | Me | Ph | o-ClC6H4 | 5k 70 | 86 |
| 13 | Me | Ph | o-ClC6H4 | 5k 62 | 86g |
| 14 | Me | p-MeC6H4 | Ph | 5l 50 | 83 |
| 15 | Ph | Ph | nPr | 5m 56 | 91 |
The poor solubility of our C3-symmetric catalyst 1a in organic solvents enabled its easy recovery. Therefore, to determine the recycling ability of catalyst, 1a was recovered after the catalytic process by simple precipitation from the reaction mixture with the addition of ethyl ether and then was reused in the Michael addition of 2a with 3a under the optimized reaction conditions (Table 3). As summarized in Table 3, recycled 1a was carried through at least six runs of Michael addition that simply involved transfer of the recovered catalyst to a new reaction vessel followed by the addition of substrate and solvent. It is noteworthy that in each case the catalyst was recovered in high yield (89–92%) after the reaction was completed and maintained its catalytic activity even after six cycles (95–99% ee).
| Cycle no. | Recovery rate (%) | Yield 5a (%)b | ee (%)c |
|---|---|---|---|
| a All reactions were carried out with 0.75 mmol 2a, 1.5 mmol 3a in 5 mL of THF in the presence of 5 mol% 1a for 24 h. b Isolated yield. c ee values were determined by HPLC analysis on 5a using a chiralpac AD column. | |||
| 1 | — | 76 | >99 |
| 2 | 92 | 71 | 98 |
| 3 | 89 | 73 | 96 |
| 4 | 90 | 65 | 97 |
| 5 | 91 | 69 | 95 |
| 6 | 91 | 70 | 96 |
Conclusions
A C3-symmetric cinchonine-squaramide 1a was identified as the best catalyst for the asymmetric Michael addition of nitroalkenes to ketosulfones. By comparison with previous reports, the present work offers several distinct improvements: (1) the C3-symmetric cinchonine-squaramide can promote this Michael addition reaction leading to chiral cyclic nitrones in good yields and excellent enantioselectivities. (2) Wider scope of the substrates concerning both nitroalkenes and ketosulfones were examined. In particular, p-fluoro and p-chloro substituted nitroalkenes provided only one enantiomer without further crystallization. (3) The C3-symmetric catalyst 1a can be recovered and reused for six times without loss the activity and selectivity.Experimental section
General information
Analytical-grade solvents and β-ketosulfone were purchased, and used as received. Nitroalkenes were prepared by a nitro aldol/dehydration reaction between nitromethane and the aldehyde.19 Catalysts 1a–c were obtained in a three-step synthesis from commercially available cinchonine and squaric acid.18a Racemic samples were prepared with 1,4-diazobicyclo-[2.2.2] octane (DABCO) as the catalyst. 5a–d and 5l–m were prepared as described previously.9 NMR spectra were measured at 400 MHz for 1H spectra and 100 MHz for 13C spectra and calibrated from residual solvent signal. Enantiomeric excesses (ee) were determined by HPLC analysis with Daicel Chiralpak AD-H, IB, or Chiralcel OD-H columns, as indicated. Column chromatography purification was performed using 230–400 mesh silica gel.:petroleum = 1:2) to give nitrone 5.
(3R, 4S)-3-(2-Chlorophenyl)-5-phenyl-4-(phenylsulfonyl)-3,4-di-hydro-2H-pyrroline-1-oxide (5a): [α]20D = + 8.1 (c = 0.3, CHCl3), 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.2 Hz, 2H), 7.74 (d, J = 7.3 Hz, 2H), 7.57 (t, 1H), 7.45–7.37 (m, 3H), 7.30–7.25 (m, 3H), 7.19 (m, 2H), 7.10 (d, J = 4 Hz, 1H), 4.96 (s, 1H), 4.78 (m, 2H), 4.02 (dt, 1H). 13C NMR (100 MHz, CDCl3) δ: 137.9, 137.1, 134.5, 133.2, 130.4, 129.5, 129.3, 129.1, 128.2, 128.0, 127.7, 127.4, 126.8, 75.0, 69.7, 35.0. HPLC analysis on a Chiralpack AD column (15% i-PrOH in hexanes; flow rate = 0.9 mL min−1; λ = 220 nm; t minor = 65.5 min, t major = 70.5 min, ee > 99%).
(3R, 4S)-3,5-Diphenyl-4-(phenylsulfonyl)-3,4-dihydro-2H-pyrro-line-1-oxide (5b): [α]20D = + 16.0 (c = 0.2, CHCl3), 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.54 (d, J = 7.6 Hz, 1H), 7.38–7.18 (m, 10H), 4.86 (s, 1H), 4.68 (dd, J = 8.0 Hz, 1H), 4.28 (d, J = 8.0 Hz, 1H), 4.14 (bd, J = 12 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 141.1, 136.9, 133.5, 130.2, 129.6, 129.2, 129.0, 128.8, 128.2, 128.1, 127.4, 126.1, 69.6, 37.7, 29.6. HPLC analysis on a Chiralpack IB-H column (10% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ = 220 nm; t minor = 34.8 min, t major = 40.4 min, ee > 99%).
(3R, 4S)-3-(4-Chlorophenyl)-5-phenyl-4-(phenylsulfonyl)-3,4-di-hydro-2H-pyrroline-1-oxide (5c): [α]20D = −1.5 (c 0.1, CH2Cl2), 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8 Hz, 2H), 7.67 (d, J = 4 Hz, 2H), 7.55 (t, J = 8 Hz, 1H), 7.37–7.20 (m, 7H), 7.14 (d, J = 8.0 Hz, 2H), 4.80 (s, 1H), 4.69 (d, J = 8.0 Hz, 1H), 4.28 (d, J = 8 Hz, 1H), 4.09 (br, J = 16 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 139.4, 136.8, 134.3, 129.8, 129.3, 129.0, 128.2, 127.6, 127.4, 69.9, 37.2, 28.9. HPLC analysis on a Chiralpack IB-H column (10% i-PrOH in hexanes; flow rate = 1 mL min−1; λ = 220 nm; t major = 37.5 min, t minor = 52.2 min, ee > 99%).
(3R, 4S)-3-(4-Fluorophenyl)-5-phenyl-4-(phenylsulfonyl)-3,4-di-hydro-2H-pyrroline-1-oxide (5d): [α]20D = −2.5 (c 0.1, CH2Cl2), 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 4 Hz, 2H), 7.67 (d, J = 8 Hz, 2H), 7.54 (t, J = 8.0 Hz, 1H), 7.38–7.15 (m, 7H), 7.02 (t, J = 8.0 Hz, 2H), 4.81 (s, 1H), 4.69 (dd, J = 8.0 Hz, 1H), 4.29 (d, J = 8.0 Hz, 1H), 4.09 (dd, J = 8, 16 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 163.8, 160.9, 136.8, 134.5, 133.7, 130.3, 129.2, 129.0, 128.2, 128.0, 127.9, 127.5, 127.4, 116.7, 116.4, 76.7, 70.0, 37.1. HPLC analysis on a Chiralpack IB column (10% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ = 220 nm; t minor = 36.0 min, t major = 46.5 min, ee > 99%).
3-[2,4-Bis(chloro)phenyl]-5-phenyl-4-(4-tolysulfonyl)-3,4-dihyd-ro-2H-pyrroline-1-oxide (5e): [α]20D = −7.69 (c 0.26, EtOAc), 1H NMR (400 MHz, CDCl3) δ 7.80 (br, 2H), 7.45 (d, J = 8.0 Hz, 1H), 7.37–6.99 (m, 3H), 6.97 (dd, J = 8.0 Hz, 1H), 4.24 (br, 1H), 4.13–4.12 (m, 2H), 3.80–3.70 (m, 1H), 2.49 (s, 3H), 2.37 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 136.7, 135.0, 130.2, 129.9, 129.8, 129.7, 129.1, 129.0, 128.9, 128.2, 127.7, 71.6, 66.7, 31.9, 22.6. HPLC analysis on a Chiralpack AD-H column (10% i-PrOH in hexanes; flow rate = 1 mL min−1; λ = 220 nm; t major = 25.6 min, t minor = 31.4 min, ee = 98%). HRMS Calcd for C18H17Cl2NO3S: 398.0373 Found: 398.0379.
3-[2,4-Bis(chloro)phenyl]-5-phenyl-4-(phenylsulfonyl)-3,4-dihy-dro-2H-pyrroline-1-oxide (5f): [α]20D = + 3.86 (c = 0.44, EtOAc), 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 4.0 Hz, 2H), 7.57 (t, J = 8.0 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.39 (t, J = 7.8 Hz, 2H), 7.32 (dd, J = 15.8, 8.6 Hz, 3H), 7.19 (dd, J = 8.4, 2.0 Hz, 1H), 7.03 (d, J = 8.0 Hz, 1H), 4.88 (s, 1H), 4.78 (dd, J = 8.0 Hz, 1H), 4.70 (d, J = 8.0 Hz, 1H), 4.00 (d, J = 12.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 134.6, 130.5, 130.2, 129.3, 129.1, 128.4, 128.3, 127.7, 127.3, 74.9, 69.4, 34.7. HPLC analysis on a Chiralpack IB-H column (10% i-PrOH in hexanes; flow rate = 1 mL min−1; λ = 220 nm; t major = 34.6min, t minor = 46.1 min, ee = 90%). HRMS Calcd for C17H15Cl2NO3S: 384.0221 Found: 384.0222.
3-[2,4-Bis(chloro)phenyl]-5-methyl-4-(4-phenylsulfonyl)-3,4-dihydro-2H-pyrroline-1-oxide (5g): [α]20D = −7.27 (c 0.22, EtOAc), 1H NMR (400 MHz, CDCl3) δ 7.96 (br, 1H), 7.79 (m, 1H), 7.75–7.51 (m, 3H), 7.36 (d, 1H), 7.24–7.07 (m, 1H), 6.97 (br, 1H), 4.25 (s, 1H), 4.20–4.11 (m, 2H), 3.80–3.75 (m, 1H), 2.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 136.6, 135.0, 130.2, 129.9, 129.8, 129.7, 129.1, 129.0, 128.9, 127.8, 71.6, 66.6, 31.9, 22.6. HPLC analysis on a Chiralpack AD-H column (10% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ = 220 nm; t minor = 32.8 min, t major = 43.8 min, ee = 90%). HRMS Calcd for C22H17Cl2NO3S: 446.0373 Found: 446.0379.
3-[2-Chlorophenyl]-5-methyl-4-(4-tolysulfonyl)-3,4-dihydro-2H-pyrroline-1-oxide (5h): [α]20D = −1.77 (c 0.30, EtOAc), 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.26–7.23 (m, 3H), 6.97 (d, J = 8.0 Hz, 1H), 4.43 (br, 1H), 4.14 (s, 1H), 4.10 (s, 1H), 3.80 (br, 1H), 2.47 (s, 3H), 2.25 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 133.0, 132.8, 130.4, 129.0, 127.8, 127.5, 126.5, 66.7, 31.9, 29.7, 22.6. HPLC analysis on a Chiralpack IB-H column (10% i-PrOH in hexanes; flow rate = 1 mL min−1; λ = 220 nm; t major = 42.1 min, t minor = 53.1 min, ee = 80%). HRMS Calcd for C18H18ClNO3S: 364.0772 Found: 364.0769.
(3R, 4S)-5-Phenyl-4-(phenylsulfonyl)-3-(thiophen-2-yl)-3,4-dihy-dro-2H-pyrroline-1-oxide (5i): [α]20D = −36.90 (c 0.1, CHCl3), 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 8.0 Hz, 2H), 7.55 (t, J = 8.0 Hz, 2H), 7.36–7.19 (m, 7H), 6.98–6.89 (m, 1H), 4.97 (s, 1H), 4.72 (dd, J = 8.0, 4.0 Hz, 1H), 4.61 (d, J = 4.0 Hz, 1H), 4.12 (d, J = 4.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 143.4, 136.9, 134.6, 133.4, 130.3, 129.3, 129.0, 128.2, 127.6, 127.3, 125.1, 124.7, 70.7, 63.7, 34.0, 29.7. HPLC analysis on a Chiralpack AD-H column (30% i-PrOH in hexanes; flow rate = 0.8 mL min−1; λ = 220 nm; t minor = 41.5 min, t major = 54.6 min, ee = 81%).
(3R, 4S)-3-[3, 5-Bis (trifluouomethyl) phenyl]-5-phenyl-4-(phen-ylsulfonyl)-3, 4-dihydro-2H-pyrroline-1-oxide (5j): [α]20D = + 10 (c = 0.1, CHCl3), 1H NMR (400 MHz, CDCl3) δ 7.88 (t, J = 8.0 Hz, 3H), 7.59 (m, 4H), 7.57 (t, J = 8.0 Hz, 1H), 7.37 (t, J = 8.0 Hz, 3H), 7.21–7.31 (m, 2H), 4.83–4.76 (m, 1H), 4.50 (d, J = 8.0, 1H), 4.16 (brd, J = 16.0 Hz, 1H), 4.11 (dt, J = 8.0, 4.0Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 143.3, 138.8, 136.4, 134.8, 133.3, 133.0, 130.6, 129.4, 129.1, 128.3, 127.3, 126.7, 122.5, 122.5, 69.1, 37.3, 29.7. HPLC analysis on a Chiralpack AD-H column (20% i-PrOH in hexanes; flow rate = 0.5 mL min−1; λ = 220 nm; t major = 12.9 min, t minor = 16.5 min, ee = 80%).
(3R, 4S)-3-(2-Chlorophenyl)-5-methyl-4-(4-phenylsulfonyl)-3,4-dihydro-2H-pyrroline-1-oxide (5k): [α]20D = −10.8 (c 0.46, EtOAc), 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 7.9 Hz, 2H), 7.73 (s, 1H), 7.62 (t, J = 7.7 Hz, 2H), 7.34 (d, J = 8.0 Hz, 1H), 7.25–7.13 (m, 2H), 6.97 (s, 1H), 4.43 (s, 1H), 4.28 (m, 2H), 3.85 (m, 1H), 2.23 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 138.8, 137.2, 136.7, 136.6, 134.8, 132.9, 130.4, 129.8, 129.5, 129.0, 127.8, 127.4, 66.8, 36.0, 29.6, 13.0. HPLC analysis on a Chiralpack AD-H column (10% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ = 220 nm; t major = 40.9 min, t minor = 53.8 min, ee = 86%). HRMS Calcd for C17H16ClNO3S: 350.0621 Found: 350.0612.
(3R, 4S)-5-methyl-3-phenyl-4-tosyl-3,4-dihydro-2H-pyrroline-1-oxide (5l): [α]20D = −21.8 (c 0.3, CHCl3), 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.2 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 7.29 (s, 3H), 7.01 (d, J = 5.6 Hz, 2H), 4.34 (s, 1H), 4.08 (s, 2H), 3.86 (d, J = 7.9 Hz, 1H), 2.49 (s, 3H), 2.20 (d, J = 21.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 170.31, 146.31, 140.56, 133.72, 130.48, 129.51, 128.90, 128.18, 126.05, 81.15, 38.61, 29.69, 21.78, 14.10. HPLC analysis on a Chiralpack IB column (10% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ = 220 nm; t major = 47.5 min, t minor = 54.0 min, ee = 86%).
(3R, 4S)-3-phenyl-4-(phenylsulfonyl)-5-propyl-3,4-dihydro-2H-pyrroline-1-oxide (5m): [α]20D = −10.8 (c 0.34, EtOAc), 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 7.1 Hz, 2H), 7.65 (d, J = 7.4 Hz, 2H), 7.55 (t, J = 7.5 Hz, 1H), 7.44–7.30 (m, 3H), 7.29 (d, J = 4.5 Hz, 2H), 4.22 (dd, J = 14.0, 7.5 Hz, 1H), 3.69 (d, J = 14.1 Hz, 1H), 3.01 (q, J = 7.3 Hz, 1H), 2.09 (d, J = 32.1 Hz, 1H), 1.67 (dt, J = 14.4, 7.3 Hz, 4H), 1.01 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 137.60, 134.45, 130.23, 129.25, 129.01, 128.16, 127.51, 73.97, 68.50, 34.26, 29.71, 27.65, 10.69. HPLC analysis on a Chiralpack OD-H column (8% i-PrOH in hexanes; flow rate = 1.0 mL min−1; λ= 220 nm; t minor = 59.3 min, t major = 76.0 min, ee = 91%).
Acknowledgements
This work is supported by the National Nature Science Foundation of China (20972121, 81172935, 91017005), the Ministry of Education of China (NCET-10-0625), and the Open Research Fund Program of the State Key Laboratory of Virology of China (2011002). Hubei Key Laboratory of Molecular Imaging is also gratefully acknowledged.References
- (a) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 12, 1877–1894 CrossRef; (b) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS.
- (a) J. Christoffers, Synlett, 2001, 723–732 CrossRef CAS; (b) D. Enders, K. Lüttgen and A. A. Narine, Synthesis, 2007, 959–980 CrossRef CAS; (c) J. Christoffers, G. Koripelly, A. Rosiak and M. Rossle, Synthesis, 2007, 1279–1300 CrossRef CAS.
- (a) D. Enders, S. F. Muller, G. Raabe and J. Runsink, Eur. J. Org. Chem., 2000, 879–892 CrossRef CAS; (b) P. Mauleon and J. C. Carretero, Org. Lett., 2004, 6, 3195–3198 CrossRef CAS; (c) P. Mauleon and J. C. Carretero, Chem. Commun., 2005, 4961–4963 RSC; (d) S. Mosse and A. Alexakis, Org. Lett., 2005, 7, 4361–4364 CrossRef CAS.
- (a) S. Sulzer-Mosse, M. Tissot and A. Alexakis, Org. Lett., 2007, 9, 3749–3751 CrossRef CAS; (b) Q. Zhu and Y. Lu, Org. Lett., 2008, 10, 4803–4806 CrossRef CAS; (c) T. Llamas, R. G. Arrayas and J. C. Carretero, Angew. Chem., Int. Ed., 2007, 46, 3329–3332 CrossRef CAS; (d) J.-N. Desrosiers and A. B. Charette, Angew. Chem., Int. Ed., 2007, 46, 5955–5957 CrossRef CAS.
- (a) P. Mauleon, I. Alonso, M. R. Rivero and J. C. Carretero, J. Org. Chem., 2007, 72, 9924–9935 CrossRef CAS; (b) J.-N. Desrosiers, W. S. Bechara and A. B. Charette, Org. Lett., 2008, 10, 2315–2318 CrossRef CAS.
- (a) R. C. F. Jones and J. N. Martin, inSynthetic applications of 1,3-dipolar cycloaddition chemistry toward heterocycles and natural products, The Chemistry of Heterocyclic Compoundsvol. 59, Wiley, New York, 2002, pp. 1–81 Search PubMed; (b) P. Merino, in Science of synthesis, vol. 27, ed. A. Padwa, Thieme, Stuttgart, 2004, pp. 511–580 Search PubMed; (c) F. Cardona and A. Goti, Angew. Chem., Int. Ed., 2005, 44, 7832–7835 CrossRef CAS; (d) J. Revuelta, S. Cicchi, A. Goti and A. Brandi, Synthesis, 2007, 485–504 CAS; (e) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247–12275 CrossRef CAS; (f) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem.–Eur. J., 2009, 15, 7808–7821 CrossRef CAS; (g) M. Pineiro and V. D. Pinho e Melo, Eur. J. Org. Chem., 2009, 5287–5307 CrossRef CAS; (h) C. Najera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567–4581 RSC; (i) M. Nielsen, C. B. Jacobsen, N. Holub, M. W. Paixão and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 2668–2679 CrossRef CAS; (j) A.-N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC.
- (a) D. Enders, S. F. Muller, G. Raabe and J. Runsink, Eur. J. Org. Chem., 2000, 879–892 CrossRef CAS; (b) S. Mosse and A. Alexakis, Org. Lett., 2005, 7, 4361–4364 CrossRef CAS.
- P. H. Bos, A. J. Minnaard and B. L. Feringa, Org. Lett., 2008, 10, 4219–4222 CrossRef CAS.
- O. G. Mancheño, P. Tangen, R. Rohlmann, R. Fröhlich and J. Alemán, Chem. Eur. J., 2011, 17, 984–992 CrossRef.
- S.-G. Kim, K.-H. Kim, J. Jung, S. K. Shin and K. H. Ahn, J. Am. Chem. Soc., 2002, 124, 591–596 CrossRef CAS.
- K. H. Ahn, H.-Y. Ku, Y. Kim, S.-G. Kim, Y. K. Kim, H. S. Son and J. K. Ku, Org. Lett., 2003, 5, 1419–1422 CrossRef CAS.
- L. H. Gade and S. Bellemin- Laponnaz, Chem.–Eur. J., 2008, 14, 4142–4152 CrossRef CAS.
- (a) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632 RSC; (b) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS; (c) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS.
- (a) S. E. Gibson and M. P. Castaldi, Chem. Commun., 2006, 3045–3062 RSC; (b) A. Z. Gonzalez and F. D. Toste, Org. Lett., 2010, 12, 200–203 CrossRef CAS; (c) K. Murai, S. Fukushima, S. Hayashi, Y. Takahara and H. Fujioka, Org. Lett., 2010, 12, 964–966 CrossRef CAS; (d) D. M. Du, T. Fang, J. X. Xu and S. W. Zhang, Org. Lett., 2006, 8, 1327–1330 CrossRef CAS; (e) J. Zhou and Y. Tang, J. Am. Chem. Soc., 2002, 124, 9030–9031 CrossRef CAS; (f) M. C. Ye, J. Zhou and Y. Tang, J. Org. Chem., 2006, 71, 3576–3582 CrossRef CAS; (g) J. Zhou and Y. Tang, Chem. Soc. Rev., 2005, 34, 664–676 RSC; (h) P. D. Cao, Y. Y. Zhou, X. L. Sun, J. C. Zheng, Z. W. Xie and Y. Tang, Angew. Chem., Int. Ed., 2010, 49, 4463–4466 CrossRef CAS.
- M. T. Reetz, H. C. Guo, J. A. Ma, R. Goddard and R. J. Mynott, J. Am. Chem. Soc., 2009, 131, 4136–4142 CrossRef CAS.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS.
- (a) L. Dai, S. X. Wang and F. E. Chen, Adv. Synth. Catal., 2010, 13, 2137–2141 CrossRef; (b) H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS; (c) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153–156 CrossRef CAS; (d) J. W. Lee, T. H. Rhu, J. S. Oh, H. Y. Bae, H. B. Jang and C. E. Song, Chem. Commun., 2009, 7224–7226 CAS; (e) W. Yang and D. M. Du, Org. Lett., 2010, 12, 5450–5453 CrossRef CAS; (f) Z. Dong, X. Q. Jin, P. C. Wang, C. Min, J. Zhang, H. B. Zhou and C. Dong, ARKIVOC, 2011,(ix), 367–380 CAS; (g) R. I. Storer, C. Aciroa and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC; (h) J. Aleman, A. Parra, H. Jiang and K. A. Jorgensen, Chem.–Eur. J., 2011, 17, 6890–6899 CrossRef CAS; (i) D. Q. Xu, Y. F. Wang, W. Zhang, S. P. Luo, A. G. Zhong, A. B. Xia and Z. Y. Xu, Chem.–Eur. J., 2010, 16, 4177–4180 CrossRef CAS.
- (a) C. Min, X. Han, Z. Q. Liao, X. F. Wu, H.-B. Zhou and C. Dong, Adv. Synth. Catal., 2011, 353, 2715–2720 CrossRef CAS; (b) X. F. Wu, C. Min, H.-B. Zhou and C. Dong, Tetrahedron: Asymmetry, 2011, 22, 1640–1643 CrossRef CAS.
- P. J. Black, G. Cami-Kobeci, M. G. Edwards, P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Org. Biomol. Chem., 2006, 4, 116–125 CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: NMR and HPLC spectra of compounds 5. See DOI: 10.1039/c2ra21162a/ |
| This journal is © The Royal Society of Chemistry 2012 |