Synthesis and Li-storage behavior of CrN nanoparticles
B.
Das
,
M. V.
Reddy
,
G. V.
Subba Rao
and
B. V. R.
Chowdari
*
Department of Physics, National University of Singapore, 117542, Singapore. E-mail: phychowd@nus.edu.sg; Fax: +65-67776126; Tel: +65-65162956
First published on 7th August 2012
Abstract
Bulk CrN nanoparticles are prepared by the thermal decomposition of a Cr–urea complex in a flowing NH3 + N2 atmosphere and characterized by X-ray diffraction (XRD) and high resolution-transmission electron microscopy (HR-TEM) along with selective area electron diffraction (SAED) techniques. The Li-cycling performance of bulk CrN is evaluated by galvanostatic cycling and cyclic voltammetry on the cells with Li metal as counter electrode in the voltage range of 0.005–3.0 (3.5) V at ambient temperature. When cycled at 60 mA g−1 (0.1 C) up to 3.0 V, the composition 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:15 (active material:carbon:binder) showed a first-cycle reversible capacity of 635 (±10) mA h g−1 (1.6 moles of Li). The reversible capacity of 500 (±10) mA h g−1 (1.23 moles of Li) is stable between 10 and 80 cycles. At 0.5 C, it showed a stable capacity of 350 (±10) mA h g−1 for 40 cycles and the original capacity is regained when cycled at 0.1 C rate after 160 cycles. The coulombic efficiency is found to be >96% in the range of 20–80 cycles. The low impedance at the discharge potential <1.3 V and high impedance at charge potential 3.0 V evaluated from the impedance spectra (EIS) showed the decomposition and formation of CrN during the 1st cycle. The apparent DLi+ obtained from EIS is in the range, 0.73–3.6 (±0.1) × 10−14 cm2 s−1 during the first-cycle.
:30:15 (active material:carbon:binder) showed a first-cycle reversible capacity of 635 (±10) mA h g−1 (1.6 moles of Li). The reversible capacity of 500 (±10) mA h g−1 (1.23 moles of Li) is stable between 10 and 80 cycles. At 0.5 C, it showed a stable capacity of 350 (±10) mA h g−1 for 40 cycles and the original capacity is regained when cycled at 0.1 C rate after 160 cycles. The coulombic efficiency is found to be >96% in the range of 20–80 cycles. The low impedance at the discharge potential <1.3 V and high impedance at charge potential 3.0 V evaluated from the impedance spectra (EIS) showed the decomposition and formation of CrN during the 1st cycle. The apparent DLi+ obtained from EIS is in the range, 0.73–3.6 (±0.1) × 10−14 cm2 s−1 during the first-cycle.
Introduction
Lithium ion batteries (LIBs) are considered as the most promising answer to the emerging demands for effective energy storage devices. But, they need improvements with respect to energy density and power density to find applications in electric and hybrid electric vehicles (EV/HEVs). To achieve this goal, a lot of research effort is going on to find high capacity anode materials as an alternative to graphite (specific capacity; theor.: 372 mA h g−1), which is presently being used as the anode for commercial LIBs.1–5 In this respect, metal nitrides are considered as promising anodes due to their low potential vs. Li-metal and large reversible capacity based on conversion reaction, i.e.,| MN + 3Li+ + 3e− → M0 + Li3N (M = Metal) | (1) |
Also, the electrochemically formed Li3N during the first discharge, which has a layered crystal structure, provides excellent conducting pathways for reversible Li-cycling.6,7 The ternary metal nitrides of chemical formula, Li3−xMxN (M = Co, Fe, Ni, Cu),8–10 which have similar crystal structures as Li3N were studied as prospective anode materials. The composition, Li2.6Co0.4N showed superior electrochemical performance with capacity ∼700–800 mA h g−1. Ternary metal nitride, LiNiN was studied by Cabana et al.11 and they showed ∼350 mA h g−1 at the end of the 40th cycle when cycled in the voltage range, 0–1.65 V at 0.05 C-rate. Binary metal nitrides in the form of thin films have also been explored as prospective anode materials for LIBs.12–16 We reported the cycling performance of nanoflake CoN thin film prepared by RF magnetron sputtering and showed a high capacity of ∼950 mA h g−1 at 0.59 C-rate when cycled in the voltage range, 0.005–3.0V vs. Li up to 80 cycles and a capacity of ∼690 mA h g−1 at 6.6 C-rate up to 50 cycles.15 The electrochemical performance of a few binary metal nitrides in powder form has also been reported. Recently, Yamada et al.17 reported the electrochemical performance of Li3−xFexN in the form of powder, which showed a maximum reversible capacity of 700 mA h g−1 for the composition x = 0.2 when cycled in the range of 0.05–1.3 V vs. Li/Li+. However successive cycles showed capacity fading, and <450 mA h g−1 was observed at the end of the 5th cycle. Palacín et al.18 reported the electrochemical performance of Ni3N prepared by ammonolysis of different precursors. They showed that Ni3N prepared from decomposition of nickel amide showed a larger reversible capacity. But, it showed capacity fading and delivered ∼600 mA h g−1 at the end of the 5th cycle.
The binary metal nitride, CrN has received considerable attention as an anode material, as it can deliver a theoretical capacity of 1218 mA h g−1 (consumption of 3 moles of Li). Sun and Fu16 studied thin film CrN as an anode material and reported a second cycle discharge capacity of 1200 mA h g−1 at current density, 28 μA cm−2 when cycled in the voltage range of 0.0–3.5 V. However, there is no literature report on Li-cycling studies on bulk CrN.
In the present study, we prepared the CrN nanoparticles by nitridation of a Cr–urea complex in a flowing NH3 + N2 atmosphere and for the first time studied the Li-cycling performance of bulk CrN nanoparticles. A reversible capacity of ∼500 mA h g−1 (∼1.23 moles of Li) at 0.1 C-rate stable up to 80 cycles was observed. A good C-rate capability is shown by CrN nanoparticles. At 0.5 C, it showed a stable capacity of ∼350 mA h g−1 at the end of 40 cycles and the original capacity is regained when cycled at 0.1 C-rate after 160 cycles. For the first time, we calculated the apparent Li-diffusion co-efficient (DLi+) of CrN from the impedance data and it is in the range, 0.73–3.6 (±0.1) × 10−14 cm2 s−1 during the first-cycle. The results are compared with those reported on the thin films of CrN.
Experimental section
Synthesis of Cr-precursor and bulk CrN particles
For the preparation of the precursor material, Cr[OC(NH2)2]6Cl3, 0.04 mole of chromium chloride (CrCl3·6H2O) (98%, Sigma Aldrich) was dissolved in absolute ethanol to get a concentrated solution named as Sol-A for clarity. Then, the Sol-A was added dropwise into saturated urea/ethanol solution (0.24 mole of urea (CO(NH2)2, purity, 99% Aldrich) dissolved in 20 mL of ethanol) with the help of a burette at 60–70 °C with stirring, until the final salt/urea molar ratio reached 1:6. The obtained green precipitate of Cr–urea coordinated compound was separated by filtering and dried at 80 °C. The synthesis procedure of Cr[OC(NH2)2]6Cl3 precursor was reported by Qiu and Gao.19
For the synthesis of CrN nanoparticles, 2.0 g of the Cr–urea precursor material was used for each batch and was loaded in an alumina boat. The boat was placed in a tubular furnace (Carbolite, UK) with flowing NH3 (15%) + N2 (85%). The gas mixture was allowed to pass through the furnace for 30 min to remove the air in it before the furnace was turned on, and the heating rate was kept at 1 °C min−1 before reaching the set temperature. The Cr–urea precursor was heated at 600 °C for 2 h and the solid black colored sample was collected, after cooling down the furnace to room temperature in the flow of NH3 (15%) + N2 (85%) gas. The sample was ground to fine powder and used for further characterization.
The structure and morphology of the CrN nanoparticles were characterized by X-ray diffraction (XRD) using Philips X'PERT MPD unit (Cu-Kα radiation), high resolution transmission microscopy (HR-TEM) and selected area electron diffraction (SAED) (JEOL JEM 3010 operating at 300 kV). For electrochemical measurements, the composite electrode was fabricated by mixing the active material, polymer binder (Kynar 2801) and conducting carbon (Super P, MMM) in the mass ratio of 70:15:15 (or 55:30:15). N-methyl-pyrrolidinone (NMP) was used as the solvent for the binder to get a homogeneous slurry. A 15 μm thick film was prepared by coating the homogeneously mixed slurry onto the etched Cu-foil (10 μm thick, Alpha Industries Co. Ltd., Japan) using doctor blade technique. The film was dried overnight at 70–80 °C to evaporate the NMP. The thick film was cut into circular discs to form the electrodes which were vacuum dried for 12 h at ∼70 °C prior to cell fabrication and inserted to an Ar-filled glove box (MBraun, Germany) for cell assembly. The level of H2O and O2 content was maintained <1 ppm inside the glove box. The Li-metal foil (Kyokuto Metal Co., Japan) was cut in to circular discs (16 mm diameter) and used as the counter electrodes. The glass microfiber filter (GF/F) (Whatman Int. Ltd, Maidstone, England) was used as the separator and 1M LiPF6 dissolved in ethylene carbonate (EC) + dimethyl carbonate (DMC) in the ratio of 1:1 volume (Merck) as the electrolyte. More details on the cell fabrication are described elsewhere.20,21 The cells were aged for 12 h before performing the electrochemical measurements to confirm the percolation of electrolyte into the active material. The galvanostic cycling and cyclic voltammetry were carried out at room temperature using a multichannel battery tester (model SCN, Bitrode, USA) and Macpile II system (Biologic, France), respectively. The electrochemical impedance spectroscopy (EIS) measurements were carried out on the freshly prepared cell at open circuit potential (OCV) in the frequency range of 0.18 MHz to 3 mHz with ac signal amplitude of 10 mV by using the computer controlled Solartron Impedance/gain-phase analyzer (model SI 1255) coupled with a computer controlled battery test unit (model 1470) at room temperature (RT).
Results and discussions
Structure and morphology
Fig. 1 shows the X-ray diffraction (XRD) pattern of CrN. The compound shows lines characteristic of cubic crystal structure with the Fm3m space group. The lattice parameter, calculated by the Rietveld refinement (Topas R 2.1 software) (2000 Bruker AXS, Germany) is: a = 4.151(2) Å in good agreement with the value of 4.140(2) Å given in the JCPDS file (card No. 76-2494). The TEM photograph of CrN nanoparticles shows a particle size of ≤30 nm with mixed type morphology of both cube and sphere (Fig. 2a). The HR-TEM lattice image shows the interplanar d-spacings of 2.42 and 2.10 (±0.02) Å, and can be assigned to the Miller indices (111) and (200), respectively (Fig. 2b). The selected area electron diffraction (SAED) pattern of the CrN nanoparticles shown in Fig. 2c, indicates diffuse rings with few bright spots. The d-values corresponding to these concentric rings were evaluated by measuring the rings from the center and the assigned Miller indices are shown.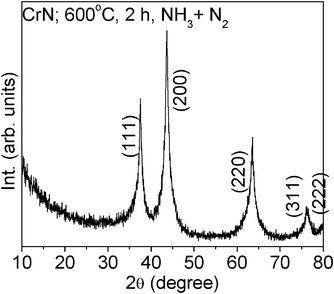 | ||
| Fig. 1 XRD pattern of CrN nanoparticles prepared at 600 °C, 2 h, NH3 + N2 atm. | ||
 | ||
| Fig. 2 (a) TEM photograph of CrN nanoparticles. (b) HR-TEM lattice image of CrN nanoparticles. The interplanar d-spacings are indicated by arrows. (c) SAED pattern of CrN nanoparticles. Scale bars are shown. | ||
The crystallite size of CrN nanoparticle was calculated by using the Scherrer's formula, D = K λ/(β1/2 cosθ), where D is crystallite size, K is Scherrer constant (0.89), λ is the wavelength of Cu-Kα- radiation (1.54 Å), β1/2 is the full width at half maximum (FWHM) in radians of the XRD peak and θ is the scattering (Bragg) angle.22,23 The instrumental broadening was calculated by using the nano-TiO2 (anatase) (Evonik Degussa; 99.5%) of ∼25 nm particle size as standard and was found to be 0.08°. For TiO2, it is assumed that the crystallite size is of the same order as particle size. The crystallite sizes were calculated by using β1/2 (FWHM) of two or three high intensity peaks and the corresponding θ values in the XRD patterns. The value is found to be 8 (±2) nm for the CrN nanoparticles.
Electrochemical characterizations
To evaluate the electrochemical behavior, the CrN nanoparticles were tested as the cathode in a cell against Li metal, which was used as the counter electrode. Fig. 3 shows the galvanostatic discharge–charge profile of CrN nanoparticles of the composition 55:30:15, at a current of 60 mA g−1 (0.1 C; assuming 1C = 600 mA g−1) in the voltage range of 0.005–3.0 V, up to 50 cycles. During the first discharge reaction, the voltage drops from the open circuit voltage (OCV) of ∼3.0 V to ∼1.5 V, followed by small voltage plateaus at ∼1.2, ∼0.75 and ∼0.6 V. These voltage plateaus are ascribed to the different electrochemical reactions of Li+ with CrN nanoparticles leading to crystal structure destruction and formation of Cr metal and Li3N as per eqn (1). No Li+ intercalation to CrN is noticed, as there are no empty sites where Li+ can be stored (CrN has Cubic crystal structure). The overall capacity observed at the end of the deep discharge is ∼1490 mA h g−1 (∼3.6 moles of Li per mole of CrN). The observed capacity at the end of 0.005 V, is due to the crystal structure destruction of CrN and during the first-charge reaction, no significant voltage plateaus are seen until 3.0 V, but only a continuous sloping profile, which can be ascribed to capacitive like behavior of CrN during charge cycles. The overall first-charge capacity observed at the end of 3.0 V is ∼635 mA h g−1 (∼1.6 moles of Li), with the irreversible capacity loss (ICL) ∼855 mA h g−1 (∼2.1 moles of Li). The ICL during the first-cycle is due to the solid electrolyte interface (SEI) formation and due to the formation of a polymeric-like layer on metal particles under deep discharge conditions.15,23–29
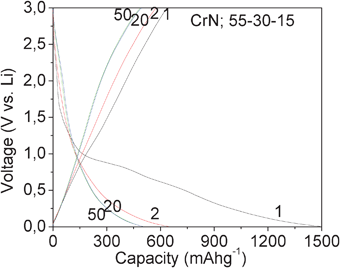 | ||
| Fig. 3 Galvanostatic discharge–charge profiles of CrN nanoparticles. 1–50 cycles. The numbers indicate cycle number. The data are collected at 24 °C. Voltage range, 0.005–3.0 V vs. Li, at a current rate of 60 mA g−1 (0.1 C). Electrode composition: 55:30:15. | ||
The voltage vs. capacity profile during the second discharge reaction is different from the first discharge reaction, indicating different electrochemical reactions. The capacity vs. voltage profile shows a continuous decrease in potential till ∼0.6 V, with a voltage plateau noticed at ∼0.25 V. The overall capacity at the end of the second-discharge is ∼645 mA h g−1 (∼1.6 moles of Li). The voltage vs. capacity profile during second-charge reaction is analogous to the first-charge indicating a similar electrochemical process. A high reversible capacity of ∼575 mA h g−1 (∼1.41 moles of Li) is observed at the end of the second-charge. Similar voltage vs. capacity profiles are observed during 2–50 cycles showing good reversibility. At the end of the 50th cycle, a reversible capacity of ∼500 mA h g−1 (∼1.23 moles of Li) is observed.
The capacity vs. cycle number plots of CrN nanoparticles of different CrN to carbon percentages and at different cut-off voltages, at a current of 60 mA g−1 (0.1 C) are shown in Fig. 4a. The composition 70:15:15 (CrN:Super P carbon:PVDF) shows a first-discharge capacity of ∼875 mA h g−1 (∼2.15 moles of Li), which is less than the theoretical capacity, i.e., consumption of 3 moles of Li (1218 mA h g−1) (Fig. not shown). This could be due to the incomplete electrochemical reaction i.e. partial crystal structure destruction of CrN. The low conductivity of CrN does not allow the Li+ to penetrate to the bulk, but electrochemically react with the surface of the CrN particles. The initial decrease in capacity is noticed until the 10th cycle and remained stable at ∼275 mA h g−1 (∼0.6 moles of Li) up to 40 cycles (Table 1). When the carbon content in the composite electrode was increased to 30 wt % i.e., 55:30:15, a dramatic improvement in the electrochemical performance is noticed. For the composition 55:30:15 at 60 mA g−1, the first-discharge capacity is observed as ∼1490 mA h g−1 (∼3.6 moles of Li), which is higher than the theoretical capacity. This is due to the increase in both electronic and ionic conductivity of CrN by addition of extra carbon and complete electrochemical reaction is observed leading to full crystal structure destruction as per eqn (1). Similar crystal structure destruction was noticed for CrN thin film during the first-discharge cycle.16 The capacity vs. cycle number profile remained similar to the previous composition, but resulted in higher capacity values. The initial decrease in capacity is noticed until the 10th cycle and remained stable at ∼500 mA h g−1 (∼1.23 moles of Li) up to 80 cycles (Table 1). Sun and Fu16 reported that the CrN thin film when cycled to an upper cut-off voltage of 3.5 V showed higher capacity, but with capacity fading of 0.5% per cycle. We have also noticed higher capacity for CrN nanoparticles when cycled to 3.5 V. The first-discharge capacity observed is ∼1475 mA h g−1 (∼3.6 moles of Li), whereas the first-charge is ∼915 mA h g−1 (∼2.3 moles of Li). The observed capacity is almost near to the reversible capacity noticed in the case of CrN thin film.16 A similar capacity vs. cycle number profile is noticed until the 20th cycle with sudden fall of capacity up to the 40th cycle. The reason for sudden capacity fading is not clear yet.
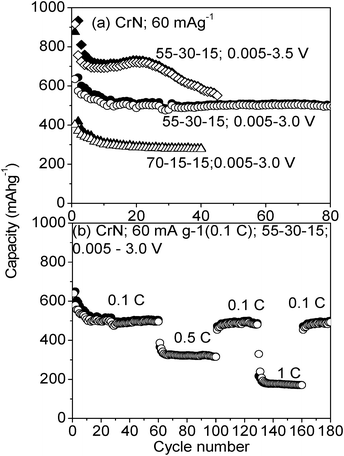 | ||
| Fig. 4 Capacity vs. number of cycles for CrN nanoparticles at 24 °C. Electrode composition, current rate and voltages range are shown. Open symbol: charge capacity; closed symbol: discharge capacity. | ||
| Voltage range; Electrode composition | Theoretical capacity (mA h g−1) | Observed capacity values; (±5) (mA h g−1) | |||||
|---|---|---|---|---|---|---|---|
| 1st disch. | 1st charge | 40th disch. | 40th charge | 80th disch. | 80th charge | ||
| 0.005–3.0 V; | 875 | 400 | 280 | 275 | — | — | |
| (70:15:15) |
(2.15 Li) | (1.0 Li) | (0.7 Li) | (0.6 Li) | |||
| 0.005–3.0 V; | 1218 | 1490 | 635 | 500 | 495 | 500 | 495 |
| (55:30:15) |
(3 Li) | (3.6 Li) | (1.6 Li) | (1.23 Li) | (1.2 Li) | (1.23 Li) | (1.2 Li) |
| 0.005–3.5 V; | 1475 | 915 | 590 | 580 | — | — | |
| (55:30:15) |
(3.6 Li) | (2.3 Li) | (1.5 Li) | (1.4 Li) | |||
The rate capability of CrN is performed in order to evaluate the feasibility of using it as prospective anode materials and is shown in Fig. 4b. When cycled at 0.5 C, the nano-CrN showed a reversible capacity of ∼350 mA h g−1 (∼0.86 moles of Li) stable up to 40 cycles. At 1 C-rate, it shows a capacity of ∼200 (±5) mA h g−1 (∼0.5 moles of Li) stable up to 30 cycles. The initial capacity of ∼500 mA h g−1 is even regained, when the current rate is changed to 0.1 C after 160 cycles. This confirms the good electrochemical reversibility of CrN and there is no capacity degradation noticed when cycled at different current rates. Hence, we can conclude that, CrN can be a prospective anode material. In all cases, the coulombic efficiency is 96–98%.
The cyclic voltammetry (CV) plots of CrN nanoparticles were measured in the potential range 0.005–3.5 V up to 6 cycles at 58 μV s−1 to complement the galvanostatic cycling studies and also to know the redox potentials and are shown in Fig. 5. The Li metal is used as the counter and reference electrode. During the first cathodic sweep (negative scan) from OCV (∼3.0 V) to 0.005 V, two cathodic peaks are noticed at 0.65 and 0.42 V, followed by a minor shoulder peak at ∼0.15 V. These cathodic peaks could be due to different electrochemical reduction processes of CrN with Li and formation of Cr metal particles and Li3N as per eqn (1). The corresponding anodic peaks are broad and are at ∼1.0 V and ∼2.5 V (which is not so prominent) when charged to 3.5 V. The unusual type hump is noticed in the range of ∼3.2 V to 3.5 V and the reason is not clearly understood. It is believed that the Li3N is decomposed in excess of 2.0 V due to the slow kinetics and the low conductivity of CrN itself. So, the peaks at∼0.65 V and ∼2.5 V could be due to the reduction and oxidation of CrN. In the second cathodic sweep, the peaks observed at ∼0.65 and 0.42 V observed in the first cathodic sweep, are not seen, but with a continuous fall in voltage until 0.005 V. The CVs from 2–6 cycles are similar and overlap with each other indicating good reversibility.
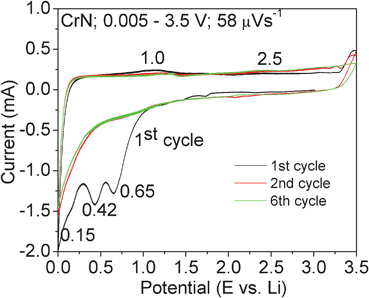 | ||
| Fig. 5 Cyclic voltammograms of CrN nanoparticles. 1st, 2nd and 6th cycle are shown. Scan rate, 58 μV s−1. Li metal anode was the counter and reference electrode. Numbers represent the potentials in volts. Electrode composition: 55:30:15. | ||
Electrochemical impedance spectroscopy
The electrochemical impedance spectroscopy (EIS) studies were carried out on CrN nanoparticles at selected voltages in the range of 0.005–3.0 V vs. Li, at the current of 60 mA g−1 during the 1st cycle. The cells were discharged or charged to the selected voltage values, relaxed for 2 h at that voltage and the impedance spectra were measured. Fig. 6 shows the Nyquist plots (Z′ vs. −Z′′) during the 1st cycle at various voltage values. The impedance data were analyzed by fitting to an equivalent electrical circuit shown in Fig. 6c similar to the circuits reported in the literature.20,22,30–32 It consists of the electrolyte resistance (Re), surface film (Rsf) and charge transfer (Rct) resistances, a constant phase element, CPEi (instead of pure capacitance, due to the observation of a depressed semicircle in the spectra) along with diffusional components like Warburg impedance (Ws). The symbols in Fig. 6 are the experimental data whereas the continuous lines represent the fitted curves. The derived impedance parameters at various voltages are given in Table 2.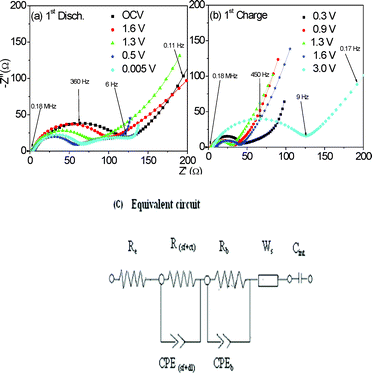 | ||
| Fig. 6 Nyquist plots (Z′ vs. −Z′′) of CrN nanoparticles at different voltages. (a) During the 1st discharge reaction from OCV (2.4) to 0.005 V; (b) 1st charge reaction from 0.3 to 3.0 V. Symbols represents experimental data and continuous lines represent the fitted curve using the equivalent circuit of Fig. 6c. Geometric area of the electrode is 2 cm2. | ||
| First-discharge cycle | ||||||
| Parameter values: | 2.4 | 1.6 | 1.3 | 0.5 | 0.005 | |
| Cell voltage, V vs. Li | ||||||
| R (sf+ct) (±5) Ω | 104 | 81 | 60 | 48 | 56 | |
| R b (±5) Ω | 80 | 87 | ||||
| CPE (sf+dl) (±5) μF | 40 | 25 | 33 | 42 | 43 | |
| CPE b (±2) mF | 13 | 12 | ||||
| α (±0.02) | 0.75 | 0.86 | 0.84 | 0.81 | 0.80 | |
| Frequency; fL (±3) mHz | — | — | — | 7.1 | 3.6 | |
| App. diff. coeff. (DLi+, (±0.2) × 10−14) cm2 s−1 | — | — | — | 1.4 | 0.73 | |
| First-charge cycle | ||||||
| Parameter values: | 0.3 | 0.9 | 1.3 | 1.6 | 3.0 | |
| Cell voltage, V vs. Li | ||||||
| R (sf+ct) (±5) Ω | 33 | 28 | 26 | 30 | 114 | |
| R b (±5) Ω | 64 | |||||
| CPE (sf+dl) (±5) μF | 35 | 96 | 100 | 100 | 25 | |
| CPE b (±2) mF | 25 | |||||
| α (±0.02) | 0.89 | 0.72 | 0.70 | 0.67 | 0.74 | |
| Frequency; fL (±3) mHz | 11 | 18 | — | — | — | |
| App. diff. coeff. (DLi+, (±0.2) × 10−14) cm2 s−1 | 2.2 | 3.6 | — | — | — | |
The fresh cell (OCV ∼2.4 V) shows a single semicircle in the frequency region 0.18 MHz–6 Hz, followed by a Warburg type slope in the low frequency region. The fitted value of impedance is 104 (±5) Ω, attributed mainly to the surface-film resistance (Rsf). The associated capacitance (CPEsf) is 40 (±5) μF. The impedance spectra measured at the voltages of 1.6 and 1.3 V also show only a single semicircle, similar to the spectrum at OCV (Fig. 6a). Here, the curve fitting was carried out using R(sf+ct) combination since electronic and ionic charge transfer are involved in the discharge process. As can be seen from Table 2, the R(sf+ct) values decrease to 81 and 60 (±5) Ω at 1.6 and 1.3 V, respectively. The corresponding CPE(sf+dl) (dl = double layer) range from 25–33 (±5) μF in the voltage range 1.6–1.3 V. The spectra measured at 0.5 V and 0.005 V differ from the spectra at higher voltages and show two large-diameter semicircles indicating the onset of contribution from the bulk resistance (Rb) in addition to R(sf+ct). Accordingly, the Rb and CPEb components in the circuit (Fig. 6c) were used to fit the spectra. The fitted values of R(sf+ct) and Rb are: 48 and 80 (±5) Ω, and 56 and 87 (±5) Ω respectively, at the voltages 0.5 and 0.005 V. The corresponding CPE(sf+dl) values are 42 and 43 (±5) μF. The extracted CPEb are fairly large and are 13 and 12 (±2) mF (Table 2).
The impedance of the constant phase element, Z′CPE is related to the angular frequency, ω by the relation, ZCPE = 1/[Ci (jω)α], where j = √ − 1 and Ci is the capacitance and α is a constant. The value of α (<1) is an estimate of the degree of distortion from the pure capacitor behavior. As can be seen from Table 2, the values of α (degree of distortion, <1) range from 0.75 to 0.86 during the first-discharge process. The Nyquist plots during the first-charge cycle at various voltages are shown in Fig. 6b, and they resemble the spectra during the discharge cycle. The R(sf+ct) values remain almost constant at 30 (±5) Ω in the voltage range, 0.3–1.6 V, and increases to 114 (±5) Ω at the voltage 3.0 V. The corresponding CPE(sf+dl) values show an increase from 35 to 100 μF in the voltage range, 0.3–1.6 V, but decreases to 25 μF at 3.0 V. The α values range from 0.67 to 0.89 depending on the voltage (Table 2).
The apparent Li-ion diffusion coefficient (DLi+) for CrN nanoparticles are calculated from the EIS data by using the following eqn (2)22,33,34
| DLi+ = π fL L2 | (2) |
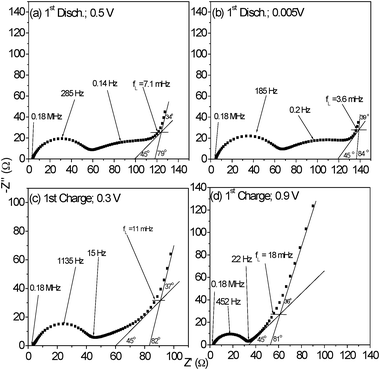 | ||
| Fig. 7 Selected impedance spectra of CrN nanoparticles redrawn from Fig. 6a and b in an expanded scale, for the determination of the limiting frequency (fL). First-discharge: (a) at 0.5 V and (b) at 0.005 V. First-charge: (c) at 0.3 V and (d) at 0.9 V. The fL values and selected frequencies are shown. | ||
We can conclude that the decrease in the impedance value during the discharge below 1.3 V and increase at 3.0 V during charging can be ascribed to decomposition and reformation of CrN nanoparticles and supports the observed galvanostatic cycling data (Fig. 3). The apparent DLi+ obtained from EIS is in the range, 0.73 − 3.6 (±0.1) × 10−14 cm2 s−1 during the first-cycle.
Conclusions
The CrN nanoparticles were prepared by the thermal decomposition of a Cr–urea complex in flowing NH3 + N2 gas and characterized by X-ray diffraction (XRD), high resolution transmission electron microscopy (HR-TEM) along with selective area electron diffraction (SAED) techniques. The Li-cycling performance of CrN nanoparticles was evaluated by galvanostatic cycling and cyclic voltammetry on the cells with Li metal as counter electrode in the voltage range of 0.005–3.0 (3.5) V at ambient temperature. When cycled at 60 mA g−1 (0.1 C), the CrN electrode composition 70:15:15, showed a first-charge reversible capacity of 400 (±10) mA h g−1 (∼1.0 moles of Li) and remained stable at 275 (±10) mA h g−1 (∼0.6 moles of Li) in the range of 20–40 cycles. The CrN electrode with composition 55:30:15, showed better performance compared to composition 70:15:15, when cycled at 60 mA g−1 (0.1 C). The first-cycle reversible capacity of 635 (±10) mA h g−1 (1.6 moles of Li) slowly decreases to ∼500 (±10) mA h g−1 (∼1.23 moles of Li) after 10 cycles and remains stable up to the end of the 80th cycle. It also showed a good rate capability when cycled at different current rates. At 0.5 C, the CrN showed a stable capacity of 350 (±10) mA h g−1 for 40 cycles and the original capacity was regained when cycled at 0.1 C-rate. The high and stable capacity was obtained due to the presence of increased carbon content, which increased the electronic and ionic conductivity of CrN. When the cut off voltage is increased to 3.5 V, CrN showed a high reversible capacity, but with a drastic fall in the range of 20–40 cycles. The coulombic efficiency for both compositions is found to be >96%. The low impedance at the discharge potential <1.3 V and high impedance at charge potential 3.0 V showed the decomposition and formation of CrN during the 1st cycle and supports the observed galvanostatic cycling. The apparent DLi+ obtained from EIS is in the range, 0.73 − 3.6 (±0.1) × 10−14 cm2 s−1 during the first-cycle.
Acknowledgements
Authors thank the Defence Advanced Research Projects Agency (DARPA), USA (Grant no. R-144-000-226-597). BKD thanks the NUS for Research scholarship and MVR thanks the National Research Foundation (NRF), Singapore for the research grant (No R-144-000-295-281 and R-143-000-360-281).References
- Lithium Batteries: Science and Technology, ed. G.-A. Nazri, G. Pistoia, Kluwer, Academic Publ., New York, 2003 Search PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS.
- Y. Q. Sun, Q. O. Wu and G. Q. Shi, Energy Environ. Sci., 2011, 4, 1113 CAS.
- L. F. Nazar, G. Goward, F. Leroux, M. Duncan, H. Huang, T. Kerr and J. Gaubicher, Int. J. Inorg. Mater., 2001, 3, 191 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170 CrossRef CAS.
- D. H. Gregory, P. M. O'Meara, A. G. Gordon, J. P. Hodges, S. Short and J. D. Jorgensen, Chem. Mater., 2002, 14, 2063 CrossRef CAS.
- T. Lapp, S. Skaarup and A. Hooper, Solid State Ionics, 1983, 11, 97 CrossRef CAS.
- J. B. Ducros, S. Bach, J. P. P.-Ramos and P. Willmann, Electrochem. Commun., 2007, 9, 496 CrossRef.
- J. B. Ducros, S. Bach, J. P. P.-Ramos and P. Willmann, J. Power Sources, 2008, 175, 517 CrossRef CAS.
- S. Bach, J. P. P.-Ramos, J. B. Ducros and P. Willmann, Solid State Ionics, 2009, 180, 231 CrossRef CAS.
- J. Cabana, Z. Stoeva, J. J. Titman, D. H. Gregory and M. R. Palacin, Chem. Mater., 2008, 20, 1676 CrossRef CAS.
- Y. Wang, Z. W. Fu, X. L. Yue and Q. Z. Qin, J. Electrochem. Soc., 2004, 151, E162 CrossRef CAS.
- Q. Sun and Z.-W. Fu, Electrochim. Acta, 2008, 54, 403 CrossRef CAS.
- Q. Sun and Z.-W. Fu, Electrochem. Solid-State Lett., 2008, 11, A233 CrossRef CAS.
- B. Das, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Solid State Ionics, 2009, 180, 1061 CrossRef CAS.
- Q. Sun and Z.-W. Fu, Electrochem. Solid-State Lett., 2007, 10, A189 CrossRef CAS.
- A. Yamada, S. Matsumoto and Y. Nakamura, J. Mater. Chem., 2011, 21, 10021 RSC.
- F. Gillot, J. Oró-Solé and M. R. Palacín, J. Mater. Chem., 2011, 21, 9997 RSC.
- Y. Qiu and L. Gao, Mater. Res. Bull., 2003, 38, 1551 CrossRef CAS.
- B. Das, M. V. Reddy, C. Krishnamoorthi, S. Tripathy, R. Mahendiran, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2009, 54, 3360 CrossRef CAS.
- B. Das, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Solid State Electrochem., 2008, 12, 953 CrossRef CAS.
- B. Das, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2011, 21, 1171 RSC.
- B. Das, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2012, 22, 17505 RSC.
- S. Grugeon, S. Laruelle, L. Dupont and J.-M. Tarascon, Solid State Sci., 2003, 5, 895 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J.-M. Tarascon, J. Electrochem. Soc., 2002, 149, A627 CrossRef CAS.
- M. V. Reddy, Z. Beichen, L. J. Nicholette, Z. Kaimeng and B. V. R. Chowdari, Electrochem. Solid-State Lett., 2011, 14, A79 CrossRef CAS.
- Y. Yu, C.-H. Chen, J.-L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 44, 7085 CrossRef CAS.
- M. N. Obrovac, R. A. Dunlap, R. J. Sanderson and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A576 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- F. Nobili, R. Tossici, R. Marassi, F. Croce and B. Scrosati, J. Phys. Chem. B, 2002, 106, 3909 CrossRef CAS.
- M. D. Levi and D. Aurbach, J. Phys. Chem. B, 1997, 101, 4630 CrossRef CAS.
- D. Aurbach, A. Nimberger, B. Markovsky, E. Levi, E. Sominski and A. Gedanken, Chem. Mater., 2002, 14, 4155 CrossRef CAS.
- B. Garcia, J. Farcy, J. P. Pereira-Ramos and N. J. Baffier, J. Electrochem. Soc., 1997, 144, 1179 CrossRef CAS.
- P.-J. Cho, E.-D. Jeong and Y.-B. Shim, Bull. Korean Chem. Soc., 1998, 19, 39 CAS.
| This journal is © The Royal Society of Chemistry 2012 |