DOI:
10.1039/C2RA21123H
(Paper)
RSC Adv., 2012,
2, 7456-7461
Nontoxic, nonvolatile, and highly efficient osmium catalysts for asymmetric dihydroxylation of alkenes and application to one mol-scale synthesis of an anticancer drug, camptothecin intermediate†
Received
4th June 2012
, Accepted 11th June 2012
First published on 13th June 2012
Abstract
Novel polymer-incarcerated osmium (PI Os) catalysts have been developed. The Os catalysts were shown to be nontoxic and nonvolatile, and stable for several months in air. Asymmetric dihydroxylation of alkenes using these Os catalysts proceeded smoothly to afford the corresponding diols in high yields with high enantioselectivities. The catalysts could be recovered and reused. One mol-scale preparation of a key intermediate for camptothecin, an anticancer drug, has also been successfully demonstrated by using the catalysts.
Introduction
In modern organic synthesis based on Green Sustainable Chemistry (GSC),1 highly toxic reagents and catalysts are avoided even if their activities are very high. Examples are lead and mercury reagents/catalysts, and more recently organotin(IV) compounds. A similar case is osmium tetroxide (OsO4), which catalyses dihydroxylation of alkenes by oxidants such as hydrogen peroxide. While OsO4 is very toxic and volatile, OsO4-catalyzed dihydroxylation of alkenes is particularly useful when applied to asymmetric variants. The asymmetric dihydroxylation of alkenes using catalytic amounts of OsO4 and biscinchona alkaloids such as 1,4-bis(9-O-dihydroquinidinyl)phthalazine [(DHQD)2PHAL] provides optically active vicinal diols very efficiently.2 Although several processes have gained wide acceptance of this asymmetric dihydroxylation that could be applied to the total synthesis of natural products, pharmaceuticals, fine chemicals, etc. on preparative scales,3 few fruitful industrial applications have been accomplished because of the toxicity of OsO4 and the difficulty in separating and recovering the osmium.4 To address these issues, several groups have investigated immobilization of chiral ligands onto soluble and insoluble polymers or inorganic supports.5 However, recovery and reuse of the osmium (Os) is still difficult, presumably due to relatively weak interactions between Os and ligands. On the other hand, we have developed a novel immobilized OsO4, microencapsulated (MC) OsO4, where OsO4 was immobilized onto polystyrene-based polymers by the microencapsulation technique.6 While MC OsO4-catalyzed dihydroxylation of alkenes including asymmetric variants proceeded smoothly in high yields with high selectivities without significant leaching of Os, solvents employed in these reactions were limited. Although other immobilized OsO4 catalysts have also been developed,7 similar issues are reported, and to the best of our knowledge, large-scale synthesis including industrial applications has not been disclosed. Here, we report novel, nontoxic, and nonvolatile polymer-supported Os catalysts, which can be recovered and reused without significant loss of activity in asymmetric dihydroxylation of alkenes. The catalysts were applied to one mol-scale synthesis of an anticancer drug, camptothecin intermediate.
Results and discussion
Preparation of polymer-incarcerated osmium (PI Os) catalysts
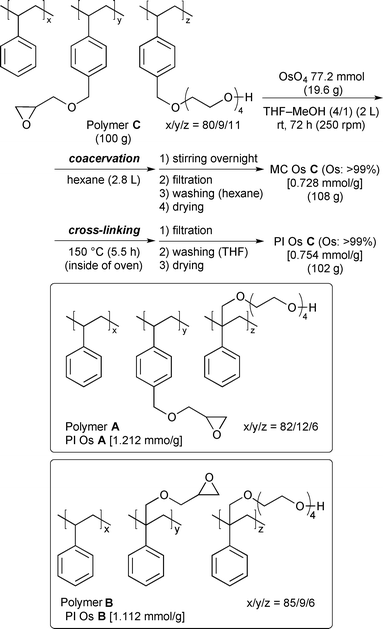
The new Os catalysts were prepared based on our polymer-incarcerated (PI) method.8 The PI method is based on two procedures; microencapsulation and cross-linking. While the MC technique is efficient for keeping metal(0) nanoclusters stable, interaction between metal nanoclusters and benzene rings in polymers is enough strong to prevent aggregation, but not too strong to deactivate catalysts. On the other hand, MC catalysts are dissolved in some solvents, and in those cases, recovery and reuse are difficult. In order to address this issue, cross-linking parts were introduced in PI catalysts. The PI catalyst can be used in most solvents without leaching of metal sources. In addition to the feature of solvent tolerance, metal(0) nanoclusters are “locked up” in the polymer network and cannot aggregate any more to keep high reactivity. The PI method was previously used mainly for immobilization and stabilization of metal nanoclusters such as Pd, Pt, and Au, etc. We intended to use this method for the immobilization of OsO4 (Scheme 1). First, OsO4 was added to a solution of copolymers A–C8 in THF–MeOH (4/1), and the mixture was stirred at room temperature. The colour of the mixture gradually turned from pale yellow to black. After stirring for 72 h, hexane was added dropwise to form black microencapsulated osmium (MC Os) catalysts, and this suspension was stirred overnight. The solvents were removed by decantation, and the resulting MC Os A–C were washed with hexane several times and dried in vacuo. MC Os A–C were then heated at 150 °C for 5–5.5 h in an oven, filtered, and washed with THF to afford PI Os A–C, which were insoluble in almost all organic solvents and water.
Appearance, volatility, and toxicity of PI Os
The PI Os catalysts were black powders (Fig. 1d), and it was found that there was no sublimation of the Os component from the PI Os catalysts. This is remarkable because OsO4 is very volatile. Indeed, OsO4 is colourless to pale yellow crystals, but sublimes rapidly at room temperature. For example, a few crystals of OsO4 were placed on cottons in a sealed sample case, and even after 90 s, dark yellow sublimation was observed (Fig. 1a).10 After 2 h, no solid existed and the yellow colour became darker (Fig. 1b) and darker (after 6 h, Fig. 1c). On the other hand, no change was observed for PI Os C even after 24 h. Indeed, PI Os is stable for several months in air, and no sublimation of the Os component is observed.
 |
| | Fig. 1 Appearance and volatility analysis of OsO4 (a–c) and PI Os C (d).10 | |
Next, the toxicity of these PI Os catalysts was examined. We conducted the acute toxicity assays, and mice received an oral administration of OsO4, OsO2, or PI Os C. Three out of five mice died after receiving OsO4 (100 mg kg−1). This result is consistent with the literature on chemical substances (LD50, 162 mg kg−1).11 Administration of OsO2 (300 mg kg−1) led to delayed toxicity (dead after three days, 1/5 mice; dead after a week, 1/5 mice). However, five out of five mice were alive without any apparent defects at least a week after receiving PI Os C (300 mg kg−1). Next, to confirm deposited Os in organs, some organs were excised from each of the mice treated with OsO4, OsO2, and PI Os C and analysed by XRF analysis after homogenization (see ESI† for details). Deposited Os was detected in the oesophagus (33.7 ppm), stomach (780.7 ppm), and lung (17.4 ppm) of OsO4 (300 mg kg−1)-treated mice. By contrast, no Os deposit was observed in the organs of OsO2 (300 mg kg−1)- or PI Os C (300 mg kg−1)-treated mice. In the case of PI Os C, administration of a higher amount of catalysts (3000 mg kg−1) was tested and again no Os deposit was observed. These findings indicate that PI Os has no acute toxicity and no adsorbent action on organs.
XAFS analysis of PI Os
We next examined the active site structure of PI Os. An X-ray absorption technique was applied to investigate the electronic state and local structure of Os species in the catalysts. For the X-ray absorption near-edge structure (XANES) of PI Os C and the black precipitate from a methanol solution of OsO4 (see ESI† for details), the peak positions of the spectra are not close to those for either OsO4 or K2OsO4, which contain Os8+ and Os6+, but are identical to that for OsO2, which contains Os4+ (Fig. 2). These results indicated that the oxidation state was reduced from Os8+ in OsO4 as the starting material to Os4+ in the catalyst sample during the catalyst preparation.
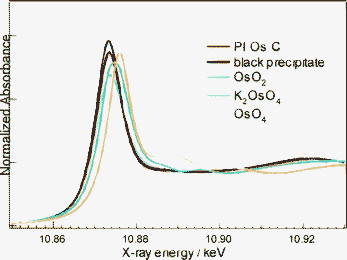 |
| | Fig. 2 Os L3-edge XANES spectra of PI Os and reference compounds. | |
Nonlinear curve-fitting analyses of the Fourier-filtered EXAFS of the first neighbour around Os atoms (Table 1) show that the coordination numbers and interatomic distances for the Os–O bonds in the catalyst were estimated as 2.0 and 1.86 Å for the short Os–O bond, and 4.0 and 2.03 Å for the long Os–O bond, which is consistent with the values for the OsO2 reference sample. These results demonstrate that the Os species in the catalyst would have a similar local structure to that in OsO2. The curve-fitting analysis also indicates that the local structure of the black precipitate (see ESI†) is fundamentally the same as that of the catalyst. Therefore, it can be proposed that the starting material OsO4 was reduced to OsO2 with methanol during the encapsulation step in the polymer backbone (see Scheme 1).
Table 1 Results of curve-fitting analyses for the first neighbour of the Os atom in the samples
| Sample |
Shell |
Coordination numbera |
Interatomic distance (Å) |
Δσ2b |
|
The errors in coordination number and interatomic distance are ±20% and ±0.01 Å.
Δσ2: The difference of Debye–Waller factor from that for OsO4.
See ESI†.
|
| OsO4 |
Os–O |
4.0 |
1.70 |
0 |
| OsO2 |
Os–O |
2.0 |
1.85 |
0.00089 |
| Os–O |
4.0 |
2.02 |
0.00316 |
| PI Os C |
Os–O |
2.0 |
1.86 |
0.00021 |
| Os–O |
4.0 |
2.03 |
0.00205 |
| Black precipitatec |
Os–O |
2.0 |
1.87 |
−0.00134 |
| Os–O |
4.0 |
2.03 |
0.00069 |
Application of PI Os catalysts to asymmetric dihydroxylation and the synthesis of a key intermediate of camptothecin
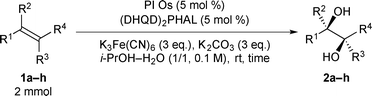
We then conducted asymmetric dihydroxylation of alkenes 1a–1h using PI Os A–C (Table 2). When PI Os A was used as a catalyst for asymmetric dihydroxylation of α-methylstyrene 1a, although the reaction proceeded smoothly to afford 2-phenyl-1,2-propanediol 2a in good yield with good enantiomeric excess (ee), slightly higher leaching of the Os was observed by X-ray fluorescence (XRF) analysis (entry 1). By contrast, PI Os B and C gave the same catalytic activity and selectivity as that of PI Os A with less leaching of the Os (entries 2 and 3). We selected PI Os C because of the availability of the polymer. Furthermore, we examined the concentration of the substrate and the ratio of iPrOH and H2O; however, no improvement was observed.12 Styrene 1b and β-substituted styrene derivatives 1c and 1d worked well under the standard conditions to afford the corresponding diols 2b–2d in high yields with high enantioselectivities (entries 4–6). In the cases of 2-phenylcyclohexene 1e and ethyl cinnamate 1f, t-BuOH in place of iPrOH was found to be effective (entries 7 and 8). It is noteworthy that aliphatic olefins 1g and 1h also reacted smoothly to give the desired diols 2g and 2h in high yields with high to excellent enantioselectivities (entries 9 and 10).
| Entry |
Substrate |
PI Os |
Time (h) |
Yield (%)a |
ee (%)b |
Leaching (%)c |
|
Yield of the isolated product after purification.
The ee was determined by HPLC analysis.
Leaching of the Os was determined by XRF analysis.
Results in 2nd run and 3rd run.
1 eq. of MeSO2NH2 was added.
In t-BuOH–H2O (1/1, 0.1 M).
Determined by HPLC analysis of bisbenzoate.
|
| 1 |
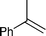
|
1a
|
A
|
5 |
2a, 94 |
92 |
5 |
| 2 |
B
|
2 |
2a, 87 |
91 |
1 |
| 3 |
C
|
5 |
2a, 88 (92, 91)d |
92(92, 91)d |
1 (3, 2)d |
| 4 |
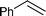
|
1b
|
C
|
3 |
2b, 81 |
95 |
< 1 |
| 5e |

|
1c
|
C
|
5 |
2c, 91 |
97 |
2 |
| 6e |

|
1d
|
C
|
10 |
2d, 96 |
99 |
< 1 |
| 7ef |
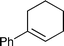
|
1e
|
C
|
21 |
2e, 87 |
97 |
2 |
| 8ef |

|
1f
|
C
|
24 |
2f, 80 |
>99.5 |
3 |
| 9 |

|
1g
|
C
|
4.5 |
2g, 82 |
82 |
1 |
| 10e |

|
1h
|
C
|
5 |
2h, 92 |
97g |
1 |
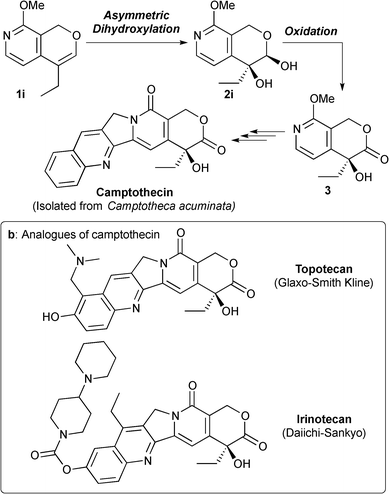
Next, we employed PI Os catalysts for the synthesis of a key intermediate of camptothecin.13 Camptothecin is a cytotoxic quinoline alkaloid that inhibits the DNA enzyme topoisomerase I.14 Since the discovery of camptothecin, many analogues have been synthesized and two of them, topotecan15 and irinotecan16 (Fig. 3b), are used in clinics for the treatment of cancers. In 1994, Fang and co-workers reported a practical application of Sharpless asymmetric dihydroxylation to the synthesis of key intermediate 3 produced in Comins's asymmetric synthesis of camptothecin (Fig. 3a).17
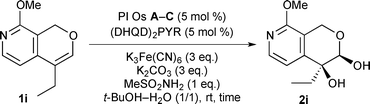
We examined asymmetric dihydroxylation of olefin 1i using PI Os catalysts (Table 3). In this investigation, hydroquinidine-2,5-diphenyl-4,6-pyrimidinediyl diether [(DHQD)2PYR]18 was used as a chiral ligand. When PI Os A was used as a catalyst, although the reaction proceeded smoothly to afford the desired diol 2i in good yield with good ee, significant leaching of the Os was observed by XRF analysis (entry 1). On the other hand, PI Os B or C gave the same catalytic activity and selectivity as that of PI Os A with less leaching of the Os (entries 2 and 3). Encouraged by these results, we next investigated the concentration of the substrate for reduction of the amount of solvents for large-scale production. However, when the reaction was carried out at higher concentration, the ee dropped (entry 4). In particular, in a 10 mmol-scale reaction, significant loss of the ee was observed (entry 6). On the other hand, in the case of 0.1 M, the catalyst could be recovered and reused without loss of activity and selectivity (entry 5). Therefore, we determined the conditions as shown in entry 5 to be the optimal reaction conditions and increased the reaction scale to 100 mmol. As a result, high yield and good ee were obtained (entry 7). At this time, we also tried to recover the chiral ligand from the crude mixture, and it is noted that 97% of (DHQD)2PYR could be recovered.
| Entry |
PI Os |
Scale of 1i (mmol) |
Conc. (M) |
Time (h) |
Yield (%)a |
ee (%)b |
Leaching (%)c |
|
Yield of the isolated product after purification.
The ee was determined by HPLC analysis after oxidation of diol 2i.
Leaching of the Os was determined by XRF analysis.
Results in 2nd run and 3rd run.
Recovery of (DHQD)2PYR.
|
| 1 |
A
|
2 |
0.1 |
13 |
92 |
88 |
20 |
| 2 |
B
|
2 |
0.1 |
13 |
87 |
85 |
5 |
| 3 |
C
|
2 |
0.1 |
16 |
90 |
85 |
1 |
| 4 |
C
|
2 |
0.2 |
25 |
90 |
84 |
3 |
| 5 |
C
|
10 |
0.1 |
20 |
92 (99, 97)d |
86 (85, 85)d |
1 (1, 2)d |
| 6 |
C
|
10 |
0.2 |
20 |
99 (quant, 95)d |
80 (77, 76)d |
<1 (1, 1)d |
| 7 |
C
|
100 |
0.1 |
20 |
97[97]e |
84 |
2 |
With these good results in hand, we further scaled up and conducted 1 mol-scale asymmetric dihydroxylation of olefin 1i (Scheme 2). After optimization of the large-scale preparation, the dihydroxylation reaction proceeded smoothly and after 16 h, full conversion of olefin 1i was observed. To quench the reaction, 316 g of sodium thiosulfate (Na2S2O3) and 2 L of isopropyl alcohol (iPrOH) were added, and the mixture was further stirred for 10 min. PI Os C was recovered (97%) by simple filtration and washed with iPrOH and H2O. Although a very small amount of Os leaching was detected in the filtrates, the amounts could be suppressed to minimum level. A part of the organic phase was treated with a catalytic amount of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and sodium hypochlorite (NaOCl) to determine the enantioselectivity of the product (86% ee).19 Diol 2i and the chiral ligand were readily separated by precipitation (twice) and column chromatography on silica gel. The desired diol 2i was isolated in 97% yield (234 g) with a small amount of contamination of methanesulfonamide (93% purity) and 97% of (DHQD)2PYR was recovered. It is noted that satisfactory results were obtained in 1 mol-scale preparation of the key intermediate of camptothecin, an anticancer drug.
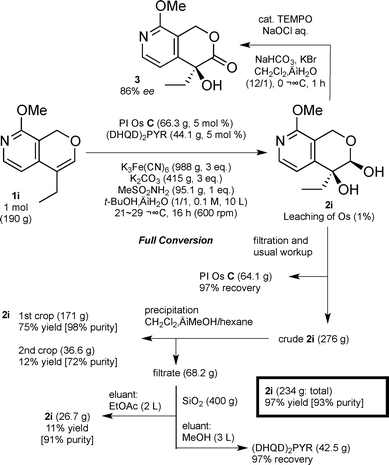 |
| | Scheme 2 One mol-scale asymmetric dihydroxylation of olefin 1i using PI Os C.20 | |
Conclusions
In summary, we have developed novel polymer-incarcerated (PI) Os catalysts. Several important findings are disclosed here: (1) The PI method, which was previously used mainly for immobilization of metal nanoclusters, was successfully used for the immobilization of OsO4; (2) No sublimation of the Os component was observed from PI Os, which was shown to be stable for several months in air; (3) It was shown from experiments with mice that PI Os has no acute toxicity and no absorbent action on organs; (4) XANES and EXAFS analyses of PI Os indicated that the active site structure of PI Os was similar to that of OsO2, which could be formed from OsO4 with methanol during the preparation of the catalyst;21 (5) PI Os showed excellent catalytic activities for asymmetric dihydroxylation of a variety of alkenes; (6) In particular, PI Os was applied to one mol-scale asymmetric synthesis of a key intermediate for camptothecin, an anticancer drug, and high yield and good ee were obtained; The catalyst could be recovered and the Os leaching was suppressed to minimum level. For a long time OsO4 has been avoided in spite of its usefulness in organic synthesis. We now conclude that the new osmium catalysts (PI Os catalysts) presented here are safe and clean. Applications to the large-scale preparation of pharmaceutical intermediates may also be possible. Moreover, the catalysts and concepts shown in this study may be applicable to other reagents and catalysts, which have high activities but are avoided due to high toxicity and volatility.
Acknowledgements
This work was partially supported by ERATO (JST), NEDO (Green-Sustainable Chemical Process Project), a Grant-in-Aid for Science Research from the Japan Society for the Promotion of Science (JSPS) and Global COE Program (Chemistry Innovation through Cooperation of Science and Engineering), the University of Tokyo, MEXT, Japan. The X-ray absorption experiments were performed with the approval of the Photon Factory Program Advisory Committee (Proposal No. 2003G250, 2005G204, and 2008G510). We thank Dr Tasuku Ishida for his contribution at the initial stage of this work.
References
-
(a)
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed;
(b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS;
(c) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS;
(d) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC;
(e) P. T. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
-
(a) K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS;
(b) W. Amberg, Y. L. Bennani, R. K. Chadha, G. A. Crispino, W. D. Davis, J. Hartung, K.-S. Jeong, Y. Ogino, T. Shibata and K. B. Sharpless, J. Org. Chem., 1993, 58, 844–849 CrossRef CAS.
-
(a) H. C. Kolb, M. S. Van Nieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS;
(b)
R. A. Johnson and K. B. Sharpless, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Weinheim, 2nd edn, pp. 357–398 Search PubMed.
- While the use of K2OsO2(OH)4 instead of OsO4 was reported,2a the toxicity issue still remains. In addition, due to some other reasons, OsO4 is still used for asymmetric dihydroxylation. See for example: M. M. Ahmed, M. S. Mortensen and G. A. O'Doherty, J. Org. Chem., 2006, 71, 7741–7746 CrossRef CAS.
-
(a) Selected examples: C. Bolm and A. Gerlach, Eur. J. Org. Chem., 1998, 21–27 CrossRef CAS;
(b) P. Salvadori, D. Pini and A. Petri, Synlett, 1999, 1181–1190 CrossRef CAS;
(c) S. Kobayashi and M. Sugiura, Adv. Synth. Catal., 2006, 348, 1496–1504 CrossRef CAS;
(d) B. B. Lohray, A. Thomas, P. Chittari, J. R. Ahuja and P. K. Dhal, Tetrahedron Lett., 1992, 33, 5453–5456 CrossRef CAS;
(e) D. Pini, A. Petri and P. Salvadori, Tetrahedron: Asymmetry, 1993, 4, 2351–2354 CrossRef CAS;
(f) C. E. Song, E. J. Roh, S.-g. Lee and I. O. Kim, Tetrahedron: Asymmetry, 1995, 6, 2687–2694 CrossRef CAS;
(g) H. Han and K. D. Janda, Tetrahedron Lett., 1997, 38, 1527–1530 CrossRef CAS;
(h) E. Nandanan, A. Sudalai and T. Ravindranathan, Tetrahedron Lett., 1997, 38, 2577–2580 CrossRef CAS;
(i) C. E. Song, J. W. Yang and H.-J. Ha, Tetrahedron: Asymmetry, 1997, 8, 841–844 CrossRef CAS;
(j) A. Petri, D. Pini, S. Rapaccini and P. Salvadori, Chirality, 1999, 11, 745–751 CrossRef CAS;
(k) H. Li, Y.-Y. Luk and M. Mrksich, Langmuir, 1999, 15, 4957–4959 CrossRef CAS;
(l) A. Mandoli, D. Pini, A. Agostini and P. Salvadori, Tetrahedron: Asymmetry, 2000, 11, 4039–4042 CrossRef CAS;
(m) H. M. Lee, S.-W. Kim, T. Hyeon and B. M. Kim, Tetrahedron: Asymmetry, 2001, 12, 1537–1541 CrossRef CAS;
(n) I. Motorina and C. M. Crudden, Org. Lett., 2001, 3, 2325–2328 CrossRef CAS;
(o) B. M. Choudary, N. S. Chowdari, K. Jyothi, N. S. Kumar and M. L. Kantam, Chem. Commun., 2002, 586–587 RSC;
(p) Y.-Q. Kuang, S.-Y. Zhang and L.-L. Wei, Synth. Commun., 2003, 33, 3545–3550 CrossRef CAS;
(q) M. S. DeClue and J. S. Siegel, Org. Biomol. Chem., 2004, 2, 2287–2298 RSC;
(r) Y. S. Park, C. H. Jo, H. Y. Choi, E. K. Kwon and C. E. Song, Bull. Korean Chem. Soc., 2004, 25, 1671–1675 CrossRef CAS;
(s) R.-t. Cha, S.-y. Wang and S.-l. Cheng, J. Appl. Polym. Sci., 2008, 108, 845–849 CrossRef CAS.
-
(a) Selected examples: S. Nagayama, M. Endo and S. Kobayashi, J. Org. Chem., 1998, 63, 6094–6095 CrossRef CAS;
(b) S. Kobayashi, M. Endo and S. Nagayama, J. Am. Chem. Soc., 1999, 121, 11229–11230 CrossRef CAS;
(c) S. Kobayashi, T. Ishida and R. Akiyama, Org. Lett., 2001, 3, 2649–2652 CrossRef CAS;
(d) T. Ishida, R. Akiyama and S. Kobayashi, Adv. Synth. Catal., 2003, 345, 576–579 CrossRef CAS;
(e) T. Ishida, R. Akiyama and S. Kobayashi, Adv. Synth. Catal., 2005, 347, 1189–1192 CrossRef CAS;
(f) R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS;
(g) S. Kobayashi and R. Akiyama, Chem. Commun., 2003, 449–460 RSC;
(h) See also: S. Kobayashi and S. Nagayama, J. Am. Chem. Soc., 1998, 120, 2985–2986 CrossRef CAS.
-
(a) Selected examples: B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, Angew. Chem., Int. Ed., 2001, 40, 4620–4623 Search PubMed;
(b) B. M. Choudary, N. S. Chowdari, K. Jyothi and M. L. Kantam, J. Am. Chem. Soc., 2002, 124, 5341–5349 CrossRef CAS;
(c) L. C. Branco and C. A. M. Afonso, Chem. Commun., 2002, 3036–3037 RSC;
(d) C. E. Song, D.-U. Jung, E. J. Roh, S.-g. Lee and D. Y. Chi, Chem. Commun., 2002, 3038–3039 RSC;
(e) S. V. Ley, C. Ramarao, A.-L. Lee, N. Østergaard, S. C. Smith and I. M. Shirley, Org. Lett., 2003, 5, 185–187 CrossRef CAS;
(f) A.-L. Lee and S. V. Ley, Org. Biomol. Chem., 2003, 1, 3957–3966 RSC;
(g) J. H. Miao, J. H. Yang, L. Y. Chen, M. Y. Huang and Y. Y. Jiang, Chin. Chem. Lett., 2003, 14, 1008–1011 CAS;
(h) Q. Liu, Z. Zhang, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. A: Chemical, 2004, 224, 213–216 CAS;
(i) L. C. Branco and C. A. M. Afonso, J. Org. Chem., 2004, 69, 4381–4389 CrossRef CAS;
(j) K. J. Kim, H. Y. Choi, S. H. Hwang, Y. S. Park, E. K. Kwueon, D. S. Choi and C. E. Song, Chem. Commun., 2005, 3337–3339 RSC;
(k) B. S. Lee, S. Mahajan and K. D. Janda, Tetrahedron Lett., 2005, 46, 4491–4493 CrossRef CAS;
(l) S. M. Reddy, M. Srinivasulu, Y. V. Reddy, M. Narasimhulu and Y. Venkateswarlu, Tetrahedron Lett., 2006, 47, 5285–5288 CrossRef CAS;
(m) K.-i. Fujita, M. Yamazaki, T. Ainoya, T. Tsuchimoto and H. Yasuda, Tetrahedron Lett., 2010, 51, 8536–8543 CrossRef;
(n) S. Qiu, J. Sun, Y. Li and L. Gao, Mater. Res. Bull., 2011, 46, 1197–1201 CrossRef CAS.
-
(a) Selected examples: R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3412–3413 CrossRef CAS;
(b) H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151–4154 CrossRef CAS;
(c) H. Miyamura, R. Matsubara and S. Kobayashi, Chem. Commun., 2008, 2031–2033 RSC;
(d) C. Lucchesi, T. Inasaki, H. Miyamura, R. Matsubara and S. Kobayashi, Adv. Synth. Catal., 2008, 350, 1996–2000 CrossRef CAS;
(e) H. Miyamura, M. Shiramizu, R. Matsubara and S. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 8093–8095 CrossRef CAS;
(f) H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776–778 RSC;
(g) H. Miyamura, K. Maehata and S. Kobayashi, Chem. Commun., 2010, 46, 8052–8054 RSC;
(h) K. Kaizuka, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2010, 132, 15096–15098 CrossRef CAS;
(i) T. Yasukawa, H. Miyamura and S. Kobayashi, Chem.–Asian J., 2011, 6, 621–627 CrossRef CAS;
(j) W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 3095–3103 CrossRef CAS;
(k) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550–18554 CrossRef;
(l) S. Kobayashi and H. Miyamura, Chem. Rec., 2010, 10, 271–290 CrossRef CAS . In the preparation of PI catalysts, the catalysts were heated at 120–150 °C for cross-linking. No sublimation of Os was observed in the preparation of PI Os.
- Polymer C can be obtained easily from commercially available compounds and monomers, which are synthesized by a scalable simple reaction in a single step (see ESI† for details). Furthermore, it is easy to change the ratio of the monomer for polymers A and B. PI Os A and B were prepared by using the same procedure as that of PI Os C, and >99% of the Os component was immobilized onto the polymers.
- 20 mg of OsO4 (Fig. 1a–c) and 300 mg of PI Os C (Fig. 1d) were loaded into sample cases, which were made of plastic and left them at room temperature. As a result, in the case of OsO4, colour change was observed immediately after the investigation was started (a: after 90 s, b: after 2 h, c: after 6 h). On the other hand, in the case of PI Os C, no colour change was observed even after 24 h.
- U.S. NIOSH, Registry of Toxic Effects of Chemical Substances (RTECS), RTECS No. RN1140000 (2009).
- Initial investigation of reaction conditions were studied using α-methylstyrene (1a) as model substrate. See ESI† for details.
-
(a) C. R. Hutchinson, Tetrahedron, 1981, 37, 1047–1064 CrossRef CAS;
(b) W. Du, Tetrahedron, 2003, 59, 8649–8687 CrossRef CAS.
- M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS.
- W. D. Kingsbury, J. C. Boehm, D. R. Jakas, K. G. Holden, S. M. Hecht, G. Gallagher, M. J. Caranfa, F. L. McCabe, L. F. Faucette, R. K. Johnson and R. P. Hertzberg, J. Med. Chem., 1991, 34, 98–107 CrossRef CAS.
-
(a) S. Sawada, S. Okajima, R. Aiyama, K. Nokata, T. Furuta, T. Yokokura, E. Sugino, K. Yamaguchi and T. Miyasaka, Chem. Pharm. Bull., 1991, 39, 1446–1454 CrossRef CAS;
(b) D. L. Comins, M. F. Baevsky and H. Hong, J. Am. Chem. Soc., 1992, 114, 10971–10972 CrossRef CAS.
- F. G. Fang, S. Xie and M. W. Lowery, J. Org. Chem., 1994, 59, 6142–6143 CrossRef CAS.
- G. A. Crispono, K.-S. Jeong, H. C. Kolb, Z.-M. Wang, D. Xu and K. B. Sharpless, J. Org. Chem., 1993, 58, 3785–3786 CrossRef.
- K. E. Henegar, S. W. ashford, T. A. Baughman, J. C. Sih and R.-L. Gu, J. Org. Chem., 1997, 62, 6588–6597 CrossRef CAS.
- The impurity in diol 2i was not the chiral ligand but methane-sulfonamide. The ee was determined using a small part of the crude mixture after oxidation. See ESI† for details.
- We confirmed that OsO2 and the black precipitate (see Fig. 2 and ESI†) had activity for the dihydroxylation (the yields were lower than that using PI Os, presumably because of low solubility of OsO2 and the black precipitate.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental procedures for the synthesis of polymers A–C and PI Os A–C, the asymmetric dihydroxylation of olefins including one mol scale reaction of 1i, acute toxicity assay and XAFS analysis of PI Os, as well as spectroscopic data of synthesized compounds, are available (23 pages) (PDF). See DOI: 10.1039/c2ra21123h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.