The dispersion of TiO2 modified by the accumulation of CO2 molecules in water: An effective medium for photocatalytic H2 production†
Ruixia
Liu
,
Hiroshi
Yoshida
,
Satomi
Narisawa
,
Shin-ichiro
Fujita
and
Masahiko
Arai
*
Division of Chemical Process Engineering, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan. E-mail: marai@eng.hokudai.ac.jp; Fax: (+81)-11-706-6594
First published on 13th July 2012
Abstract
The photocatalytic H2 production from water with TiO2 can be noticeably enhanced by the presence of both dense phase CO2 and a surfactant of sodium dodecyl sulfate, being larger by more than one order of magnitude than that from water with TiO2 alone. The surface of TiO2 may be modified by the accumulation of CO2 molecules assisted by the TiO2- and CO2-philic surfactant, changing its photocatalytic activity.
Introduction
Recently, H2 has been investigated as a storable, clean, and environmental-friendly fuel for future energy systems.1 Photocatalytic water splitting into H2 has been considered as a significant and attractive solution to solve the global energy and environmental problems.2 It is well known that TiO2 is a cheap, nontoxic, stable and efficient photocatalyst. However, the intrinsic activity of TiO2 is not sufficient due to the facile recombination of photo-generated electron–hole pairs, which is thermodynamically favored3–5 and TiO2 is to some extent constrained by its wide band gap (3.2 eV), which requires ultraviolet irradiation for photocatalytic activation. In order to solve these problems, many researchers have paid much attention to the modification of electronic and optical properties of TiO2, such as surface modification via organic materials/semiconductor coupling and band gap modification with metal and nonmetal dopants.6–9 In addition, various oxidizing sacrifice agents are used in order to suppress the reverse reaction of O2 and H2 forming back into H2O.10,11The present authors have been investigating the effects of dense phase CO2 in different chemical reactions, which can act as a promoter as well as a solvent. One of the significant findings is that CO2 molecules dissolved in organic substrates and solvents interact with some particular functional groups of substrates and modify their reactivity. This does promote the reaction rate and change the product selectivity in hydrogenation reactions of α,β-unsaturated aldehydes12,13 and aromatic nitro compounds,14,15 Heck coupling16 and Diels–Alder reaction.16 Furthermore, the CO2 molecules could affect the properties of supported metal particles as suggested by in situ high pressure optical absorption measurements in dense phase CO2 medium.17
Those previous works with either TiO2 or dense phase CO2 have stimulated us to investigate the possibility of synergistic effects of the two species in the production of H2 by photocatalytic water splitting. In the present work, the production of H2 has been studied with different media including TiO2, dense phase CO2, and/or surfactant. The surfactant has been used to form CO2 micelles, which are adsorbed on the surface of TiO2. This work will report an interesting result that the H2 production can be promoted by synergistic effects of dense phase CO2 and a surfactant of sodium dodecyl sulfate (SDS) by more than one order of magnitude compared to the medium with TiO2 alone.
Results and discussion
The authors tested different reaction media in the presence of dense phase CO2 and/or a surfactant of SDS. The results obtained are shown in Fig. 1. The rate of H2 production observed in the present reaction runs was small. The authors used a laboratory-designed high pressure stainless steel reactor of 50 cm3 as described in the Experimental section. The reaction mixture was illuminated by a high-pressure Hg lamp through a quartz window of 1 cm in diameter attached to the reactor. Therefore, the whole volume of reaction mixture was not necessarily illuminated. This is a main reason for the small rate of H2 production obtained. The rate of H2 production was enhanced from 93 μL g−1 h−1 to 158 μL g−1 h−1 when 3 MPa CO2 was introduced to pure water. Interestingly, the rate of H2 production was noticeably increased to 1285 μL g−1 h−1 when SDS was also added to the medium. The presence of both dense phase CO2 and SDS was indispensable for promoting photocatalytic H2 production with TiO2 in aqueous medium. When dense phase CO2 is present, the pH of the aqueous medium decreases and some acidic species such as H2CO3 and HCO3− are formed;18 the pH decreases to about 3 at CO2 pressures > 3 MPa.19,20 The pH and the formation of these acidic species may be important factors determining the rate of photocatalytic H2 production.21–24 When SDS surfactant is used, TiO2 powders can be dispersed well in water. From the results of Fig. 1, however, the acidic nature and the stable TiO2 dispersion are not responsible for the even larger H2 production in the presence of both dense phase CO2 and SDS, which should act in a synergistic way in modifying the properties of TiO2. We conducted control reaction runs using Ar instead of CO2 under the same conditions. The reactions were made with TiO2 in H2O–SDS–Ar medium at Ar pressures of 3 and 1.5 MPa. The H2 production was observed to be very small compared to that in H2O–SDS–CO2 but comparable to those in H2O and H2O–SDS media. This indicates the importance of a chemical species of CO2 for the higher H2 production observed in the H2O–SDS–CO2 medium.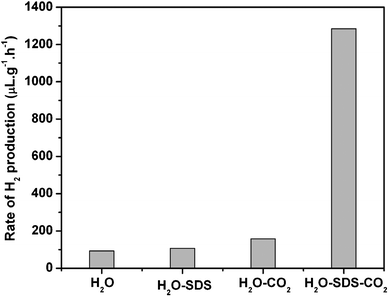 | ||
| Fig. 1 Rate of photocatalytic H2 production from water with TiO2 in different aqueous media. Reaction conditions: TiO2 100 mg, H2O 20 cm3, CO2 3 MPa, SDS 20.25 mM, 323 K, 8 h. | ||
Other surfactants, listed in Table 1, were also tested for comparison (Fig. 2). The comparison of H2 production in the presence and absence of dense phase CO2 imply that 3 MPa CO2 has some positive effect on the H2 production; however, the couples of CTAB, DTAB, and NaHS with dense phase CO2 cannot improve the H2 production so significantly as observed with SDS as shown in Fig. 1. The authors further tested a nonionic surfactant P123 and fluorinated ones of APFO and AHPFO, which were more CO2-philic than the above-mentioned ones. Unfortunately, the degradation of those surfactants was observed to occur to some extent. For example, about 23% of the initial amount of APFO decomposed under the same reaction conditions. However, no decomposition of SDS was observed and a very small fraction, less than 0.5% of the initial quantities, decomposed for the other surfactants of CTAB, DTAB, and NaHS. The TiO2 powders were also observed to be dispersed well in water in the presence of these surfactants, except for the cationic ones. However, the dispersion of TiO2 powders with SDS was more stable compared to the other surfactants; it was more difficult to separate the SDS-stabilized TiO2 powders by simple filtration. The chemical nature of SDS should be important for the enhanced H2 production.
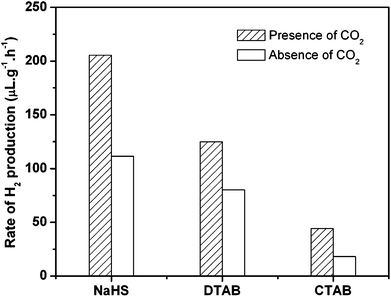 | ||
| Fig. 2 Comparison of photocatalytic H2 production from water with different surfactants in the presence and absence of dense phase CO2. Reaction conditions: TiO2 100 mg, H2O 20 cm3, CO2 3 MPa, 323 K, 8 h. The concentrations of the surfactants used are 2.5 times their CMCs. | ||
| Surfactant | Abbreviation | Molecular formula | CMCa (mM, 298 K) |
|---|---|---|---|
| a Data of critical micelle concentration (CMC) from ref. 26. | |||
| Sodium dodecyl sulfate | SDS | CH3(CH2)11OSO3Na | 8.1 |
| 1-Hexadecanesulfonic acid sodium salt | NaHS | CH3(CH2)15OSO2Na | 0.7 |
| Dodecyltrimethylammonium bromide | DTAB | CH3(CH2)11N(CH3)3Br | 15 |
| Hexadecyltrimethylammonium bromide | CTAB | CH3(CH2)15N(CH3)3Br | 0.92 |
| Poly(ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) | P123 | HO(CH2CH2O)20(CH2CH(CH3)O)70(CH2CH2O)20H | 0.052 |
| Ammonium pentadecafluorooctanoate | APFO | CF3(CF2)6COONH4 | 30–34 |
| Ammonium heptadecafluorooctanesulfonate | AHPFO | CF3(CF2)7SO3NH4 | – |
Then, the influence of either CO2 pressure or SDS concentration and the catalyst stability were further investigated for media including TiO2, dense phase CO2, and SDS. Fig. 3a shows the effect of CO2 pressure on the H2 production, which increased greatly from 365 μL g−1 h−1 at 0.5 MPa to 3185 μL g−1 h−1 at 1.5 MPa but then decreased with the pressure. Very small amounts of CO and CH4, which were less than 5 ppb against H2, were also detected under the conditions used, and the rates of CO and CH4 production deceased as CO2 pressure increased. Fig. 3b gives the effect of SDS concentration, indicating that the H2 production was maximal at a certain concentration comparable to its critical micelle concentration (CMC). The rate of CO production is independent of the concentration of SDS. The reaction was tested for longer reaction times under standard conditions. The production of H2 was observed to occur at a constant rate during the reaction up to 14 h (Fig. S1 in the ESI†); namely, the photocatalytic activity of TiO2 was durable.
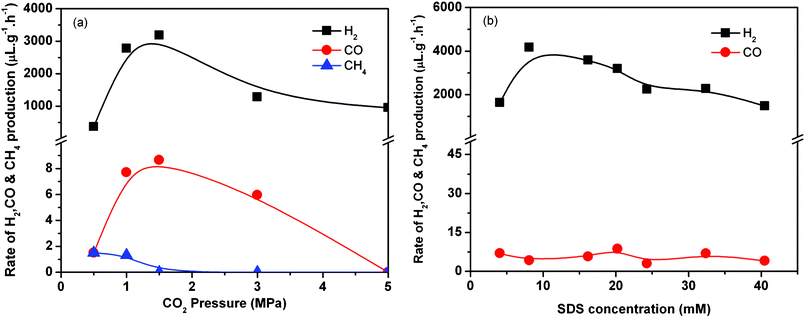 | ||
| Fig. 3 Effect of CO2 pressure (a) and SDS concentration (b) on photocatalytic H2 production in the presence of both SDS and dense phase CO2. Reaction conditions: TiO2 100 mg, H2O 20 cm3, CO2 3 MPa for (b), SDS 20.25 mM for (a), 323 K, 8 h. | ||
Those results show that the presence of both dense phase CO2 and SDS is required to enhance the photocatalytic H2 production. The noticeably larger H2 production observed (Fig. 1) should result from some synergistic effects of these two components. In situ high pressure UV/Vis spectroscopy was used to examine the properties of TiO2 in water in the presence of dense phase CO2 and surfactants. In these UV/Vis experiments, a thin film TiO2 on a quartz plate was used instead of TiO2 powders (see the Experimental section). Fig. 4a shows UV/Vis spectra collected for the TiO2 film in different aqueous solutions compressed by 3 MPa CO2. In the SDS solution, TiO2 shows absorption in a wide range of wavelengths, tailing off at larger wavelengths compared to the other surfactants. The spectrum with NaHS also has a wider absorption but to a lesser extent compared to SDS. The spectrum with DTAB is similar to that in pure water and the one with CTAB has a narrower absorption. The UV/Vis spectra were further collected at different SDS concentrations (Fig. 4b). A wider absorption spectrum was obtained at a concentration of 8.1 mM compared to those at 4.05 mM and 20.25 mM. Comparison with the results of Fig. 1, 2 and 3b suggests that a wider UV/Vis absorption is beneficial to a larger H2 production. However, similar UV/Vis spectra were obtained for the TiO2 film in aqueous solutions including SDS (20. 25 mM) compressed by CO2 at different pressures of 1–12 MPa although the H2 production was maximal at a certain pressure (Fig. 3a).
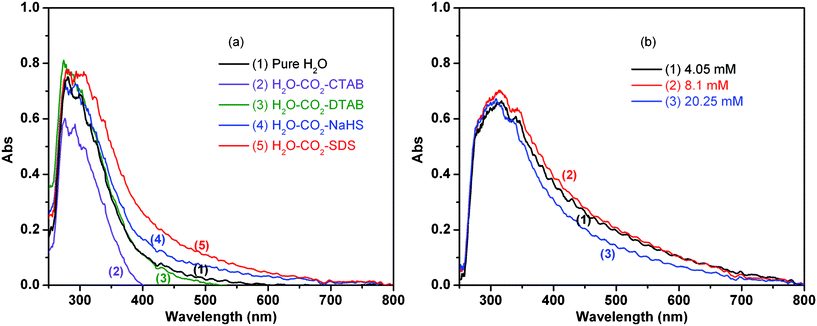 | ||
| Fig. 4 UV/Vis spectra of TiO2 film (a) in water (1) and in different aqueous solutions with CTAB (2), DTAB (3), NaHS (4), and SDS (5) at a CO2 pressure of 3 MPa. The concentrations of the surfactants used is 2.5 times their CMC. (b) Spectra for the aqueous solutions with SDS at different concentrations of (1) 4.05 mM, (2) 8.1 mM, and (3) 20.25 mM at a CO2 pressure of 3 MPa. | ||
Additional reaction experiments were made using a cutoff filter through which the reaction mixture was illuminated by light of 230–410 nm in wavelength. Three reaction systems, H2O–CO2–SDS, H2O alone, and H2O–CO2–CTAB were used. The rates of H2 production were observed to decrease in all these systems as compared to those obtained without the cutoff filter. It is important to note, however, that the extent of the decrease depends on the system examined; the rate of H2 production decreased by 63%, 33%, and 21% for H2O–CO2–SDS, H2O alone, and H2O–CO2–CTAB, respectively. Namely, the most significant decrease appeared for the most active system of H2O–CO2–SDS (–TiO2). These results also indicate the importance of the modification of TiO2 by CO2 molecules on its surface, showing the larger extension of light absorption at wavelengths >360 nm (Fig. 4a).
Although the details are still unclear about the effects of dense phase CO2 and SDS, the coexistence of these two components is indispensable for noticeably enhancing H2 production over TiO2. In the presence of SDS, CO2 molecules are emulsified in SDS micelles and those CO2 micelles should exist in contact with the surface of TiO2 powders, as illustrated in Fig. 5. The accumulation of CO2 molecules on TiO2 is assisted by the presence of SDS and this should modify its surface properties, as suggested by UV/Vis spectroscopy (Fig. 4). The surface of TiO2 may be positively charged under the reaction conditions used because its point zero charge is at a pH value of about 6.18,23 The anionic SDS surfactant is likely to have electrostatic and hydrogen bonding interactions between its sulfate head group and the positively charged surface of TiO2. In contrast, CTAB- and DTAB-containing tetraalkylammonium anions are less likely to have such interactions 25 and so TiO2 powder could not be well dispersed in the water phase in the presence of these surfactants. Another feature of SDS is the occurrence of interactions with the electron deficient CO2 molecules and the interactions should be stronger than that with the cationic surfactant. So, these TiO2– and CO2-philic natures of SDS should be crucial for the larger H2 production in the presence of dense phase CO2 and SDS. For TiO2 modified by CO2 accumulation, it is speculated that the photo-excitation generating electrons and holes may occur with light in a wider wavelength (in a wider energy) tailing off at longer wavelengths (lower energy) (Fig. 4) and/or the recombination of the formed electrons and holes may be slowed down. When the SDS concentration is increased, the CO2 micelles should occupy a larger area of the surface of TiO2; when the whole area is covered by the micelles, however, the contact of TiO2 with water molecules should become difficult. Therefore, the positive effect of SDS may be maximal at a certain concentration (Fig. 3b). The BET surface area of TiO2 used is 11 m2 g−1 and the total area of TiO2 powder used (0.10 g) is 1.1 m2. Under the conditions used, the number of SDS molecules present in water of 20 cm3 is 0.162 mmol at its CMC (8.1 mM).26 Assuming the area occupied by one SDS molecule at the surface of micelles is 0.65 nm2, the surfactant used can cover an area of 64 m2. Possibly, the amount of SDS surfactant molecules is so sufficient at CMC that a sufficient number of CO2 molecules may accumulate on the surface of TiO2 and change its properties. The influence of CO2 pressure is also maximal at a certain pressure (Fig. 3a). When CO2 pressure is raised, the number and/or volume of the micelles in contact with TiO2 could increase. Some fraction of the surface of TiO2 is required to be exposed free of CO2 micelles for the photocatalysis and so a certain pressure may give a maximum effect. The CO2 micelles, adsorbed on TiO2 and present freely in water, might work as a pool for H2.
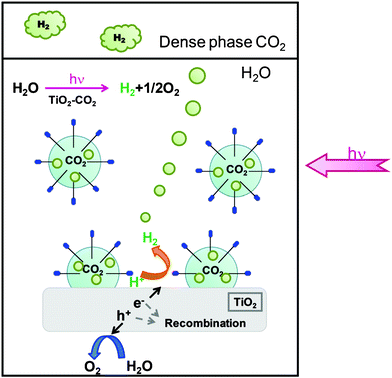 | ||
| Fig. 5 Illustration of the reaction medium including TiO2 powder, SDS surfactant, and dense phase CO2. The properties of TiO2 photocatalyst may be modified by the accumulation of CO2 molecules on its surface assisted by the surfactant. | ||
In conclusion, the photocatalytic H2 production from water with TiO2 can be noticeably enhanced by the presence of both dense phase CO2 and SDS surfactant, by more than one order magnitude compared to that in neat water. The surface properties of TiO2 photocatalyst should be modified by the accumulation of CO2 micelles (nano bubbles). This is a new type of the chemical function of dense phase CO2 as promoter and modifier. The dense phase CO2 accumulating on the surface of TiO2 would promote the photo-excitation generating electron and hole or retard the recombination of the electron and hole formed. Further experiments of photocatalytic reactions under different conditions and in situ high pressure UV/Vis and FTIR spectroscopy are in progress to clarify the synergistic effects of CO2 and SDS in our laboratory.
Experimental
General
Anatase TiO2 (particle size 217 nm, surface area 11 m2 g−1) and several types of surfactants were purchased from Aldrich and used without further purification. Distilled water (Wako) and CO2 (99.99%) were used as delivered.Photocatalytic hydrogen production
The photocatalytic reactions were carried out in a 50 cm3 stainless steel autoclave with a quartz window (diameter 1 cm), using a 500 W high-pressure Hg lamp (Ushio USH-500SC). This lamp emits predominantly in the wavelength range of 360–370 nm, 400–410 nm, and 430–440 nm. In a typical run, a weighed amount of surfactant and water (20 cm3) were added into the reactor and the mixture was stirred for 2 min. Then, the mixture was loaded with 100 mg TiO2 and further stirred for 2 min. After sealing, the reactor was flushed three times with Ar and CO2 for media without CO2 and those with CO2, respectively, to remove the air. The reactor was placed on a heating plate and bound with heating tape. The temperature was monitored by a thermocouple embedded in the reactor wall. When the reactor was heated and reached the reaction temperature of 323 K, CO2 was introduced up to the desired pressure and the reaction mixture was illuminated by turning on the lamp. After the reaction, the gases (mainly H2, CO2, CO, CH4) evolved were collected in a gas trap. The amount of H2 was determined by a gas chromatograph (Shimadzu GC-8A, molecular sieve 5A packed column, TCD detector, N2 carrier) and CO, CO2, and CH4 by another gas chromatograph (Shimadzu GC-8A, Porapak Q packed column, FID detector, N2 carrier) with a methanizer converting those gases into CH4. The reaction was also tested without TiO2 in the presence of dense phase CO2 and surfactant under illumination and with TiO2 in dark. No products were detected in these reactions. The reaction runs were repeated under the same conditions to ensure the reliability of data collection.In situ high pressure UV/Vis measurement
The properties of TiO2 in water compressed by CO2 were examined by in situ high pressure UV/Vis spectrometer (Jasco Model V-570).17 We failed to collect good spectra for TiO2 powders due to their light scattering; our UV/Vis cell did not have an integrated mirror ball to gather sufficient effluent light. So, thin TiO2 film on a quartz plate was used, which was prepared by vacuum evaporation of Ti in a thickness of 10 nm on the plate (about 5 mm × 25 mm) followed by oxidation in air at 723 K for 5 h.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) with a Grant-in-Aid for Scientific Research (B) 22360327 and for JSPS Fellows 2200038601. The authors are thankful to Prof. T. Shimada and Dr T. Nagahama for their help in the preparation of the thin Ti film sample.References
- D. Y. C. Leung, X. Fu, C. Wang, M. Ni, M. K. H. Leung, X. Wang and X. Fu, ChemSusChem, 2010, 3, 681–694 CrossRef CAS.
- M. Bowker, Green Chem., 2011, 13, 2235–2246 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M .R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwahi, K. Aoki and Y. Taga, Science, 2001, 293, 269–217 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- G. Liu, L. Wang, H. Yang, H. Cheng and G. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- A. A. Nada, M. H. Barakat, H. A. Hamed, N. R. Mohamed and T. N. Veziroglu, Int. J. Hydrogen Energy, 2005, 30, 687–691 CrossRef CAS.
- K. Hara, K. Sayama and H. Arakawa, Appl. Catal., A, 1999, 189, 127–137 CrossRef CAS.
- F. Zhao, S. Fujita, J. Sun, Y. Ikushima and M. Arai, Chem. Commun., 2004, 2326–2327 RSC.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 12590–12599 CrossRef.
- X. Meng, H. Cheng, Y. Akiyama, Y. Hao, W. Qiao, Y. Yu, F. Zhao, S. Fujita and M. Arai, J. Catal., 2009, 264, 1–10 CrossRef CAS.
- H. Yoshida, K. Kato, J. Wang, X. Meng, S. Narisawa, S. Fujita, Z. Wu, F. Zhao and M. Arai, J. Phys. Chem. C, 2011, 115, 2257–2267 CAS.
- S. Fujita, T. Tanaka, Y. Akiyama, K. Asai, J. Hao, F. Zhao and M. Arai, Adv. Synth. Catal., 2008, 350, 1615–1625 CrossRef CAS.
- M. Arai, Y. Nishiyama and Y. Ikushima, J. Supercrit. Fluids, 1998, 13, 149–153 CrossRef CAS.
- Y. Ku, W. Lee and W. Wang, J. Mol. Catal. A: Chem., 2004, 212, 191–196 CrossRef CAS.
- K. L. Toews, R. M. Shroll, C. M. Wai and N. G. Smart, Anal. Chem., 1995, 67, 4040–4043 CrossRef CAS.
- J. D. Holmes, D. C. Steytler, G. D. Rees and B. H. Robinson, Langmuir, 1998, 14, 6371–6376 CrossRef CAS.
- X. Yang, T. Xiao and P. P. Edwards, Int. J. Hydrogen Energy, 2011, 36, 6546–6552 CrossRef CAS.
- A. Hameed, M. A. Gondal, Z. H. Yamani and A. H. Yahya, J. Mol. Catal. A: Chem., 2005, 227, 241–246 CrossRef CAS.
- W. Lin, W. Yang, I. L. Huang, T. Wu and Z. Chung, Energy Fuels, 2009, 23, 2192–2196 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Chem. Soc., Faraday Trans., 1997, 93, 1647–1654 RSC.
- K. D. Dobson, A. D. Roddick-Lanzilotta and A. J. McQuillan, Vib. Spectrosc., 2000, 24, 287–295 CrossRef CAS.
- J. H. Fendler and E. J. Fendler, Catalysis in Micellar and Macromolecular Systems, Academic Press, London, 1975, pp. 20–21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21102e |
| This journal is © The Royal Society of Chemistry 2012 |