Synthesis and chemistry of a very stable but reactive diisocyanate, trioxabicyclo[3.3.1]nonadiene diisocyanate†
Ralph
Smounig
a,
Sabine
Leber
ab,
David
Kvaskoff
b,
Gert
Kollenz
*a and
Curt
Wentrup
*b
aInstitute of Chemistry, Karl-Franzens University of Graz, Heinrichstrasse 28, A-8010, Graz, Austria. E-mail: gert.kollenz@uni-graz.at
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia. E-mail: wentrup@uq.edu.au
First published on 27th June 2012
Abstract
The bisdioxine diisocyanate 5 is exceptionally stable, being formed readily in Curtius and Hofmann rearrangements. It does not hydrolyze easily but forms diurea and diurethane derivatives in high yields.
Introduction
The concave, axially chiral1 bridged bisdioxine (trioxabicyclo[3.3.1]nonadiene) diacid dichloride 3 is obtained by acid hydrolysis and subsequent chlorination of the surprisingly stable α-oxoketene 2, itself obtained by dimerization of dipivaloylketene 1.2–4 Since 3 and its derivatives are chiral, they exist as racemates. Only the S-forms are shown for clarity. The concave and sterically shielding5 tetra-tert-butylated structure of the bis-dioxine unit makes it an interesting spacer group, which has been applied successfully in syntheses of several macrocyclic systems.6–9 Some of them exhibit pronounced complexation of metal ions.8–10 Functional group manipulation is of interest in order to enhance the toolkit for generating novel macrocyclic and macromolecular systems. We now report the synthesis and chemical properties of the surprisingly stable, monomeric diisocyanate 5.There are two paths leading to the bisdioxine diisocyanate 5 (Scheme 1). (a) The bisdioxine diacid dichloride 3 and sodium azide react at room temperature in a one-pot procedure via a non-isolable diacyl azide in a Curtius rearrangement11 to give 5 in 87% yield. Here it is interesting to note that the extrusion of nitrogen occurs spontaneously without any heating or irradiation. (b) The primary amide 4 is prepared by adding dry, gaseous ammonia into a solution of the diacid dichloride 3 in dichloromethane. Treating the amide 4 with sodium hydroxide/bromine according to the Hofmann rearrangement12 surprisingly does not lead to hydrolysis to form the corresponding primary diamine, but instead the reaction again stops at the diisocyanate, which is isolated in 80% yield. The amazing stability of the diisocyanate 5 probably derives from the strong steric shielding caused by the bulky tert-butyl groups. The structural characterization of 5 is based on IR, NMR and MS data as well as the elemental analysis (see Experimental section). In particular, the 13C NMR data are in excellent agreement with previously reported data6–10 for other derivatives and thereby characterize the compound as a bisdioxine. A 13C resonance at 123 ppm and a strong IR absorption at 2295 cm−1 (calculated 2290 cm−1 at the B3LYP/6-31G** level, scaled by a factor 0.9613) unambiguously demonstrate the presence of the isocyanate function.
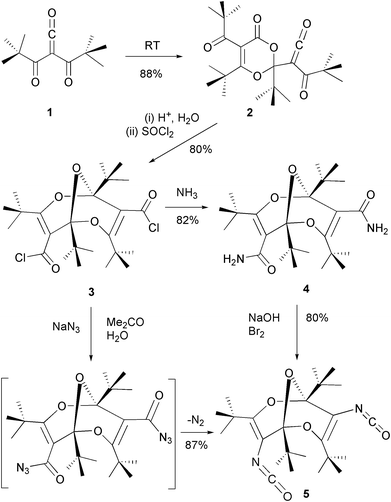 | ||
| Scheme 1 Synthesis of diisocyanate 5. | ||
In order to examine the chemical behaviour and properties of diisocyanate 5, in particular to test its suitability as a spacer molecule in macrocycle and polymer synthesis, it was first reacted with simple NH- and OH-nucleophiles, which resulted in the formation of the corresponding urea and urethane derivatives 6 and 7 in high yields (Scheme 2). In contrast, attempted cycloaddition reactions of 5 with several activated multiple bond systems, e.g. alkenes, carbodiimides, and oxoketenes, met with complete failure, presumably caused by severe steric hindrance owing to the bulky geometry of 5.
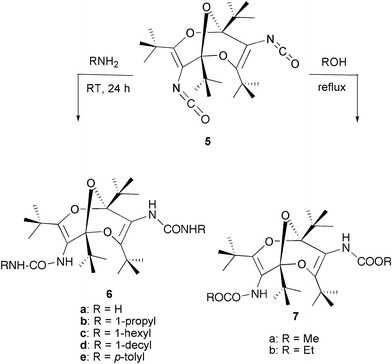 | ||
| Scheme 2 Synthesis of ureas and urethanes from 5. | ||
Calculations†
The structure and key geometrical parameters of diisocyanate 5 at the B3LYP/6-31G** level are shown in Fig. 1 and 2. The analogous calculated structures of the diethyl diester and the diacid corresponding to 3 at the same level of theory are in excellent agreement with the previously determined X-ray structures of these compounds.1 It is therefore very reasonable to assume that the calculated structure of 5 is close to reality.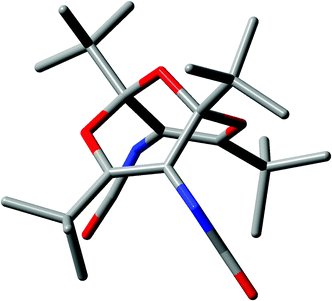 | ||
| Fig. 1 B3LYP/6-31G**-calculated structure of diisocyanate 5 (hydrogen atoms omitted for clarity). See ESI for additional views.† | ||
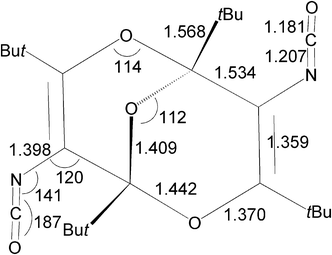 | ||
Fig. 2 Geometrical parameters for diisocyanate 5 at the B3LYP/6-31G** level. Bond lengths in Å and angles in degrees. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are twisted by an angle 11.6°, the concave angle between the C2O fragments of the two six-membered rings is 110°, and there is a C2 symmetry axis passing through the central ether bridge. See ESI for further bond lengths and angles.† C bonds are twisted by an angle 11.6°, the concave angle between the C2O fragments of the two six-membered rings is 110°, and there is a C2 symmetry axis passing through the central ether bridge. See ESI for further bond lengths and angles.† | ||
It is noteworthy that all molecules of this type exist only in the endo,endo forms shown, i.e. with the functional groups pointing away from the central ether bridge (see structure 5 in Schemes 1 and 2). Consequently, the flanking tert-butyl groups are always exo (see Fig. 1 and 2). The corresponding exo,endo and exo,exo isomers do not form stable minima at this computational level, and attempted optimization leads to ring opening (see ESI†).
It is seen in Fig. 1 that each NCO group is surrounded by three tert-butyl groups. While approach of a nucleophile to the CO moieties of the isocyanate groups is still possible, leading to the urea and urethane derivatives (Scheme 2), approach to the N atoms, as would be required in a normal [2+2] cycloaddition to an isocyanate, is highly hindered and does not occur.
Conclusion
The bisdioxine diisocyanate 5 has been prepared by Curtius and Hofmann rearrangements of the corresponding diazides and diamides. It is remarkably stable, not hydrolyzing in 1.5 N NaOH under the Hofmann rearrangement conditions. Nevertheless, it readily forms diurea and diurethane derivatives in high yields (84–94%) under mild conditions. These properties make it an interesting candidate for the preparation of macrocyclic and polymeric ureas and urethanes.Experimental
General
Elemental analyses were performed on a Carlo Erba Elemental Analyser. IR spectra were measured on a Perkin Elmer 290 in KBr, NMR spectra on Varian XL 200 or Bruker 360 MHz instruments, and mass spectra on MS-902 or ZAB-2-SEQ spectrometers. Assignments of NMR signals for bisdioxine units were made in agreement with previously reported data.6–10 Melting points are uncorrected.Preparations of dipivaloylketene 1, the dimeric oxoketene 2 and 1,3,5,7-tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene-4,8-dicarboxylic acid and dicarboxylic acid dichloride 3 were carried out according to literature procedures.1–4
1,3,5,7-Tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-diamide (4)
Dry, gaseous ammonia was passed into a solution of freshly prepared diacid dichloride 3 (200 mg, 0.42 mmol) in dry dichloromethane (20 mL) for 20 min. The solution was stirred at 20 °C for 2 h. The solvent was then evaporated, and the crude, solid residue was digested with acetonitrile (3 mL), filtered by suction, and washed twice with water. Recrystallization from acetonitrile afforded 150 mg (82%) of pure 4, mp 238–240 °C. IR (KBr): 3500, 3360–3150 (NH2), 3020–2870 (CH), 1670 (CO), 1620 (CC). 1H-NMR (CDCl3) δ 1.15, 1.35 (2s, 36H, t-Bu), 5.35, 5.95 (4H, NH2) ppm; 13C-NMR (CDCl3) δ 24.8, 29.1 (CH3, t-Bu), 37.8, 40.2 (C, t-Bu), 97.7 (bisdioxine, C-1/C-5), 105.1 (bisdioxine, C-4/-C-8), 160.0 (bisdioxine, C-3/C-7), 171.0 (CO) ppm. Anal. calcd. for C24H40N2O5: C, 66.01; H, 9.24; N, 6.42. Found: C, 66.00; H, 9.46; N, 6.18%.
1,3,5,7-Tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-diisocyanate (5)
(a) Bromine (0.12 mL; 2.4 mmol) was added dropwise at 0 °C to a solution of 480 mg (12 mmol) of sodium hydroxide in water (8 mL). Diamide 4 (436 mg; 1 mmol) was then added to the solution with stirring at −5 °C, and stirring was continued for 48 h. Sodium sulfite (100 mg) was now added, the mixture was acidified to pH 2, stirred for an additional 30 min, made alkaline with aqueous potassium hydroxide, and saturated with potassium carbonate. The resulting mixture was extracted several times with ether (altogether 200 mL), and the ethereal phase was dried over sodium sulfate. The crude, solid residue obtained after evaporation was triturated with acetonitrile. The final purification by dry column flash chromatography (silicagel 60H, eluting with n-hexane) afforded 345 mg (80%) of pure diisocyanate 5, mp 179–180 °C.(b) Sodium azide (58 mg; 0.89 mmol) was dissolved in 1 mL of water and added to an ice-cold solution of bisacid chloride 3 (200 mg; 0.42 mmol) in 5 mL of dry acetone with vigorous stirring. After 30 min, 10 mL of water was added, and the so-formed precipitate was filtered by suction, washed with water, and dried over phosphorus pentoxide. The white crystals obtained (158 mg, 87%, mp 180 °C) were analytically pure.
IR (KBr) 3040–2870 (CH), 2295 (vs, NCO), 1640 (CC) cm−1. 1H NMR (CDCl3) δ 1.11, 1.25 (2s, 36H, t-Bu). 13C NMR (CDCl3) δ 25.4, 27.7 (CH3, t-Bu), 35.8, 38.4 (C, t-Bu), 97.7 (bisdioxine, C-1/C-5), 103.5 (bisdioxine, C-4/C-8), 123.0 (NCO), 158.0 (bisdioxine, C-3/C-7) ppm. MS (FAB): 432 (M+). Anal. calcd. for C24H36N2O5: C, 66.60; H, 8.30; N, 6.48. Found: C, 66.55; H, 8.58; N, 6.36%.
1,3,5,7-Tetra-tert-butyl-2,6,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-diurea (6a)
Dry, gaseous ammonia was added into a solution of 250 mg (0.46 mmol) of diisocyanate 5 in dry dichloromethane (50 mL) during 10 min with stirring at room temperature. Stirring was continued for 2 h, and the colourless precipitate so formed was isolated by suction filtration and washed twice with water (10 mL). The crude product was recrystallized from dimethylformamide. Yield: 190 mg (8%); mp >350 °C. IR (KBr) 3500–3180 (NH2), 3010–2870 (CH); 1655 (CO), 1590 cm−1, 1H-NMR (d6-DMSO) δ 1.09, 1.18 (2s, 36H, t-Bu), 5.6 (4H, NH2), 6.25 (2H, NH) ppm. 13C-NMR (d6-DMSO) δ 26.07, 29.38 (CH3, t-Bu), 38.9, 39.3 (C, t-Bu, overlap with solvent signal); 97.6 (bisdioxine C-1/C-5), 105.1 (bisdioxine, C-4/C-8), 158.4 (bisdioxine, C-3/C-7), 159.4 (CO) ppm. Anal. calcd. for C24H42N4O5 : C, 61.78; H, 9.07; N, 12.01; Found : C, 61.75; H, 9.21; N, 11.97.
General procedure for reaction of diisocyanate 5 with amines
A portion of 1.5 mmol of the amine is added to a solution of 250 mg (0.46 mmol) of diisocyanate 5 in dry dichloromethane (5 mL), and the mixture is stirred for 24 h at 20 °C. The precipitate so formed is separated by suction and washed two twice with toluene (3 mL). The crude product is triturated with acetonitrile (5 mL) for 20 min with stirring, then filtered by suction and recrystallized from dimethylformamide.1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-dipropylurea (6b)
Yield: 230 mg (90%); mp >350 °C; IR (KBr) 3460–3280 (NH), 3040–2840 (CH), 1690, 1640 (CO). 1550 (CC) cm−1; 1H-NMR (d6-DMSO) δ 0.75–2.75 (m, 46H, t-Bu, CH2, CH3), 2.75 (t, 4H, NCH2); 13C-NMR (d6-DMSO) δ 26.12, 29.37 (CH3, t-Bu), 38.9, 39.2 (C, t-Bu, overlap with solvent signal), 97.8, 98.0 (bisdioxine C-1/C-5, rotamers), 104.4, 104.7 (bisdioxine C-4/C-8, rotamers), 157.8, 158.2 (bisdioxine, C-3/C-7, rotamers), 160.0, 160.7 (CO, rotamers) ppm; Anal. calcd. for C30H54N4O5: C, 65.42; H, 9.88; N, 10.17; found: C, 65.18; H, 10.00; N, 9.83.
1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-dihexylurea (6c)
Yield: 254 mg (84%); mp >350 °C. IR (KBr) 3460–3280 (NH), 3040–2870(CH), 1690 (w), 1640 (s,CO), 1550 cm−1. 1H-NMR (d6-DMSO) δ 0.8–1.6 (m, 58H, t-Bu, CH2, CH3), 2.75 (t, 4H, CH2), 7.8 (4H, NH) ppm. Anal. calcd. for C36H66N4O5: C, 68.10; H, 10.48; N, 8.82. Found: C, 68.02; H, 10.44; N, 8.74.1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-didecylurea (6d)
Yield: 280 mg (94%); mp >350 °C. IR (KBr) 3460–3280 (NH), 3140–2870 (CH), 1680(w), 1630 (s,CO), 1550 cm−1; 1H-NMR (d6-DMSO) δ 0.8–1.6 (m, 74H, t-Bu, CH2, CH3), 2.72 (t, 4H, CH2); Anal. calcd. for C44H82N4O5: C, 70.73; H, 11.06; N, 7.50. Found: C, 70.72; H, 11.27; N, 7.44.1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-di-p-toluidylurea (6e)
Yield: 280 mg (94%); mp >350 °C. IR (KBr) 340–3290 (NH), 2940–2870 (CH), 1695(w), 1640 (s,CO), 1550 cm−1; 1H-NMR (d6-DMSO) δ 0.95–1.3 (m, 36H, t-Bu), 2.2, 2.25 (2s, CH3, rotamers), 6.9–7.4 (m, 8H, Ar), 8.0 (2H, NH), 8.4 (2H, NH) ppm. Anal. calcd. for C38H54N4O5: C, 70.54; H, 8.68; N, 8.66. Found: C, 70.36; H, 8.63; N, 8.74.General procedure for reactions of diisocyanate 5 with alcohols
A solution of 250 mg (0.46 mmol) diisocyanate 5 in 5 mL of dry methanol (or ethanol) and 1.5 mL of dry triethylamine is refluxed for 2 h. After cooling to room temperature the precipitate formed is separated by suction and recrystallized from acetonitrile.1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-dimethylurethane (7a)
Yield: 200 mg (88%); mp 168–170 °C. IR (KBr) 3260–3140 (NH), 3040–2840 (CH), 1700 (CO), 1640 cm−1; 1H-NMR (CDCl3) δ 1.05, 1.1 (2s, 36H, t-Bu), 3.65 (s, 6H, CH3) ppm. Anal. calcd. for C26H44N2O7: C, 62.90; H, 8.87; N, 5.64. Found: C, 63.07; H, 9.05; N, 5.51.1,3,5,7-Tetra-tert-butyl-2,5,9-trioxabicyclo[3.3.1]nona-3,7-diene 4,8-diethylurethane (7b)
Yield: 205 mg (85%); mp 173–175 °C. IR (KBr) 3260–3140 (NH), 3020–2860 (CH), 1700 (CO) cm−1; 1H-NMR (CDCl3) δ 1.05–1.25 (m, 42H, t-Bu, CH3), 3.4 (t, 4H, CH2), 4.8 (NH) ppm.Acknowledgements
This work was supported by the Karl Franzens Universität Graz and The University of Queensland. SL gratefully acknowledges the award of a Visiting Scholar stipend at UQ.References
- J. Kremsner, B. C. Wallfisch, F. Belaj, G. Uray, C. O. Kappe, C. Wentrup and G. Kollenz, Eur. J. Org. Chem., 2008, 3382 CrossRef CAS.
- C. O. Kappe, R. A. Evans, C. R. L. Kennard and C. Wentrup, J. Am. Chem. Soc., 1991, 113, 4234 CrossRef CAS.
- C. O. Kappe, G. Faerber, C. Wentrup and G. Kollenz, J. Org. Chem., 1992, 57, 7078 CrossRef CAS.
- C. O. Kappe, G. Kollenz, W. M. F. Fabian, C. Wentrup and G. Faerber, J. Org. Chem., 1993, 58, 3361 CrossRef CAS.
- S. Leber, G. Kollenz and C. Wentup, Beilstein J. Org. Chem., 2012, 8, 738 CrossRef CAS.
- B. C. Wallfisch, T. Egger, W. Heilmayer, C. O. Kappe, C. Wentrup, K. Gloe, F. Belaj, G. Klintschar and G. Kollenz, Supramol. Chem., 2002, 14, 383 CrossRef CAS.
- R. Smounig, C. O. Kappe, C. Wentrup and G. Kollenz, Monatsh. Chem., 2003, 134, 509 CrossRef CAS.
- V. A. Chebanov, C. Reidlinger, H. Kanaani, C. Wentrup, C. O. Kappe and G. Kollenz, Supramol. Chem., 2004, 16, 121 CrossRef.
- P. Haberz, F. Belaj, K. Gloe, C. Wentrup and G. Kollenz, Supramol. Chem., 2012, 24, 279 CrossRef CAS.
- W. Heilmayer, B. C. Wallfisch, C. O. Kappe, C. Wentrup, K. Gloe and G. Kollenz, Supramol. Chem., 2003, 15, 375 CrossRef CAS.
- P. A. S. Smith, Organic Reactions, 1946, 3, 337 Search PubMed.
- E. S. Wallis and J. F. Lane, Organic Reactions, 1946, 3, 267 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details. See DOI: 10.1039/c2ra21073h |
| This journal is © The Royal Society of Chemistry 2012 |