Nanoparticle-mediated internalisation and release of a calcium channel blocker†
Cameron W.
Evans
ab,
Helena M.
Viola
c,
Diwei
Ho
a,
Livia C.
Hool
c,
Sarah A.
Dunlop
b,
Melinda
Fitzgerald
*b and
K. Swaminathan
Iyer
*a
aSchool of Chemistry and Biochemistry, The University of Western Australia, Crawley WA 6009, Australia. E-mail: swaminatha.iyer@uwa.edu.au; Tel: +61 8 6488 4470
bExperimental and Regenerative Neurosciences, School of Animal Biology, The University of Western Australia, Crawley WA 6009, Australia. E-mail: lindy.fitzgerald@uwa.edu.au; Tel: +61 8 6488 2353
cSchool of Anatomy, Physiology and Human Biology, The University of Western Australia, Crawley WA 6009, Australia
First published on 25th July 2012
Abstract
Lomerizine is a calcium channel blocker that selectively blocks L- and T-type Ca2+ channels, but it is effectively insoluble under physiological conditions. Herein we show that lomerizine can be released from a nanoparticle-based carrier by intracellular protonation in PC12 cells, suppressing Ca2+ influx in these cells in response to glutamate.
Neurotrauma describes a physical injury to the central nervous system (CNS), usually acute and caused by mechanical insult.1 In addition to the primary injury, the accumulation of neurotransmitters (particularly the excitatory amino acid glutamate) and changes in the intracellular calcium concentration [Ca2+]i may be toxic to neuronal and glial cells.2 Subsequent indirect cell death is associated with a loss of Ca2+ homeostasis and is termed secondary degeneration;3–5 preventing the death of these intact but vulnerable cells is a key objective in neurotrauma treatment.6 Secondary degeneration is initiated within minutes of injury, and develops over several weeks following the initial insult.7,8 There is an important window of therapeutic opportunity during this time to protect cells that were spared by the initial event,9 because even rescuing a small proportion of neurons can have significant benefits in maintaining function.10 Lomerizine is a calcium channel-blocking drug that protects against secondary degeneration following neurotraumatic injury. Treatment is required for weeks to months after injury, but lomerizine has limited aqueous solubility and is rapidly excreted. In this work, an extended release mechanism for lomerizine is investigated by encapsulating the drug in multifunctional polymer nanospheres.
Lomerizine (KB-2796, 1-[bis(4-fluorophenyl)methyl]-4-(2,3,4-trimethoxybenzyl)piperazine dihydrochloride) is a calcium channel blocker that selectively blocks L- and T-type Ca2+ channels, but not N-type channels.11 Additionally, lomerizine may reduce the formation of reactive oxygen species in dissociated cerebellar neurons.12 In accordance with its calcium channel-blocking abilities, lomerizine protects against H2O2-induced death,13 glutamate-induced toxicity,14 and glutamate-induced cell death15 in rat hippocampal neuron cultures; protects against hypoxia in rat retinal ganglion cell cultures;16 improves recovery after ischaemic spinal cord damage in rabbits;17,18 protects neurons in visual centers of the brain after retinal damage by intravitreal injection of N-methyl-D-aspartate (NMDA) in mice;19 protects against cerebral hypoxia/ischaemia20 and retinal ischaemia/reperfusion15 in rats; and provides limited protection against secondary degeneration following partial optic nerve injuries in rats.21,22
Nevertheless, the administration of lomerizine for neuroprotection has some drawbacks. Regular doses of lomerizine are required, because the half-life of the drug is short (elimination half-life 108 h in humans)23 compared to the optimal duration of treatment, which exceeds one month.7 Also, 24 h after oral administration of lomerizine in rats, 51.7% of the dose was excreted in the faeces, and 4.1% was excreted in urine,24 suggesting that lomerizine uptake by the gastrointestinal system is not particularly efficient. In previous in vivo studies of neuroprotection, therefore, lomerizine was administered orally at least once a day at doses of 10–30 mg kg−1 in rats and rabbits. A delivery system using nanoparticles could assist in improving the bioavailability of the drug, and also enable sustained release over time. Nanoparticles have been shown to increase the availability of poorly soluble drugs, protect therapeutic agents against degradation, provide dosing over extended periods, and distribute drugs specifically to target tissues.25,26
In this report, we use magnetic, rhodamine B (RhB)-labelled fluorescent poly(glycidyl methacrylate) (PGMA) nanospheres for the intracellular delivery of lomerizine. The uptake of these nanospheres, shown in Fig. 1a, has been demonstrated previously in PC12 rat neural progenitor cells.27 Functionalisation of the nanospheres with polyethylenimine (PEI) was found to be necessary for cellular uptake (Fig. 1b, c), but although PEI can induce toxic changes, the nanospheres used here do not reduce cell viability or escape from endosomes.27
 | ||
Fig. 1 Polymer nanospheres were prepared and characterised, and were internalised by PC12 cells. (a) The nanospheres contain magnetite nanoparticles (inset) and rhodamine B fluorescent dye (scale bar 500 nm). (b) PEI-modified nanospheres are internalised by PC12 cells (orange, RhB, nanospheres; blue, Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342, nuclei; DIC overlay; 100 ×/1.40; scale bar 20 μm). (c) Transmission electron microscopy (TEM) shows nanospheres incubated with PC12 cells are contained within intracellular vesicles after 72 h (scale bar 200 nm). (d) Nanosphere size distributions as measured by dynamic light scattering; PEI-modified nanospheres (LNP+PEI and ENP) show a slight increase in size compared to unmodified nanospheres (LNP−PEI). 342, nuclei; DIC overlay; 100 ×/1.40; scale bar 20 μm). (c) Transmission electron microscopy (TEM) shows nanospheres incubated with PC12 cells are contained within intracellular vesicles after 72 h (scale bar 200 nm). (d) Nanosphere size distributions as measured by dynamic light scattering; PEI-modified nanospheres (LNP+PEI and ENP) show a slight increase in size compared to unmodified nanospheres (LNP−PEI). | ||
The preparation and characterisation (fluorescence spectra, Figure S1 and magnetometry, Figure S2) of the nanospheres used in this work is described in the ESI.† For the present work, three nanosphere preparations were made. The first contained no drug (ENP), the second contained lomerizine but was not functionalised with PEI (LNP−PEI), and the third contained lomerizine and was PEI-functionalised (LNP+PEI). The mean hydrodynamic diameter of the unmodified nanospheres was 210 nm (FWHM 120 nm) as measured by dynamic light scattering (Fig. 1d), with a small increase in size associated with PEI attachment. The encapsulation efficiency of lomerizine in nanospheres was 7.1 ± 1.3%. Drug release from the nanospheres into phosphate-buffered saline (PBS) was assessed at physiological and slightly acidic pH (Fig. 2). The rate and quantity of lomerizine released from the polymer nanospheres decreased with increasing pH; at normal physiological pH (7.4) the release of lomerizine was below the detectable limit at all time points. In these experiments, the concentration of lomerizine in the sink reached a maximum level after several hours; this may reflect saturation of the sink solution that would be unlikely to occur in vivo. The extent of the lomerizine release from the modified nanospheres was reduced by PEI on the particle surface. This was perhaps a result of steric hindrance or electrostatic interactions between the drug and PEI, both of which carry a positive charge in acidic solution.
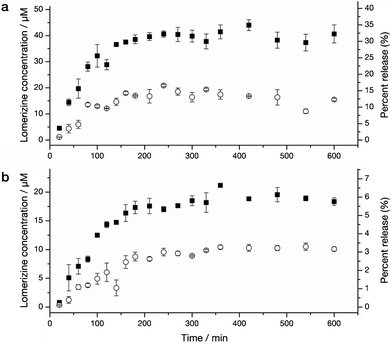 | ||
| Fig. 2 Lomerizine release from nanospheres with and without PEI modification (LNP ± PEI) over time. Filled symbols show release at pH 5, and open symbols the release at pH 6. In both cases, no release of lomerizine was detectable by high pressure liquid chromatography (HPLC) at pH 7.4 (not shown). (a) Release from nanospheres without modification (LNP−PEI). (b) Release from nanospheres with PEI modification (LNP+PEI). Error bars denote standard error (SE).. | ||
We have previously demonstrated that PEI-modified polymer nanospheres localize in endosomes within 24 h (Fig. 1c).27 The pH in early endosomes is mildly acidic and falls below 5 as the endosomes mature,28 so the release of lomerizine from nanospheres could be triggered under these conditions. Here, we administered lomerizine to PC12 cells using nanospheres, and measured the intracellular calcium concentrations after glutamate challenge in order to determine whether the intracellular release of lomerizine could be employed for neuroprotection. PC12 cells express both N- and L-type voltage gated calcium channels (VGCCs), but not T-type channels,29,30 and in undifferentiated PC12 cells, glutamate exposure (1–10 mM) over 24 h causes oxidative stress and increased [Ca2+]i.31,32 Glutamate-associated toxicity in PC12 cells has been shown to be independent of typical NMDA receptors,33 and the PC12 cell line is therefore suitable for assessing the L-type calcium channel blockade by lomerizine.
Cells receiving nanosphere treatments were incubated with nanospheres containing lomerizine, with or without modification with PEI (LNP+PEI and LNP−PEI respectively), or nanospheres containing no lomerizine but with PEI modification (ENP). These treatment groups were compared with cells treated with lomerizine only (i.e., the free drug) at 1 μM. In all cases, nanospheres were added at 20 μg mL−1, which was estimated to yield a concentration of lomerizine similar or lower than that of the free drug, based on the data in Fig. 2. The results are shown in Fig. 3. Compared to resting cells (‘untreated’), [Ca2+]i was significantly increased in cells that were treated with empty nanospheres (ENP) and exposed to glutamate (p ≤ 0.01). Lomerizine delivered using PEI-modified nanospheres (LNP+PEI), or added as the free drug dissolved in 0.01% DMSO, resulted in a significant (p ≤ 0.001) decrease in [Ca2+]i in the presence of glutamate; both routes were equally effective. Nanospheres containing lomerizine but without PEI modification (LNP−PEI) did not reduce [Ca2+]i (p > 0.05) compared to cells treated with empty nanospheres. This was presumably because these LNP−PEI nanospheres were not internalised by cells and were not exposed to the acidic endosomal environment. Taken together, these results suggest that lomerizine release from nanospheres requires a drop in pH and that PEI-modified nanospheres may be a suitable vehicle for the intracellular release of this drug.
![The intracellular calcium concentration is reduced in cells treated with lomerizine delivered using nanospheres following glutamate challenge. (a) Both lomerizine delivered using nanospheres modified with PEI (LNP+PEI) and 1 μM free drug caused similar reductions in the intracellular calcium [Ca2+]i after exposure to glutamate (10 mM, 24 h). Lomerizine-loaded nanospheres without PEI modification (LNP−PEI) do not release lomerizine and therefore [Ca2+]i is not reduced after glutamate injury. All statistical significances relative to ENP (vehicle); * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA with Bonferroni post hoc correction; error bars denote SE. (b) Representative traces showing differences in Fura-2 fluorescent ratios (R = F340/F380) recorded in single PC12 cells after nanosphere treatment with either LNP+PEI or ENP and 24 h exposure to glutamate; ionomycin was added at 5 min, and Rmax was determined from the subsequent [Ca2+]i rise.](/image/article/2012/RA/c2ra21058d/c2ra21058d-f3.gif) | ||
| Fig. 3 The intracellular calcium concentration is reduced in cells treated with lomerizine delivered using nanospheres following glutamate challenge. (a) Both lomerizine delivered using nanospheres modified with PEI (LNP+PEI) and 1 μM free drug caused similar reductions in the intracellular calcium [Ca2+]i after exposure to glutamate (10 mM, 24 h). Lomerizine-loaded nanospheres without PEI modification (LNP−PEI) do not release lomerizine and therefore [Ca2+]i is not reduced after glutamate injury. All statistical significances relative to ENP (vehicle); * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA with Bonferroni post hoc correction; error bars denote SE. (b) Representative traces showing differences in Fura-2 fluorescent ratios (R = F340/F380) recorded in single PC12 cells after nanosphere treatment with either LNP+PEI or ENP and 24 h exposure to glutamate; ionomycin was added at 5 min, and Rmax was determined from the subsequent [Ca2+]i rise. | ||
Internalisation of polymer nanospheres is not a requirement for lomerizine release, however, as drug release will occur in any environment with a lowered pH. After CNS injury, vascular interruption causes a switch to anaerobic glycolysis in the brain; this results in a decrease in pH in the intercellular space known as acidosis. The pH falls about 1 unit during ischaemia, and can drop below pH 6.0 during severe ischaemia or under hyperglycemic conditions.34 In this situation, we would expect lomerizine release from the nanospheres with the dose being dependent on the extent of the injury. Therefore, we suggest that the nanospheres we have described will be useful in either their PEI-modified or unmodified forms, and that intracellular delivery or release into the intercellular space could be achieved.
We have demonstrated the use of multifunctional polymer nanoparticles to facilitate the intracellular delivery of a calcium channel blocker in vitro. Release of lomerizine from nanospheres will likely assist in improving the bioavailability of this drug and may provide a means for extended release under slightly acidic conditions. Neurotrauma treatment often requires surgery immediately after injury (to repair vascular damage, for example, or for debridement and decompression following spinal cord injury), so nanospheres could potentially be administered directly at injury sites at this time. In such a case, nanospheres would not need to cross the blood–brain barrier, which is a significant obstacle in using nanoparticles to treat the CNS.35 Lomerizine alone is not sufficient to prevent the secondary damage arising as a result of neurotrauma21 and combinatorial therapies will probably be needed to target not only the neurochemically-induced changes, but also to initiate and provide a permissive environment for axonal regrowth.9 A nanoparticle-based approach could be beneficial for this purpose with potential for the delivery of multiple therapeutic strategies simultaneously.
Acknowledgements
PGMA was provided by Dr Bogdan Zdyrko and Prof. Igor Luzinov of Clemson University. The TEM image in Fig. 1c was acquired by Benjamin Padman, Dr Jeremy Shaw, and Prof. Martin Saunders, of the Centre for Microscopy, Characterisation & Analysis at the University of Western Australia. We also thank Tristan Clemons and John Murphy for their assistance. This work was funded by the Australian Research Council (ARC), the National Health & Medical Research Council (NHMRC) of Australia, and Perpetual (Philanthropy Australia) as administrators of the Sir Harry Secombe Trust.References
- Y. T. Kim, J. M. Caldwell and R. V. Bellamkonda, Biomaterials, 2009, 30, 2582–2590 CrossRef CAS.
- T. K. McIntosh, M. Juhler, R. Raghupathi, K. E. Saatman and D. H. Smith, in Traumatic Brain Injury, ed. D. W. Marion, Thieme, New York, 1999 Search PubMed.
- C. Giaume, F. Kirchhoff, C. Matute, A. Reichenbach and A. Verkhratsky, Cell Death Differ., 2007, 14, 1324–1335 CrossRef CAS.
- Q. Jiang and P. K. Stys, J. Neurochem., 2000, 74, 2101–2107 CrossRef CAS.
- A. Camins, N. Crespo-Biel, F. Junyent, E. Verdaguer, A. M. Canudas and M. Pallàs, Curr. Drug Metab., 2009, 10, 433–447 CrossRef CAS.
- J. T. E. Richardson, Clinical and Neuropsychological Aspects of Closed Head Injury, Psychology Press, Hove, 2000 Search PubMed.
- M. Selt, C. A. Bartlett, A. R. Harvey, S. A. Dunlop and M. Fitzgerald, Brain Res. Bull., 2010, 81, 467–471 CrossRef CAS.
- M. J. Crowe, J. C. Bresnahan, S. L. Shuman, J. N. Masters and M. S. Beattie, Nat. Med., 1997, 3, 73–76 CrossRef CAS.
- R. Ellis-Behnke, Med. Clin. North Am., 2007, 91, 937–962 CrossRef CAS.
- B. A. Sabel, Restor. Neurol. Neurosci., 1999, 15, 177–200 CAS.
- T. Watano, H. Hara and T. Sukamoto, Jpn. J. Pharmacol., 1997, 75, 209–213 CrossRef CAS.
- N. Akaike, H. Ishibashi, H. Hara, Y. Oyama and T. Ueha, Brain Res., 1993, 619, 263–270 CrossRef CAS.
- M. Ishii, R. Iizuka, Y. Kiuchi, Y. Mori and S. Shimizu, Mol. Cell. Biochem., 2011, 358, 1–11 CrossRef CAS.
- H. Hara, K. Yokota, M. Shimazawa and T. Sukamoto, Jpn. J. Pharmacol., 1993, 61, 361–365 CrossRef CAS.
- N. Toriu, A. Akaike, H. Yasuyoshi, S. Zhang, S. Kashii, Y. Honda, M. Shimazawa and H. Hara, Exp. Eye Res., 2000, 70, 475–484 CrossRef CAS.
- H. Yamada, Y. N. Chen, M. Aihara and M. Araie, Brain Res., 2006, 1071, 75–80 CrossRef CAS.
- V. Danielisová and M. Chavko, Neurochem. Res., 1994, 19, 1503–1507 CrossRef.
- V. Danielisová and M. Chavko, Exp. Neurol., 1998, 149, 203–208 CrossRef.
- Y. Ito, S. Nakamura, H. Tanaka, K. Tsuruma, M. Shimazawa, M. Araie and H. Hara, CNS Neurosci. Ther., 2010, 16, 103–114 CrossRef CAS.
- H. Hara, A. Ozaki, M. Yoshidomi and T. Sukamoto, Arch. Int. Pharmacodyn. Ther., 1990, 304, 206–218 CAS.
- M. Fitzgerald, C. A. Bartlett, L. Evill, J. Rodger, A. R. Harvey and S. A. Dunlop, Exp. Neurol., 2009, 216, 219–230 CrossRef CAS.
- Z. Karim, A. Sawada, H. Kawakami, T. Yamamoto and T. Taniguchi, Curr. Eye Res., 2006, 31, 273–283 CrossRef.
- H. Hara, T. Morita, T. Sukamoto and F. M. Cutrer, CNS Drug Rev., 1995, 1, 204–226 CrossRef CAS.
- T. Kawashima, O. Satomi and N. Awata, J. Pharmacobio-Dyn., 1991, 14, 449–459 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS.
- R. A. Siegel and M. J. Rathbone, in Fundamentals and Applications of Controlled Release Drug Delivery, ed. J. Siepmann, R. A. Siegel and M. J. Rathbone, Controlled Release Society, 2012, pp. 19–43 Search PubMed.
- C. W. Evans, M. Fitzgerald, T. D. Clemons, M. J. House, B. S. Padman, J. A. Shaw, M. Saunders, A. R. Harvey, B. Zdyrko, I. Luzinov, G. A. Silva, S. A. Dunlop and K. S. Iyer, ACS Nano, 2011, 5, 8640–8648 CrossRef.
- J. Mercer, M. Schelhaas and A. Helenius, Annu. Rev. Biochem., 2010, 79, 803–833 CrossRef CAS.
- D. L. Lewis, H. J. De Aizpurua and D. M. Rausch, J. Physiol., 1993, 465, 325–342 CAS.
- H. Liu, R. Felix, C. A. Gurnett, M. De Waard, D. R. Witcher and K. P. Campbell, J. Neurosci., 1996, 16, 7557–7565 CAS.
- Q. Q. Mao, X. M. Zhong, C. R. Feng, A. J. Pan, Z. Y. Li and Z. Huang, Cell. Mol. Neurobiol., 2010, 30, 1059–1066 CrossRef CAS.
- N. Li, B. Liu, D. E. Dluzen and Y. Jin, J. Ethnopharmacol., 2007, 111, 458–463 CrossRef CAS.
- D. Schubert, H. Kimura and P. Maher, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8264–8267 CrossRef CAS.
- Z. G. Xiong, X. M. Zhu, X. P. Chu, M. Minami, J. Hey, W. L. Wei, J. F. MacDonald, J. A. Wemmie, M. P. Price, M. J. Welsh and R. P. Simon, Cell, 2004, 118, 687–698 CrossRef CAS.
- G. A. Silva, Nat. Rev. Neurosci., 2006, 7, 65–74 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental methods, fluorescence spectra, and SQUID magnetometry. See DOI: 10.1039/c2ra21058d |
| This journal is © The Royal Society of Chemistry 2012 |