Eggshell membrane: a natural biotemplate to synthesize fluorescent gold nanoparticles
Parukuttyamma Sujatha
Devi
*a,
Suparna
Banerjee
a,
Sebanti Roy
Chowdhury
b and
Gopinatha Suresh
Kumar
*b
aNano-Structured Materials Division, CSIR-Central Glass and Ceramic Research Institute, 196 Raja SC Mullick Road, Kolkata, 700 032, India. E-mail: psujathadevi@cgcri.res.in/psujathadevi@gmail.com; Fax: +91 33 2473 0957; Tel: +91 33 2483 8082 Ext 3487
bBiophysical Chemistry Laboratory, Chemistry Division, CSIR-Indian Institute of Chemical Biology, 4 Raja SC Mullick Road, Kolkata, 700 032, India. E-mail: gskumar@iicb.res.in/gskumar@csiiicb.in; Fax: +91 33 2472 3967; Tel: +91 33 2499 5723
First published on 20th September 2012
Abstract
In this article we report a simple, environmentally friendly, and naturally available protein based biotemplate, viz. the chicken eggshell membrane, truly one of nature's gifts, for preparing highly fluorescent gold nanoparticles in solution at ambient conditions in a single step. We propose this as a simple green method to prepare gold nanoparticles both in solution and on the membrane for various applications. A stable colloidal solution of fluorescent Au nanoparticles has been formed in situ during the reduction. The gold nanoparticles, consisting of sizes below 20 nm, were highly fluorescent and exhibited intense blue emission around 437 ± 5 nm. Red emission around 630 ± 5 nm was also observed for the solution. The novelty of this technique lies in the unique properties of this membrane, such as high resistance to pH, safety in handling, and abundant availability in nature. The most important feature of this method is the easy abundance of the source material, which enables cost-effective preparation of gold nanoparticles. In addition to preparing nanoparticles of metals for various applications, this could be used as a very simple method to recover precious metals from chemical and industrial waste.
Introduction
The synthesis of noble metal nanoparticles, especially gold nanoparticles of different shapes and sizes, for application in information storage, biological labeling, medical diagnosis, controlled drug delivery, sensors and catalysis, has been explored in detail recently.1–9 Among the noble metal nanoparticles, gold has become a major part of many scientific and technological developments due to its peculiar properties arising out of surface plasmon resonance (SPR), which is highly sensitive to composition, size and morphology of the particles. It is worth mentioning the exponential growth in research into the synthesis and applications of gold nano particles. The most common route for synthesizing gold nanoparticles is the ‘chemical reduction’ based synthesis using a suitable reducing agent, which has been widely explored. A large number of chemical techniques are available for the preparation of gold nanoparticles.10–19 Increase in the use of hazardous and toxic chemicals in the chemical process prompted scientists to explore alternative approaches for cleaner green processes. Thus, green synthesis became a topic of considerable importance for the synthesis of gold nanoparticles.20–31 The bio-instigated methods to prepare nanoparticles of metals is a potential environmentally friendly alternative method, albeit relatively poorly exploited. From Lactobacillus strains obtained from buttermilk to live Alfalfa plants, many biological species have been exploited for the synthesis of gold nanoparticles.32 Human cancerous and normal cells also yielded gold nanoparticle formation upon incubation with gold chlorate and showed difference in reduction behavior.33 Vitamins were also exploited for synthesizing nanoparticles, one such example being the synthesis of gold and platinum nanoparticles using Vitamin C.34Thus, gold nanoparticles derived by the use of bacteria, yeast, algae, DNA, enzymes, plant extracts, carbon nanotubes and other biotemplates exhibited incredible properties in relation to their size and shape.20–34 However, although most of these techniques produced gold nanoparticles of variable shapes and sizes, very few resulted in fluorescent gold nanoparticles. There have been few recent advances in preparing fluorescent metal nanoparticles using biotemplates, proteins and peptides.35–39 Notwithstanding these advances, the need to develop better environmentally benign, non-toxic, and easily available biomaterials for the preparation of nanoparticles continues to be an area of active exploration. Therefore, it would be of great interest to experiment and identify new, naturally occurring biotemplates to prepare fluorescent gold nanoparticles.
We report herein a novel biotemplate method using an environmentally friendly and natural biomembrane, viz. the chicken eggshell membrane (ESM), truly one of nature's gifts for preparing highly fluorescent gold nanoparticles in a single step at ambient conditions. ESM is constituted of a double layer membrane in the eggshell, the composition of which is mainly water-insoluble glycoproteins such as collagen (types I, V and X), and amino acids like glycine alanine and uronic acid.40,41 Wong et al., reported the presence of Type I and Type V proteins in the inner and outer shell membranes40 where as Arias et al., observed the presence of Type X collagen in the egg shell membrane.41 A few known major applications of ESM are as a matrix for the recovery of heavy metals, as a template for hierarchically ordered macroporous materials, and for biosensing of enzyme immobilized Au-ESM.42–45
Results and discussion
Optical properties
Separate control experiments were performed with ESM, buffer and HAuCl4 solutions alone, but no change in colour was observed for either the membrane or the gold solution, whereas within 5–8 h after the ESM was treated with chloroauric acid solution, the deep yellow solution became light yellow to colourless, indicating the complete disappearance of the Au3+ ions from the solution. The disappearance of the yellow color and subsequent appearance of the pink/blue color indicated the reduction of HAuCl4 to Au0. In contrast to the characteristic red colour of the spherical gold particles, the colloidal suspension of membrane reduced gold particles was pink for lower concentrations and blue for higher concentrations. The characteristic pink to blue colour of the colloidal solution was the most convincing visual evidence for the formation of Au nanoparticles. Another noticeable effect was that the colloidal solution of Au particles formed from a 10−2 M solution was stable for 15–20 days after which particle aggregation leading to the formation of long wires occurred as a result of dendritic fractal growth.To gain further insights into the reduction process and particle formation, the in situ reactions were probed by continuously monitoring the ultraviolet-visible (UV-Vis) absorption and fluorescence spectral changes during the reaction from time to time for a week to ten days. In Fig. 1, the absorption spectral changes of a 10−4 M solution of HAuCl4 after impregnation with ESM are presented. We observed a strong absorption maximum at 295 nm from the initial Au3+ solution and a concomitant reduction in intensity of the same was noticed with time, indicating the removal of gold ions from the solution. This was completed within 6 h. A simultaneous colour change of the membrane from white to yellow was noticed, suggesting the adsorption of Au3+ on the membrane surface. The solution colour slowly changed from yellow and became colorless. The absorption band around 300 nm slowly disappeared within 5–8 h and another band at 275 nm started to grow, indicating the complete adsorption of Au3+ on the ESM followed by the reduction of Au3+ to Au0 (Fig. 1). The concomitant appearance of a weak, broad, visible band in the 500–750 nm range was also evident after 6 days. By contrast, the spectral changes during the interaction of ESM with 10−2 M concentrated HAuCl4 solution were distinctly different. In Fig. 2, the absorption spectral changes of a 10−2 M solution of HAuCl4 after impregnation with ESM are presented. Initially, a reduction in the intensity of the 294 nm band corresponding to Au3+ ions was noticed (not shown), followed by a minor red shift to around 305 nm. The concomitant appearance of a broad visible band in the 500–750 nm range with a peak maximum around 580 ± 5 nm was noticed. The spectra recorded at room temperature as a function of time exhibited the systematic appearance of this broad band with a maximum around 580 nm, which red shifted to 593 nm with time (Fig. 2). An indication of a weak but clear shoulder around 520 ± 5 nm, corresponding to the characteristic absorption peak of spherical gold particles of size below 25 nm, was also evident in the series of spectra (Fig. 2). The observed colour change in the solution is evident from the photograph shown in the inset of Fig. 2. The prominent peak observed around 585 ± 5 nm in the colored solution corresponds to the in-plane Surface Plasmon Band (SPB) of gold particles of unusual size and shape such as triangular, hexagonal or dodecahedral gold plates. The appearance of another band beyond 900 nm in the spectra, most likely due to the onset of longitudinal (out of plane) surface plasmon resonance, was also evident, which was beyond the limit of detection of our spectrophotometer. After 7 days, the SPB intensity remained invariant, indicating the complete reduction of Au ions and subsequent formation of Au nanoparticles. After 10 days, the intensity of the SP band started to diminish, indicating a probable particle aggregation. When the dilute solution was used, the observed spectral change was not so interesting, since most of the particles were adsorbed and formed on the membrane itself. Localized surface plasmon resonance (SPR) of gold nanoparticles is due to the collective oscillation of 6S electrons in the conduction band in resonance with the incident radiation, resulting in enhanced optical properties. Gold exhibits size dependent plasmon absorption by confining the size to dimensions smaller than the electron mean free path (∼50 nm). Daniel and Astruc reported that the SPB is absent for gold nanoparticles when the size is below 2 nm, as well as for bulk gold.2 They also reported SPB maxima at 517, 520, 521, 533 and 575 nm, respectively, for 9, 15, 22, 48 and 99 nm sized gold nanoparticles. Moreover, the SP band maximum also depends on the shape of the particles. The observation of a weak shoulder around 520 ± 5 nm and a strong band at 580 ± 5 nm confirms the presence of gold nanoparticles of variable size and shapes in ESM treated solution.
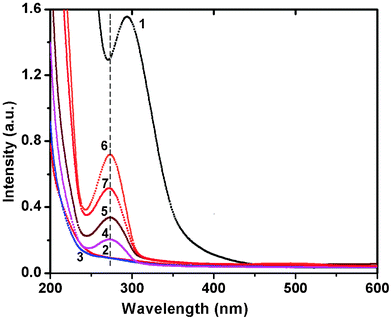 | ||
| Fig. 1 UV-Vis spectra of the reaction of ESM with chloroauric acid solution (10−4 M), recorded as a function of time. Curves (1) blank HAuCl4 (2) 1 h (3) 6 h (4) 1 day (5) 3 days (6) 6 days and (7) 7 days. | ||
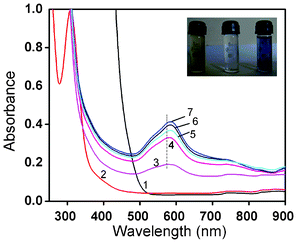 | ||
| Fig. 2 UV-Vis spectra of the reaction of ESM with chloroauric acid solution (10−2 M), recorded as a function of time. Curves (1–7) represent blank HAuCl4 and the spectral changes observed for 1–6 days. The inset shows the stable colloidal dispersion and the starting solution (yellow). | ||
So far, we have discussed only the changes that occurred in solution, but not on the membrane itself. As indicated before, a simultaneous colour change of the membrane from white to yellow was noticed after impregnation of the ESM in chloroauric acid solution, indicating the adsorption of Au3+ on the membrane. Slowly, the membrane surface changed colour from white to pink (10−4 M) (Fig. 3a) or blue (10−2 M) (Fig. 3b), depending on the concentration of the particles on the membrane. In Fig. 3c, the coloured membranes of Au obtained by impregnation of the membrane in 10−6, 10−4 and 10−2 M HAuCl4, respectively, for 7 days are shown, and the change in colour can be clearly seen. In the first case hardly any change in colour was noticed. When the concentration of the solution was 10−4 M, then the formation rate was slow, as evident from the slow reduction of Au3+ noticed in the solution, but the colour change was faster on the membrane (Fig. 3). The strong absorption band observed around 274 nm in the case of 10−4 M could be due to the presence of gold nanoparticles or clusters of size below 20 nm. At a higher concentration of 10−2 M, the changes were very fast, both in solution and on the membrane (Fig. 3), indicating the signature of formation of Au nanoparticles exhibiting plasmonic absorption. The first observation of colour change on the membrane confirms that the adsorbed aurous ions in situ are reduced to Au0 and deposited on the membrane. Meanwhile, due to the porous nature of the membrane, the loosely bound particles are released into the solution, resulting in a colour change for the solution. The immobilized coloured membranes dried at 80 °C appeared lustrous yellow in colour, indicating the presence of bulk gold.
 | ||
| Fig. 3 Photographs of (a) bare eggshell membrane and pink colored impregnated eggshell membrane with 10−4 M solution, (b) blue colored impregnated eggshell membrane with 10−2 M metal ion solution and (c) membranes impregnated with 10−6 (1), 10−4 (2) and 10−2 M HAuCl4 (3) solutions for 7 days. | ||
To verify whether the nanoparticles formed in solution exhibited fluorescence, we have monitored the emission and excitation spectra of the colloidal solutions of Au particles. The control solution of HAuCl4 had no visible fluorescence in the range studied (350–700 nm). A very strong blue luminescence with excitation and emission maxima at 335 and 440 nm, respectively, is clearly seen from the blue coloured colloidal solution (Fig. 4). We have also recorded the dependence of excitation wavelength on the emission intensity at 440 nm (Fig. 5 and Fig. 6). These dispersions exhibited fluorescence by excitation over a range of wavelengths from 330 nm to 400 nm, as evident from Fig. 5 and 6. Time dependent fluorescence studies were conducted on a 10−2 M solution of HAuCl4 during the reduction process (Fig. 7a and b). Similarly to the report of Roy et.al.,38 the ESM induced gold particles also exhibited interesting fluorescent properties, such as large Stokes shift (105 nm). The solution containing the Au nanoparticles exhibited intense blue emission around 437 ± 5 nm, corresponding to the Au6S to 6P intraband transition. The blue emission from gold nanoparticles around 440 nm has been reported to be due to the presence of isolated spherical Au nanoparticles of size below 20 nm.46–48 Zheng et al. reported the synthesis of water soluble blue emitting gold nanodots and confirmed the blue emission due to the presence of the Au8 magic cluster.49,50 Our observation clearly confirms that in situ reduction in presence of ESM could in fact produce fluorescent Au nanoparticles of size below 20 nm in solution. On excitation around 585 nm, a second emission with a band around 630 ± 5 nm was also observed in our samples (Fig. 7b).
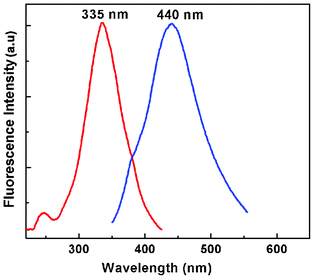 | ||
| Fig. 4 Excitation and emission spectra of ESM induced gold nanoparticle colloidal solution at room temperature (HAuCl4, 10−2 M). | ||
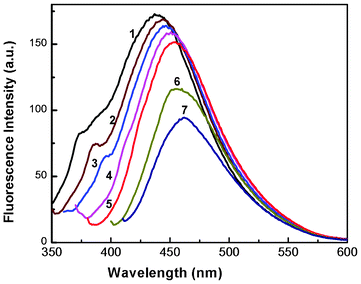 | ||
| Fig. 5 Fluorescence spectra of a colloidal solution of Au nanoparticles at different excitation wavelengths. Curves (1–7) represent excitation wavelengths of 330, 340, 350, 360, 370, 390 and 400 nm, respectively (HAuCl4, 10−4 M). | ||
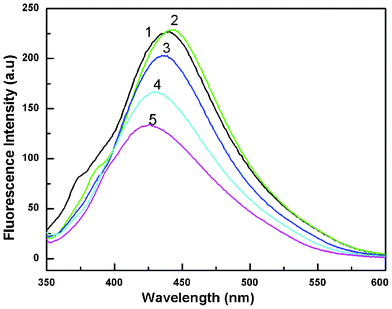 | ||
| Fig. 6 Fluorescence spectra of a colloidal solution of Au nanoparticles at different excitation wavelengths. Curves (1–5) represent excitation wavelengths of 330, 340, 350, 360 and 370 nm, respectively (HAuCl4, 10−2 M). | ||
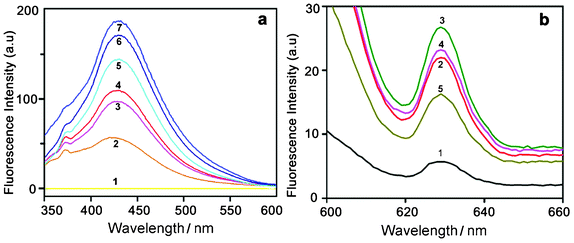 | ||
| Fig. 7 (a) Time dependent fluorescence spectra of HAuCl4 solution (10−2 M) recorded with an excitation wavelength of 330 nm. Curves (1–7) represent control HAuCl4 and the spectral changes for 2 to 7 days. (b) Fluorescence spectra recorded at an excitation wavelength of 585 nm. Curves (1–5) represent the spectral changes for 2, 3, 4, 5 and 7 days, respectively. | ||
Photoluminescence from Au nanoparticles has been reported to occur between 500–590 nm and between 410–430 nm.51,52 The range of emission wavelengths 500–590 nm corresponds to electronic transitions between the sp band just below the Fermi level and the first d band; the second emission band, 410–430 nm, is due to the recombination of electrons with holes in the lower energy band. As reported earlier by many investigators, the emission of Au nanostructures could be strongly enhanced by surface plasmon resonances occurring in nanostructures.51,52 Xie et al. reported the synthesis of red-emitting gold nanoparticles using BSA proteins and attributed it to the presence of Au25 clusters.37 Mohamed et al. reported fluorescent enhancement from Au rods and attributed this to the surface plasmon effect and surface roughness of rod shaped particles.53
It can be assumed from our emission data that the colloidal solution of gold nanoparticles formed by ESM may contain both Au8 and Au25 clusters. It is interesting to notice that the particles prepared using a lower concentration of the starting material (10−4 M) exhibited a strong absorption around 274 nm and the plasmonic absorption was almost absent or very weak in that solution. It is also exciting to note that these particles exhibited a very strong blue emission around 440 nm, but a weak NIR emission as evident from Fig. 4. On the contrary, NIR emission was strong for samples prepared from 10−2 M solution, which exhibited a strong plasmonic absorption around 580 ± 5 nm. As reported earlier, the NIR emission could be associated with the SPR peak, whereas the blue emission around 440 nm could be due to the presence of small gold clusters. Thus, we could achieve the blue emitting colloidal gold dispersion by using a dilute solution of chloroauric acid whereas a both green and red emitting colloid could be attained by using a concentrated solution.
Microstructural investigations
The size and shape of the metal nanoparticles were characterized by transmission electron microscopy (TEM). The TEM studies indicated that the particles of Au were well dispersed and more spherical in shape at the beginning of the reaction with sizes below the 20 nm range. This is consistent with the blue emission around 440 nm exhibited by isolated smaller Au particles. With time, the particles appeared to grow larger. A typical image of the Au colloidal solution (10−4 M) collected on the 7th day (Fig. 8a) showed the presence of a large number of spherical and triangular particles of size below 20 nm. The existence of smaller sized Au particles has resulted in intense blue emission around 437 ± 5 nm. Equilateral triangular plates with very smooth edges, truncated triangles, and hexagonal plates have been observed from Au solution (10−2 M) collected on the 7th day (Fig. 8b and c). The existence of such unusual shapes and sizes of nanoparticles has resulted in the appearance of a broad plasmonic absorption band in the 500–750 nm range (Fig. 2). The TEM of an isolated nano decahedron particle is shown in Fig. 8d and the SAED pattern from a collection of samples is shown in Fig. 8f. Due to the thickness of the particle, the HRTEM could be collected only from the edge of a particle, which revealed its single crystalline nature (Fig. 8e). The SAED pattern representing an intense spot pattern (Fig. 8f), collected from Fig. 8c, clearly confirmed the polycrystalline nature of the formed gold particles. The strongest spot set could be indexed to the 110 plane of the fcc gold. This confirmed that the particles formed in solution consisted of crystalline gold particles with a fcc structure. The energy dispersive analysis by X-ray (EDAX) on a single particle confirmed Au as the only element present. | ||
| Fig. 8 TEM images of the gold nanoparticles from (a) Au solution (10−4 M) collected on the 7th day, (b) Au solution (10−2 M) collected on the 7th day, (c & d) Au nanoparticles (10−2 M) collected on the 10th day, (e) HRTEM confirming the single crystalline nature of the particle in (d) and (f) SAED pattern of the particle shown in (c). | ||
The X-ray powder diffraction (XRD) analysis of the membrane further proved the crystalline nature of the metal particles formed on the membrane (Fig. 9). Natural ESM was amorphous to X-rays, whereas the metal nanoparticle immobilized membranes were highly crystalline. The XRD patterns of a pink colored membrane and a blue colored membrane are shown in Fig. 9 along with the JCPDS pattern for bulk gold. The two major reflections (111) and (200) of bulk gold with a face centered cubic structure is clearly present in the XRD spectra of the membrane. This further confirmed that the particles formed on the membrane consisted of crystalline gold particles having a fcc structure with lattice constants of 4.084 and 4.091 nm, respectively, for the dried and 10−2 M impregnated membranes, compared to 4.078 nm for bulk gold (JCPDS 04-0784). Field Emission Scanning Electron Microstructures (FESEM) of an untreated membrane and membranes with Au are shown in Fig. 10a and b. A network-like structure was observed for the ESM, indicating highly cross linked fibers and cavities (Fig. 10a). In addition to spherical particles, Au plates of hexagonal, truncated triangular and triangular geometries with very smooth edges were seen on the membrane (Fig. 10b). On heating the membrane, these nanoparticles changed to spherical particles (Fig. 10c and d). It is anticipated that due to the porous nature of the membrane, the formed particles stay on the membrane surface, giving rise to colouration (Fig. 10b and Fig. 3).
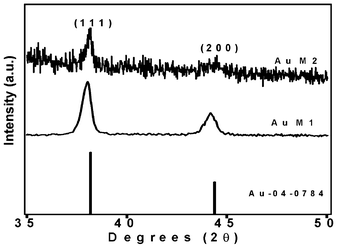 | ||
| Fig. 9 XRD patterns of Au impregnated ESM. AuM1 is 80 °C dried membrane and AuM2 is as-dried membrane impregnated with 10−2 M solution. | ||
 | ||
| Fig. 10 FESEM pictures of (a) bare ESM, (b) Au impregnated ESM and (c) and (d) 80 °C dried membrane showing spherical particles. | ||
Although the exact mechanism of the formation of gold nanoparticles by ESM is not understood, the following conclusions emerge from our results. The ESM is constituted of a double layered water insoluble membrane inside the eggshell consisting of 70% organic substance, such as proteins. The membrane structure is like an entangled thread or randomly knitted net, as revealed in the SEM picture (Fig. 10a). The lamellar structure of the membrane consisted of a thin insoluble fibrous layer of protein with numerous mesh works. The collagen proteins of type 1, V and X are known to have regularly repeating amino acid sequences with glycine, alanine, proline, lysine, arginine etc in abundance. However, the main constituents of ESM are glycine, alanine and uronic acid.40,41 The egg shell membrane also contains several bacteriolytic enzymes, such as lysozyme and N-acetyl glucosaminidase.54 Such a structure with a number of amino, carboxyl and carbonyl functional groups on the ESM fibres could get charged under the acidic conditions of our experiments and could bring the chloroaurate ions closer to the surface via hydrophobic interaction or hydrogen bonding resulting in surface adsorption. Once adsorbed on the membrane, the aldehyde (–CHO) and amino (–NH2) functional groups may act as reducing agents to reduce the adsorbed Au3+ ions to Au0. The size and shape of the reduced gold particles will also depend on the rate of reduction by the amino acid. Slow reduction induces particle growth and fast reduction forms finer particles. Aspartate induced reduction of gold has resulted in large plates, as observed by Shao et al.55 Liu et al. also reported the formation of micrometer sized gold crystals by a slow reduction method.56 The other amino acids such as lysine, tryptophan and arginine produce only spherical particles.55
Basu et al. reported a protein mediated autoreduction of gold salts to gold nanoparticles using the protein bovine serum albumin.57 Guo et al., on the other hand, showed the ability of aniline to form fibrous aggregates of gold nanoparticles.58 Tan et al. studied the comparative reduction capabilities of 20 different amino acids and reported that tryptophan was the fastest reducing amino acid.59 They also compared the binding affinity of peptides with glycine and proline groups and found the latter to be a weak binding group compared to the former. Recently, Wei et al. reported protein-directed growth of gold nanoparticles within a single crystal of a lysozyme.60 Interestingly, Selvakannan et al. demonstrated the ability of lysine to form and stabilize gold nanoparticles by acting as a capping agent.61 Dinda et al. synthesized gold nanoparticles of controllable size and shape, exhibiting a single SPR band at 533 nm, by using a conjugate of fatty acids and amino acids such as tryptophan.62 Stabilization of fluorescent gold nanoparticles (λem of 465 to 500 nm) has been observed by Bao et al., using small molecules.63 Bovine serum albumin has been successfully employed as a foaming and as a stabilizer for the synthesis of gold, silver and their alloy nanoparticles.39
As evident from the cited examples, it has been proved unequivocally that protein and peptides, where amino acids are the building blocks, could reduce Au3+ to Au0in situ, resulting in gold nanoparticles of variable size, shape and optical properties.57–63 Since collagen in the ESM contains many amino acids and enzymes such as lipozyme, it is expected to reduce the gold ions to the corresponding gold nanoparticles (eqn (1)) of variable shapes and sizes on contact with such ions, as demonstrated here.
| AuCl4− + 3e− ↔ Au0 + 4Cl− E0 = +0.99V | (1) |
The particles formed by the ESM template appeared to be different from the ones prepared by other chemical reduction techniques, as the usual chemical reduction techniques failed to produce fluorescent gold nanoparticles. The formation and stabilization of water soluble fluorescent gold nanoparticles in solution by ESM could be due to the presence of proteins on the membranes, as discussed above. In contrast to the report of Shao et al.,55 wherein fluorescent metal clusters are formed on the ESM in the presence of NaOH, we were able to demonstrate the in situ formation of fluorescent gold nanoparticles in solution without the use of any additional chemical or reducing agent. The membranes could also regulate the rate of crystal growth and its morphology by controlling the rate of availability of the reaction sites during the reduction process, resulting in fluorescent gold nanoparticles and larger particles exhibiting plasmonic absorption simultaneously.
Conclusions
The formation of Au nanoparticles of variable shapes and sizes by in situ reduction using a naturally and abundantly available biotemplate, without the use of any catalyst, surfactant, chemical reducing agent or equipment at ambient conditions is demonstrated here. Our results indicate the formation of gold particles with sizes in the nanometer to micrometer range, depending on the concentration and time of immobilization of ESM in chloroauric acid. This bio-inspired route has led to the formation of Au nanoparticles with distinct blue and red emissions, that could find applications in biolabeling and bioimaging. The gold nanoparticles consisting of size below 20 nm were highly fluorescent and exhibited intense blue emission around 437 ± 5 nm and red emission around 630 ± 5 nm, respectively. The most important features of this method are the easy abundance and economy of the source material, which enables a cost-effective preparation of the nanoparticles. The nanoparticles bound to the surface of the ESM may also be used for other applications, such as catalysis and sensor development. In addition to preparing nanoparticles of metals for various applications, this could be used as a very simple method to recover precious metals from chemical and industrial waste and research in this direction is also currently under way. The proof of concept demonstration reported here will be extended to other metal ions to understand the full potential of ESM as a template for the synthesis of metal nanoparticles and oxides.Experimental
Preparation of nanoparticles
Egg shells were carefully cleaned in distilled water. The cleaned shells were treated with dilute acetic acid to extract the membrane from the shell by soaking them for 2–4 h at room temperature. Once the calcium carbonate shell was dissolved, the membranes were carefully collected and thoroughly washed with deionized water in order to remove the albumin and then dried in air at room temperature. Extracted dried ESM was incubated in a known volume of gold chloride solution taken in a beaker and the reaction was allowed to proceed. The concentrations of the HAuCl4 solutions were varied from 0.1 mM to 0.1 M. Separate control experiments were also performed with ESM and buffer and HAuCl4 solutions alone, but no change in colour was observed for either the membrane or the gold solution. To gain an insight into the reduction process and the particle formation, the in situ reactions were probed by continuously monitoring the absorption and fluorescence spectral changes, in addition to the colour changes.Characterization of membrane and nanoparticles
All absorption spectral measurements were recorded at 20 °C on a Jasco V-660 double beam double monochromator spectrophotometer (Jasco International Company Ltd., Hachioji, Japan) in quartz cuvettes of 1 cm path length in the wavelength range 200–800 nm. Fluorescence spectra were carried out at 20 °C on a Shimadzu RF-5301PC spectrofluorimeter (Shimadzu Corporation, Kyoto, Japan), with excitation and emission slit widths of 5 nm. The room temperature powder X-ray diffraction (XRD) was carried out on the immobilized membranes for phase identification using a Philips X-ray diffractometer (PW1730) with Cu-Kα radiation at a 2θ scan rate of 2° per minute. The particle morphology and local crystallographic structure were studied by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM), respectively. The transmission electron microscopy (TEM) images were taken using a TECNAI G230 high-resolution transmission electron microscope operating at 300 kV. The samples for microscopy were prepared by depositing micro-drops of the as-formed sample on a 300 mesh carbon coated copper grid, followed by drying. Field emission scanning electron micrographs (FESEM) and scanning electron micrographs were carried out in a Field Emission Scanning Electron Microscope [SUPRATM 35 VP; Gemini Column (Carl Zeiss SMT)] and a Leo 430i (UK) scanning electron microscope.Acknowledgements
This work was supported by CSIR network program on “Nanomaterials and nanodevices for applications in health and disease” (NWP0035). PSD thanks Prof. Indranil Manna, Director, CSIR-Central Glass and Ceramic Research Institute for his patronage and Prof. Siddhartha Roy, Director, CSIR-Indian Institute of Chemical Biology for the financial support through NWP0035. SRC was supported by a project assistance-ship from NWP0036 entitled “Comparative genomics and biology of non coding RNA in the human genome”. The authors thank Prof. D. D. Sarma, Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore for fruitful suggestions.References
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. E. Edwards, Chem. Soc. Rev., 2000, 29, 27–35 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS.
- X. Wang and Y. Li, Chem. Commun., 2007, 2901–2910 RSC.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162–1194 RSC.
- R. R. Arvizo, S. Bhattacharyya, R.A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–93 CrossRef CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Siscot and A. M. Alkilaany, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS.
- G. S. Metraux, Y. C. Cao, R. Jin and C. A. Mirkin, Nano Lett., 2003, 3, 519–522 CrossRef CAS.
- Y. Xiong, B. J. Wiley and Y. Xia, Angew. Chem., Int. Ed., 2007, 46, 7157–7159 CrossRef CAS.
- J. Turkevich, P. C. Stevensen and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- T. Qiu, X. L. Wu, G. G. Siu and P. K. Chu, Appl. Phys. Lett., 2005, 87, 223115 CrossRef.
- X. Liu, N. Wu, B. H. Wunsch, R. J. Barsotti Jr. and F. Stellacci, Small, 2006, 2, 1046–1050 CrossRef CAS.
- G. De and S. Bhattacharyya, J. Mater. Chem., 2008, 18, 2816–2824 RSC.
- N. R. Jana, L. Gearheartand and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- A. D. Jennifer, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS.
- P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S. R. Sainkar, M. I. Khan, R. Ramani, R. Parischa, P. V. Ajayakumar, M. Alam, M. Sastry and R. Kumar, Angew. Chem., Int. Ed., 2001, 40, 3585–9059 CrossRef CAS.
- P. Mukherjee, S. Senapati, D. Mandal, A. Ahmad, M. I. Khan, R. Kumar and M. Sastri, ChemBioChem., 2005, 5, 461–463 Search PubMed.
- H. C. Choi, M. Shim, S. Bangsaruntip and H. Dai, J. Am. Chem. Soc., 2002, 124, 9058–9059 CrossRef CAS.
- J. L. Gardea-Torresdey, J. G. Parsons, E. Gomez, J. Peralta-Videa, H. E. Troiani, P. Santiago and M. J. Yacaman, Nano Lett., 2002, 2, 397–401 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS.
- G. S. Metraux, Y. C. Cao, R. Jin and C. A. Mirkin, Nano Lett., 2003, 3, 519–522 CrossRef CAS.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastri, Nat. Mater., 2004, 3, 482–488 CrossRef CAS.
- B. Liu, J. Xie, J. Y. Lee, Y. P. Ting and J. P. Chen, J. Phys. Chem. B, 2005, 109, 15256–15263 CrossRef CAS.
- J. M. Slocik, R. R. Naik, M. O. Stone and D. W. Wright, J. Mater. Chem., 2005, 15, 749–753 RSC.
- J. Garcia-Serrano, U. Pal, A. M. Herrera, P. Salas and C. Angeles-Chavez, Chem. Mater., 2008, 20, 5146–5153 CrossRef CAS.
- R. Baigorri, J. M. Garcia-Mina, R. F. Aroca and R.A. Alvarez-Puebla, Chem. Mater., 2008, 20, 1516–1521 CrossRef CAS.
- G. Gamez, J .L. Gardea-Torresdey, K. J. Tiemann, J. G. Parsons, I. Herrera and M. J. Yacaman, Microchem. J., 2002, 71, 193–204 CrossRef.
- A. Anshup, J. S. Venkataraman, C. Subramaniam, R. R. Kumar, S. Priya, T. R. S. Kumar, R. V. Omkumar, A. John and T. Pradeep, Langmuir, 2005, 21, 11562–11567 CrossRef.
- P. K. Vemula, U. Aslam, V. A. Mallia and G. John, Chem. Mater., 2007, 19, 138–142 CrossRef CAS.
- G. Peng, U. Tisch, O. Adams, M. Hakim, N. Shehada, Y. Y. Broza, S. Billan, R. Abdah-Bortnyak, A. Kuten and H. Haick, Nat. Nanotechnol., 2009, 4, 669–673 CrossRef CAS.
- M. Dickerson, K. Sandhage and R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS.
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- S. Roy, G. Pauli and A. Banerjee, Nanoscale, 2012, 4, 2734–2740 RSC.
- A. V. Singh, B. M. Bandgar, M. Kasture, B. L. V. Prasad and M. Sastry, J. Mater. Chem., 2005, 15, 5115–5121 RSC.
- M. Wong, M. J. C. Hendrix, K. von der Mark, C. Little and R. Stern, Dev. Biol., 1984, 104, 28–36 CrossRef CAS.
- J. L. Arias, M. S. Fernandez, J. E. Dennis and A. I. Caplan, Connect. Tissue Res., 1991, 26, 37–45 CrossRef CAS.
- R. Shoji, T. Miyazaki, T. Niinou, M. Kato and H. Ishii, J. Mater. Cycles Waste Manage., 2004, 6, 142–146 CrossRef CAS.
- D. Yang, L. Qi and J. Ma, Adv. Mater., 2002, 14, 1543–1546 CrossRef CAS.
- B. Zheng, L. Qian, H. Yuan, D. Xiao, X. Yang, M. C. Paau and M. M. F. Choi, Talanta, 2010, 82, 177–183 CrossRef CAS.
- S.-M. Lee, G. Grass, G.-M. Kim, C. Dresbach, L. Zhang, U. Gösele and M. Knez, Phys. Chem. Chem. Phys., 2009, 11, 3608–3614 RSC.
- Z. Jian, H. Liqing, W. Yongchang and L. Yimin, Phys. E., 2004, 25, 114–118 CrossRef.
- J. P. Wilcoxon, J. E. Martin, F. Parsapour, B. Wiedenman and D. F. Kelley, J. Chem. Phys., 1998, 108, 9137–9143 CrossRef CAS.
- T. Qiu, X. L. Wu, G. G. Siu and P. K. Chu, Appl. Phys. Lett., 2005, 87, 223115–3 CrossRef.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS.
- J. Zheng, C. W. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402–1-077402-4 CrossRef.
- G. T. Boyd, Z. H. Yu and Y. R. Shen, Phys. Rev. B, 1986, 33, 7923–7936 CrossRef CAS.
- E. Dulkeith, T. Niedereichholz, T. A. Klar, J. Feldmann, G. von Plessen, D. I. Gittins, K. S. Mayya and F. Caruso, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 205424–4 CrossRef.
- M. B. Mohamed, V. Volkov, S. Link and M. A. El-Sayed, Chem. Phys. Lett., 2000, 317, 517–523 CrossRef CAS.
- M. T. Hincke, J. Gautron, M. Panheleux, J. Garcia-Ruiz, M. D. McKee and Y. Nys, Matrix Biol., 2000, 19, 443–453 CrossRef CAS.
- Y. Shao, Y. Jin and S. Dong, Chem. Commun., 2004, 1104–1105 RSC.
- X. Liu, N. Wu, B. H. Wunsch, R. J. Barsotti Jr. and F. Stellacci, Small, 2006, 2, 1046–1050 CrossRef CAS.
- N. Basu, R. Bhattacharya and P. Mukherjee, Biomed. Mater, 2008, 3(3), 034105 CrossRef.
- Z. R. Guo, Y. DuanMu and N. Gu, Solid State Phenom., 2007, 121–123, 251–254 CrossRef CAS.
- Y. N. Tan, J. Y. Lee and D. I. C. Wang, J. Am. Chem. Soc., 2010, 132, 5677–5686 CrossRef CAS.
- H. Wei, Z. Wang, J. Zhang, S. House, Y. G. Gao, L. Yang, H. Robinson, L. H. Tan, H. Xing, C. Hou, I. M. Robertson, J. M. Zuo and Y. Liu, Nat. Nanotechnol., 2011, 6, 93–97 CrossRef CAS.
- P. R. Selvakannan, S. Mandal, S. Phadtare, R. Pasricha and M. Sastri, Langmuir, 2003, 19, 3545–3549 CrossRef CAS.
- E. Dinda, M. H. Rashid and T. K. Mandal, Cryst. Growth Des., 2010, 10, 2421–2433 CAS.
- Y. Bao, H-C. Yeh, C. Zhong, S. A. Ivanov, J. K. Sharma, M. L. Neidig, D. M. Vu, A. P. Shreve, R. B. Dyer, J. H. Werner and J. S. Martinez, J. Phys. Chem. C, 2010, 114, 15879–15882 CAS.
| This journal is © The Royal Society of Chemistry 2012 |