A highly active organocatalyst for the asymmetric α-aminoxylation of aldehydes and α-hydroxylation of ketones†
Xinyuan
Fan
a,
Esther
Alza
a and
Miquel A.
Pericàs
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007, Tarragona, Spain. E-mail: mapericas@iciq.es
bDepartament de Química Orgànica, Universitat de Barcelona, 08028, Barcelona, Spain
First published on 13th June 2012
Abstract
Enantiopure trans-3-trifluoromethylsulfonylamino-4-silyloxypyrrolidines efficiently catalyse the asymmetric α-aminoxylation of aldehydes. At 1% catalyst loading (solvent-free conditions) or at 2% catalyst loading (acetonitrile solution) aldehydes are completely converted in short reaction times leading to α-aminoxylation products with very high (96–99%) enantioselectivity.
Over the past decade, aminocatalysis has evolved into a key player in the asymmetric synthesis arena.1 Natural L-proline has been, by large, the archetypal aminocatalyst due to its easy availability and special bifunctional performance.2 Among its applications, the asymmetric α-aminoxylation of carbonyl compounds3 is particularly interesting since it ultimately allows the preparation of enantiopure α-hydroxycarbonyl compounds. These molecules, being important intermediates for the synthesis of biologically active natural products,4 lack efficient enolate-based procedures for its preparation.
The C2 carboxyl group present in the structure of L-proline acts both as an steric controller and as an electrophile activating group, allowing the achievement of high levels of asymmetric induction in this reaction with ketone and α-unsubstituted aldehyde substrates.5 Proline, however, needs to be used at high loading for practical reaction rates3e,f and, for this reason, substantial effort has been devoted to the development of alternative, more active catalysts for α-aminoxylation reactions.6 Among them, 4-tert-butyldimethylsilyloxy-proline (A)6b and the Maruoka chirally axial triflamide (B)6g have shown promising results. However, the development of a highly active and enantioselective catalyst, able to process aldehyde and ketone substrates in short reaction times at low catalyst loadings has remained elusive. We report in this communication substantial progress in this direction.
We have recently introduced pyrrolidines 1–37 (Fig. 1) as highly active organocatalysts for anti-selective asymmetric Mannich reactions.8 We noted that 1–3 condensed into small, readily available molecules all the key structural information of A–B, and we decided to evaluate them for the seek of high catalytic performance in α-aminoxylation reactions.
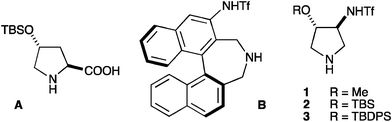 | ||
| Fig. 1 | ||
The performance of catalysts 1–3 was studied in the α-aminoxylation of propionaldehyde with nitrosobenzene in acetonitrile. The results are presented in Table 1, together with those for (R)-β-proline.8e
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | t [min] | Yield [%]b | ee [%]c |
| a All the reactions were carried out with 1 eq. PhNO, 3 eq. aldehyde and 5 mol% of the corresponding catalyst at 0 °C. b Isolated yield of reduction product. c Determined by chiral HPLC. d Reaction performed at rt. e 2 mol% 3 was used in this experiment. | ||||
| 1 | 1 | 54 | 77 | 92 |
| 2 | 2 | 47 | 87 | 92 |
| 3 | 3 | 60 | 81 | 98 |
| 4d | 3 | 25 | 66 | 97 |
| 5de | 3 | 70 | 67 | 96 |
| 6 | (R)-β-proline | 30 | 89 | 81 |
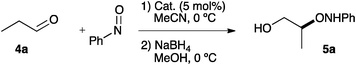
As it can be observed, pyrrolidines 1–3, bearing a Brønsted acid site at C-3 lead to high conversions in short reaction times. All of them are substantially more active than proline.3e,f The C-4 substituent clearly plays a role in the enantioselectivity, which increases with the bulk of this substituent (entries 1–3) and is clearly superior to the referable (R)-β-proline that lacks substitution at C-4 (entry 6). Interestingly, 3 can be used at room temperature with important acceleration of the reaction rate (entry 4) with practically no decrease in enantioselectivity, and catalyst loading can be reduced to 2 mol% (entry 5) while keeping reaction times short.9 There have been contradictory reports on the catalytic activity of L-proline in this reaction. Initial reports3a–d overestimated this activity, probably because of a poor understanding of the fate of nitrosobenzene in the reaction.10 To establish a direct comparison between the reactivities of L-proline and 3, two parallel experiments with 10 mol% catalyst were performed in acetonitrile-d3 at room temperature and followed by NMR (see ESI† for details). At 5 min reaction time (initial control) the reaction mediated by 3 is already complete, while any conversion can be barely appreciated in the experiment with L-proline. With this catalyst, complete conversion required ca. 60 min. It is thus clear that 3 has a catalytic activity far superior to L-proline. It is very probable that this behaviour originates from a very favourable enamine formation from 3, a pyrrolidine unsubstituted at C2 and C5.
The use of alternative reaction conditions for the α-aminoxylation reaction was next explored (Table 2). With propanal as a substrate (entries 1–7), different reaction media were explored (entries 1–4). Both chloroform and water (entries 1–2) led to very fast reactions at 5% catalyst loading. Ethanol, in turn, inhibited reaction (entry 3). We were most pleased to find that the reaction under neat conditions (entry 4) was completed in 5 min at room temperature, leading in 71% yield to α-aminoxylation product with very high enantiomeric purity (96% ee). Interestingly, catalyst loading could be reduced to 2% (entry 5) and further to 1% (entry 6) with only a small increase in reaction time and no decrease in enantioselectivity. Only when the catalyst loading was decreased to 0.5% (entry 7) reaction time had to be extended to 50 min.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | 3 [mol%] | Solvent | t [min] | Yield [%]b | ee [%]c |
| a Reactions were carried out with 1 eq PhNO and 3 eq aldehyde at room temperature. b Isolated yield of reduction product. c By HPLC using a Chiralpak AD-H column. d A similar experiment performed at 0 °C required 70 min for completion and yielded 80% product. e Using 5 eq of aldehyde. | ||||||
| 1 | Me | 5 | CHCl3 | 10 | 61d | 91 |
| 2 | Me | 5 | H2O | 15 | 53 | 95 |
| 3 | Me | 5 | EtOH | 120 | trace | nd |
| 4e | Me | 5 | neat | 5 | 71 | 96 |
| 5e | Me | 2 | neat | 6 | 69 | 96 |
| 6e | Me | 1 | neat | 10 | 78 | 97 |
| 7e | Me | 0.5 | neat | 50 | 63 | 96 |
| 8e | Et | 1 | neat | 70 | 56 | 96 |
| 9e | iPr | 1 | neat | 25 | 60 | 77 |
| 10e | n-C5H11 | 1 | neat | 60 | 52 | 90 |
These very attractive reaction conditions were also tested with a small family of α-unsubstituted aldehydes (entries 8–10). Essentially complete conversion of the limiting nitrosobenzene reactant was recorded into minutes, the corresponding α-aminoxylation products being formed with high selectivity10 and, in general, high enantioselectivity. In spite of its practical interest, the solvent-free reaction conditions have some limitation: they are dependent on the solubility of 3 in the reactant aldehyde. When this solubility is low, reaction times are comparatively longer (entries 8 and 10).
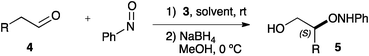
To avoid this problem, 3 was used for the α-aminoxylation of a representative family of aldehydes (4a–h) at a convenient 2 mol% loading in acetonitrile at 0 °C (Table 3). These conditions were designed for optimized yield (through minimisation of the subsequent reaction of the α-aminoxylation product with nitrosobenzene) and enhanced enantioselectivity. As it can be seen, the aminoxylation products are formed under these conditions in good to high yields and with uniformly excellent enantioselectivity. In spite of the low catalyst loading, reaction times remain reasonably low, and only 3-phenylpropanal (4h, entry 8), requires 6 h for complete conversion.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | t [h] | Yield [%]b | ee [%]c |
| a All the reactions were performed with 0.25 mmol PhNO, 0.75 mmol of aldehyde, 2 mol% of 3 and 0.25 mL of MeCN at 0 °C. b Isolated yield of reduced product. c Determined by chiral HPLC. | |||||
| 1 | Me | 5a | 5 | 85 | 98 |
| 2 | Et | 5b | 3 | 81 | 99 |
| 3 | Pr | 5c | 5 | 85 | 99 |
| 4 | n-C5H11 | 5d | 5 | 80 | 96 |
| 5 | iPr | 5e | 2 | 67 | 97 |
| 6 | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2 CHCH2 |
5f | 2 | 83 | 96 |
| 7 | CH2CH(CH2)7 |
5g | 3 | 63 | 98 |
| 8 | Bn | 5h | 6 | 66 | 98 |

The performance of 3 in the α-aminoxylation of ketone substrates was next tested (Scheme 1). It is worth mentioning here that the use of the highly active α-aminoxylation catalyst B6g with ketone substrates has not been reported, while N-(2-pyrrolidinylmethyl)triflamide6c works well with ketones but requires very high loadings (20 mol%). When the same reaction conditions as those given in Table 3 were used with ketones 6a–d, useless mixtures of α-aminoxy and α-hydroxy products 7a–d were formed. However, 7a–d could be obtained as the only reaction products in moderate to good yield and high enantiomeric purity by slow addition (syringe pump, 30 min) of excess (3 eq.) nitrosobenzene at room temperature in acetonitrile.10
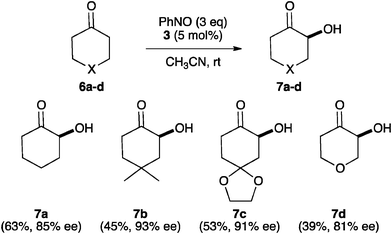 | ||
| Scheme 1 Asymmetric α-hydroxylation of ketones catalysed by 3. | ||
Catalyst 3 with the absolute configuration shown in Fig. 1 leads to α-aminoxylation products with S configuration. This is opposite to the outcome of the reactions catalysed by L-proline, and this behaviour can be rationalised on the basis of the conformational preferences of the putative enamine intermediates in the reaction (Fig. 2). According with the results obtained with 1–3 and with (R)-β-proline in the α-aminoxylation reaction (Table 1) and with 1–3 in anti-selective Mannich reactions,7 the R-oxy substituent at C-4 on the pyrrolidine ring sterically alters the conformation of the enamine with respect to L-proline. In this manner, the reacting face of the enamine is changed from Re (L-proline) to Si (1–3), as represented in Fig. 2.
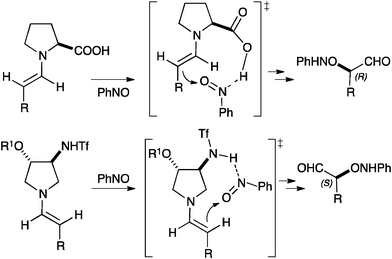 | ||
| Fig. 2 Rationalisation of the observed enantioselectivity in the α-aminoxylation of aldehydes catalysed by 1–3. | ||
With respect to stereochemical outcome, it is important to realise that 1–3 ultimately arise from the desymmetrisation11 of a meso 3-pyrroline oxide, being equally available in both enantiomeric series. In this manner, access to both enantiomers of the α-aminoxylation products can be readily secured.
In summary, we have identified an extremely efficient catalyst (3) for the asymmetric α-aminoxylation of aldehydes and for the α-hydroxylation of ketones. Aldehyde substrates with good miscibility with 3 lead to complete conversion within minutes under solvent-free conditions and at low (1 mol%) catalyst loading. For optimal yield and enantioselectivity, reactions of aldehydes are best performed in acetonitrile at 0 °C. Under these conditions, low catalyst loadings (2 mol%) are compatible with short reaction times, and α-aminoxylation products are obtained with very high (96–99%) enantioselectivity.
Acknowledgements
We thank MINECO (CTQ2008-00947/BQU), DEC (2009SGR623), and the ICIQ Foundation for financial support.References
-
(a)
A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed
; (b) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed
-
(a) B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS
-
(a) G. Zhong, Angew. Chem., Int. Ed., 2003, 42, 4247–4250 CrossRef CAS
-
(a) I. K. Mangion and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3696–3697 CrossRef CAS
- P. H.-Y. Cheong and K. N. Houk, J. Am. Chem. Soc., 2004, 126, 13912–13913 CrossRef CAS
-
(a) N. Momiyama, H. Torii and H. Yamamoto, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5374–5378 CrossRef CAS
- R. Martín-Rapún, X. Fan, S. Sayalero, M. Bahramnejad, F. Cuevas and M. A. Pericàs, Chem.–Eur. J., 2011, 17, 8780–8783 CrossRef
- H. Zhang, S. Mitsumori, N. Utsumi, M. Imai, N. Garcia-Delgado, M. Mifsud, K. Albertshofer, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2008, 130, 875–886 CrossRef CAS
- At rt, reaction of the α-aminoxylation product with nitrosobenzene leading to azoxybenzene and to α-hydroxypropanal starts to be observed (see ref. 10). Combined yield is >80%. This secondary reaction takes place to a much lesser extent with 3 than with proline under the same reaction conditions (see ref. 3f).
- D. B. Ramachary and C. F. Barbas, Org. Lett., 2005, 7, 1577–1580 CrossRef CAS
-
(a) L. E. Martínez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and HPLC chromatograms of reaction products. See DOI: 10.1039/c2ra20968c |
| This journal is © The Royal Society of Chemistry 2012 |