A panchromatic anthracene-fused porphyrin sensitizer for dye-sensitized solar cells†
James M.
Ball
a,
Nicola K. S.
Davis
b,
James D.
Wilkinson
b,
James
Kirkpatrick
ac,
Joël
Teuscher
a,
Robert
Gunning
a,
Harry L.
Anderson
*b and
Henry J.
Snaith
*a
aDepartment of Physics, University of Oxford, Clarendon Laboratory, Oxford, OX1 3PU, UK. E-mail: henry.snaith@physics.ox.ac.uk
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Oxford, OX1 3TA, UK. E-mail: harry.anderson@chem.ox.ac.uk
cMathematical Institute, University of Oxford, Oxford Centre for Collaborative Applied Mathematics, Oxford, OX1 3LB, UK
First published on 28th June 2012
Abstract
The development of ruthenium-free sensitizers which absorb light over a broad range of the solar spectrum is important for improving the power conversion efficiency of dye-sensitized solar cells. Here we study three chemically tailored porphyrin-based dyes. We show that by fusing the porphyrin core to an anthracene unit, we can extend the conjugation length and lower the optical gap, shifting the absorption spectrum into the near-infrared (NIR). All three dyes were tested in dye-sensitized solar cells, using both titanium dioxide and tin dioxide as the electron-transport material. Solar cells incorporating the anthracene-fused porphyrin dye exhibit photocurrent collection at wavelengths up to about 1100 nm, which is the longest reported for a porphyrin-based system. Despite extending the photon absorption bandwidth, device efficiency is found to be low, which is a common property of cells based on porphyrin dyes with NIR absorption. We show that in the present case the efficiency is reduced by inefficient electron injection into the oxide, as opposed to dye regeneration, and highlight some important design considerations for panchromatic sensitizers.
Introduction
Dye-sensitized solar cells (DSSCs) are a promising alternative to conventional photovoltaic technologies for solar energy conversion owing to their potentially lower fabrication costs.1 The properties of the photon-absorbing sensitizer are critical in determining DSSC performance, so optimizing the molecular design is paramount to maximizing power conversion efficiency (η). Conventional high-efficiency DSSCs have been based on polypyridyl ruthenium complexes as sensitizers.2–5 Although such dyes have broad absorption spectra, energetically compatible excited states with other cell components, relatively long-lived excited states and good stability, they can be toxic and expensive. These disadvantages have driven a search for alternatives.Porphyrins comprise a family of organic chromophores which are attractive for application in DSSCs6–23 owing to their high molar extinction coefficients, ease of modification, photochemical stability, low toxicity and potential low-cost. Porphyrin chromophores exhibit two absorption bands: the Soret- and the Q-bands which occur in the blue and red respectively. With suitable molecular control, these bands can be shifted and broadened, potentially facilitating an increase in the total photon absorption and, therefore, the achievable photocurrent in DSSCs.
Recently, Bessho et al.19 and Wang et al.20 have synthesized porphyrin dyes with electron-donating (D) and -accepting (A) moieties for intramolecular separation of the electron and hole that comprise the excited state. Devices implementing these dyes have exhibited >10% power conversion efficiency demonstrating that high efficiencies are attainable from ruthenium-free dyes. More recently still, a breakthrough in the world of DSSCs has been achieved by Yella et al.23 who have used a new cobalt electrolyte in conjunction with a porphyrin sensitizer, to deliver a record power conversion efficiency of 12.3%. However, these achievements have been made with dyes that do not absorb far enough into the NIR and further enhancement of the efficiency may be achievable by extending the size of the conjugated core, to reduce the gap between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO respectively), extending the onset of absorption to longer wavelengths.24 Towards this aim, Hayashi et al. fused an indene ring to the porphyrin core, resulting in a broadening and red-shift of the absorption bands.21 Similarly Jiao et al. synthesized a perylene-fused porphyrin, which enabled light absorption out to the NIR.22 The latter approaches have thus far yielded modest efficiencies in devices, demonstrating a need for further understanding of the mechanisms which limit light-harvesting in panchromatic porphyrin dyes.
Here we present the synthesis and characterization of three porphyrin dyes with modified molecular structures (P1–P3, Fig. 1). All three dyes are comprised of: a Zn–porphyrin core; an ethynylphenyl group linked to a carboxylic acid anchor to enhance charge-transfer from the chromophore into the metal oxide; and aryl substituents to inhibit aggregation. We compare an unsubstituted porphyrin (P1) to an anthracene-substituted porphyrin (P2), and finally to an anthracene-fused porphyrin (P3) with an extended conjugated chromophore for enhanced absorption at long wavelengths, based on a recently developed synthetic methodology.25–27 We present the electrochemical and absorption properties of the dyes and assess the impacts of these modifications on device performance. In particular, we show that devices based on P3 exhibit a remarkable photocurrent onset of ∼ 1100 nm. DSSCs based on P3 and standard TiO2 nanoparticles do not show any photocurrent generation in the NIR at 700–1100 nm, because the LUMO of the dye is too low to transfer an electron into the conduction band of the TiO2. Lowering the conduction band of the metal oxide, by using SnO2 in place of TiO2, or by doping the TiO2 with lithium ions, results in a dramatic increase in the incident photon-to-current conversion efficiency (IPCE) in the NIR spectral region. Finally we show, through simulation and transient absorbance spectroscopy, that electron injection from P3, as opposed to dye regeneration, is the inhibiting step for efficient collection of photoexcited charge carriers as a consequence of anthracene-fusion. This work highlights the importance of controlling both the energy and localization of the unoccupied molecular orbitals of porphyrin dyes when modifying their molecular and electronic structure for panchromatic absorption for use in high-efficiency DSSCs.
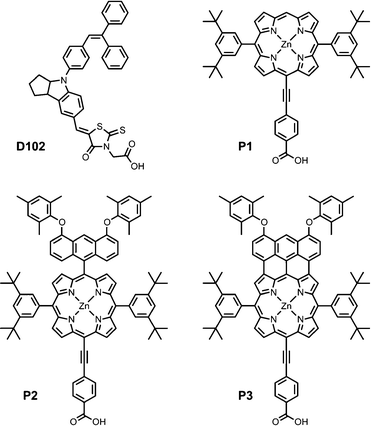 | ||
| Fig. 1 Molecular structures of the dyes studied in the present work. | ||
Methods
Porphyrin characterization
UV-vis and UV-vis-NIR spectra were in recorded in chloroform with 1% pyridine using Perkin-Elmer Lambda 20 and Perkin-Elmer Lambda 9 photospectrometers respectively. Electrochemical characterization was conducted using square-wave voltammetry (Autolab Echo-Chemie PGSTAT12) recorded in THF with 0.1 M Bu4NPF6 at a square wave frequency of 8 Hz using a glassy carbon working electrode, Pt counter electrode and Ag/AgNO3 reference electrode referenced to internal ferrocene.Molecular simulation was conducted using density functional theory (DFT).28–30 Becke's three parameter hybrid density functional (B3LYP) was used to calculate ground state geometries and molecular orbital energies. A double zeta split basis set with added polarization was used as the basis set on all atoms except the Zn, which was fitted with a Steven's effective core potential and basis sets (CEP 3-1g). Time-dependent methods were used to compute the band-gap.
Device fabrication and characterization
Devices were fabricated on fluoride-doped tin(IV) oxide (FTO) coated glass substrates which were cleaned sequentially in Hellmanex detergent (2% by volume in water), DI water, acetone and ethanol followed by exposure to O2 plasma for 10 min. Mesoporous oxide films were deposited from nanoparticle pastes by screen printing using a mesh with 120 threads/cm. TiO2 was deposited from an anatase nanoparticle paste with an average particle size of 20 nm (Dyesol, DSL 18NR-T). The SnO2 paste was prepared using a previously reported31,32 method from a nanopowder with an average particle size of ∼ 40 nm (Alfa Aesar). Thicker films were obtained by sequential screen printing with a drying step of 5 min at 150 °C between each print. The oxide films were then heated in 1 h to 500 °C for 30 min and left to cool to room temperature. After sintering, the substrates were placed in a bath of 20 mM TiCl4 in DI water for 1 h at 70 °C followed by rinsing in DI water. The substrates were then sintered for a second time to 500 °C for 30 min and left to cool to 70 °C at which point they were submerged in a bath of dye solution for 4 h in the dark at room temperature. Porphyrin dye solutions were made at 150 μM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume solvent mixture of ethanol:chloroform also containing 300 μM chenodeoxycholic acid as an aggregation inhibitor. The D102 solution was 150 μM in a 1:1 by volume mixture of anhydrous acetonitrile:tert-butanol. Devices were assembled with a thermally platinized (50 μM chloroplatinic acid in ethanol doctor bladed and annealed at 450 °C for 15 min) FTO glass counter electrode separated from the working electrode with a 25 μm Surlyn hot-melt gasket. The electrolyte was introduced through a sandblasted filling-hole in the counter electrode by vacuum infiltration and then sealed with Surlyn and a cover glass. Two non-volatile iodide/triiodide electrolyte mixtures were used with the following compositions: 0.8 M 1,3-dimethylimidazolium iodide (DMII), 0.15 M iodine, 0.5 M methylbenzimidazole and 0.1 M guanadinium thiocyanate in 3-methoxypropionitrile (MPN); 2 M lithium iodide and 0.1 M iodine in MPN. These electrolytes will be referred to henceforth as E1 and E2-Li respectively.
:1 by volume solvent mixture of ethanol:chloroform also containing 300 μM chenodeoxycholic acid as an aggregation inhibitor. The D102 solution was 150 μM in a 1:1 by volume mixture of anhydrous acetonitrile:tert-butanol. Devices were assembled with a thermally platinized (50 μM chloroplatinic acid in ethanol doctor bladed and annealed at 450 °C for 15 min) FTO glass counter electrode separated from the working electrode with a 25 μm Surlyn hot-melt gasket. The electrolyte was introduced through a sandblasted filling-hole in the counter electrode by vacuum infiltration and then sealed with Surlyn and a cover glass. Two non-volatile iodide/triiodide electrolyte mixtures were used with the following compositions: 0.8 M 1,3-dimethylimidazolium iodide (DMII), 0.15 M iodine, 0.5 M methylbenzimidazole and 0.1 M guanadinium thiocyanate in 3-methoxypropionitrile (MPN); 2 M lithium iodide and 0.1 M iodine in MPN. These electrolytes will be referred to henceforth as E1 and E2-Li respectively.
Current density-voltage (J–V) characteristics under 1 sun simulated solar illumination (100 mW cm−2, AM 1.5G) were obtained with an ABET Technologies Sun 2000 Solar Simulator and a Keithley 2400. The incident photon-to-current conversion efficiency (IPCE) was measured as a function of wavelength with a xenon lamp, monochromator (Princeton Instruments, Acton SP2150) and Keithley 2400 at 1 mW cm−2 incident power density. The apparatus for device characterization was calibrated with an NREL certified KG5 filtered Si reference diode. The solar mismatch factor was calculated to be less than 1%. The device area under illumination was defined with a metal mask as 0.25 cm2. The rear and edges of the cells were sealed with black tape to minimize reflection errors. Metal oxide thicknesses (doxide) were measured using a Veeco Dektak 150 surface profilometer.
Transient absorbance spectroscopy
Transient absorbance spectroscopy was applied to dye-sensitized “transparent” TiO2 mesoporous films (doxide = 10 μm) deposited on glass using the aforementioned techniques. Pulsed excitation (λexcitation = 510 nm, 7 ns pulse duration, 10 Hz repetition rate) of the dyed film was conducted using an Nd:YAG laser (Ekspla, NT340) with a laser fluence on the sample maintained at < 40 μJ cm−2 per pulse. The probe light, produced by a continuous wave xenon arc lamp, was first passed through a monochromator (Acton Research Corporation, SpectraPro-2150i), various optical elements, the sample, and then through a second monochromator (Acton Research Corporation, SpectraPro-2300i) before being detected by a fast photomultiplier tube module biased with 750 V (Hamamatsu, R9110). Data waves were recorded on an oscilloscope (Tektronix, DPO3054). Satisfactory signal-to-noise ratios were typically obtained by averaging over 256 laser shots. Data were smoothed using a Savitsky–Golay algorithm for presentation.Results and discussion
Porphyrin synthesis and characterization
The synthesis of porphyrin dyes P1–P3 is described in detail in the ESI.† In brief, the synthetic route for P2 and P3 is shown in Scheme 1. Anthracene-linked porphyrin 1 was synthesized by a Suzuki coupling and brominated with NBS to form 2. Sonogashira coupling of 2 with triisopropylsilylacetylene produced 3. Fusion of the linked anthracene to the porphyrin core was achieved by adding 10 equivalents of FeCl3 and 50 equivalents of ZnCl2 to 3 and stirring for 30 min followed by the addition of 10 equivalents of 1,8-bis(dimethylamino)naphthalene and a further 30 min of stirring. This sequence was repeated three times to obtain 5. Both 3 and 5 were subsequently coupled with methyl-4-iodobenzoate under the conditions indicated in Scheme 1 to form 4 and 6. Finally, hydrolysis of these porphyrin esters in basic conditions yielded P2 and P3.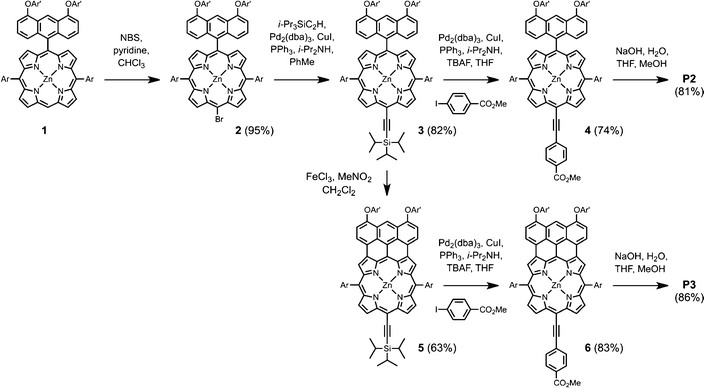 | ||
| Scheme 1 Synthetic route used to prepare porphyrin sensitizers P2 and P3 (Ar and Ar' are defined in Fig. 1). | ||
Square-wave voltammetry was carried out on the porphyrins to determine their redox potentials as shown in Fig. 2a. The first oxidation (EHOMO) and reduction (ELUMO) peaks with respect to Fc/Fc+ as well as the HOMO–LUMO energy gap (EG) of porphyrins P1–P3 are summarized in Table 1 and the energy level diagram shown in Fig. 3. P1 exhibits an electrochemical gap of 2.13 eV. P2, with the linked anthracene, shows almost identical electrochemical behavior suggesting that the linked unit makes only a minor difference to the electronic structure of the porphyrin. When fused, the electrochemical gap of the anthracene-porphyrin decreases in comparison to the unfused analogue from 2.20 eV to 1.47 eV. This is consistent with the extended conjugation of P3.
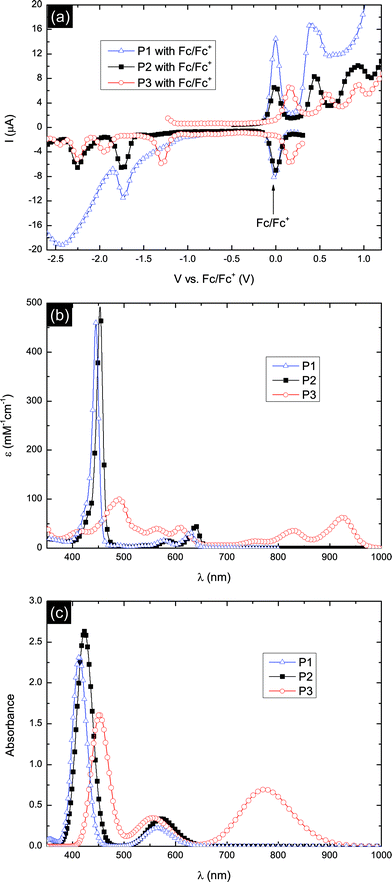 | ||
| Fig. 2 (a) Square-wave voltammetry in THF with 0.1 M Bu4NPF6 and an Ag/AgNO3 reference electrode, referenced to internal ferrocene, (b) UV-vis-NIR absorption spectra in a chloroform solution with 1% pyridine and (c) calculated UV-vis absorption spectra of P1–P3. | ||
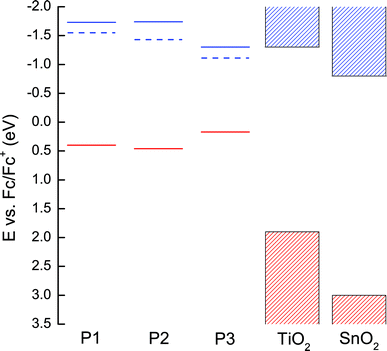 | ||
| Fig. 3 Electronic structures of P1–P3 derived from electrochemistry (full lines) and absorption spectra (dashed lines) compared to the conduction and valance bands of TiO2 and SnO2.34,35 On this energy scale occupied states are positive (red) and unoccupied states are negative (blue). | ||
| Dye | E HOMO (eV) | E LUMO (eV) | E G (eV) | Absorption peaks (nm) | |||||
|---|---|---|---|---|---|---|---|---|---|
| E-chema (vs. Fc/Fc+) | DFT (vs. vacuum) | E-chema (vs. Fc/Fc+) | DFT (vs. vacuum) | E-chema | DFT | Optical | λ Soret | λ Q | |
| a Electrochemical data were obtained from THF solutions with Fc/Fc+, 0.1 M Bu4NPF6 and an Ag/AgNO3 reference electrode. | |||||||||
| P1 | 0.40 | −5.10 | −1.73 | −2.57 | 2.13 | 2.19 | 1.95 | 440 | 618 |
| P2 | 0.46 | −5.04 | −1.74 | −2.54 | 2.20 | 2.06 | 1.89 | 453 | 639 |
| P3 | 0.17 | −4.55 | −1.30 | −2.80 | 1.47 | 1.60 | 1.28 | 490 | 924 |
The UV-vis-NIR absorption spectra of porphyrins P1–P3 are shown in Fig. 2b. Both the Soret and Q-bands of P2 are red-shifted and show an increase in intensity in comparison to the P1. In comparison to P2, the anthracene-fused porphyrin P3 exhibits a dramatic red-shift and broadening of the absorption spectrum. The Q-band peak wavelength shifts from 639 nm for P2 to 924 nm for P3. The absorption onset of ∼1000 nm is among the longest onset wavelengths investigated in DSSCs.22,33 The strong panchromatic absorption of P3 may be expected to facilitate excellent light harvesting when incorporated into DSSC devices.
The energy of an electron in the first excited state, i.e. the LUMO level, for each dye can be estimated by adding the energy of the absorption onsets34 (10% intensity of the Q-band peak) to the electrochemical oxidation potentials as shown in Fig. 3 (dashed lines). Electron injection appears to be energetically unfavorable from the first excited state of P3 when compared to the conduction band of TiO2. However, when the dyes are bound to the oxide in an environment of electrolyte within complete solar cells, this picture is likely to be modified.
Further insight into the molecular properties of dyes P1–P3 was inferred from DFT calculations. The calculated HOMO and LUMO energies are summarized in Table 1 with respect to vacuum. The simulated absorption spectra (Fig. 2c) were obtained by making a convolution of the calculated oscillator strengths for each transition to Gaussians with a width of 0.1 eV. The calculated energies of the frontier orbitals are summarized in Fig. 4. DFT correctly represents the experimental trends in band gap and frontier orbital energies, though we do note some deviation in absolute peak position, especially for the P3 Q-band.
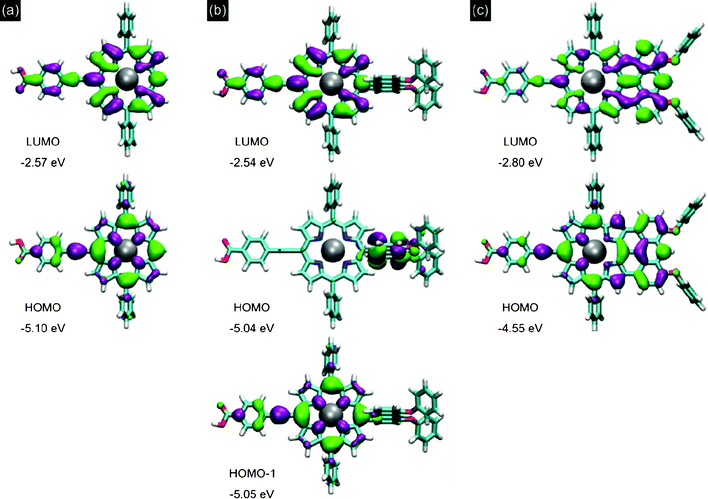 | ||
| Fig. 4 Calculated frontier molecular orbitals for (a) P1, (b) P2 and (c) P3. | ||
Fig. 4 shows the frontier molecular orbitals of each porphyrin. On P1 both the HOMO and LUMO are broadly delocalized suggesting minimal spatial separation of the charges in the excited state. Linking the anthracene group in P2 causes a shift in localization of the HOMO onto the anthracene unit. However, the HOMO-1 for P2 is found to be nearly degenerate (EHOMO − EHOMO-1 = 0.016 eV) with the HOMO and has an equivalent distribution over the porphyrin and anchor to the HOMO of P1. This is supported experimentally by the similarity in the oxidation potentials measured for P1 and P2. In P2, because the anthracene unit is perpendicular to the porphyrin core, the oscillator strength for transition from the HOMO to the LUMO is small. This means that light absorption in P2 is dominated by excitations from the HOMO-1 and explains why the spectra of P1 and P2 are qualitatively similar. Quantitatively the computed absorption spectra in Fig. 2c correctly reproduce the experimentally observed red-shift and absorption enhancement in P2 compared to P1. The influence of the anthracene linker on DSSC operation may therefore be steric rather than energetic compared to P1.
The DFT computation explains the dramatic decrease in frontier orbital energy gap for P3 as a very significant change in localization of the HOMO and LUMO. Fusing the anthracene in P3 is found to delocalize both the HOMO and LUMO onto the extended conjugated unit away from the anchoring group. Potentially, this may reduce the rate of electron injection into the oxide in comparison to P1 and P2. This suggests that a careful choice of the substituents and the positioning of the linker group are required to optimize the dye for photon-to-current conversion.
Device characterization
Initial devices were fabricated using TiO2 as the electron transport layer and a non-volatile electrolyte, E1, for dye regeneration and hole transport. The J–V characteristics under 1 sun illumination are presented in Fig. 5 and the short-circuit current density (Jsc), open-circuit voltage (Voc), fill factor (FF) and η extracted from these plots are summarized in Table 2. Control devices using an indoline-based dye, D102,36,37 exhibited power conversion efficiencies of up to 2.35%. Although improvements can be achieved through appropriate device optimization,32 this is not the topic of the present report.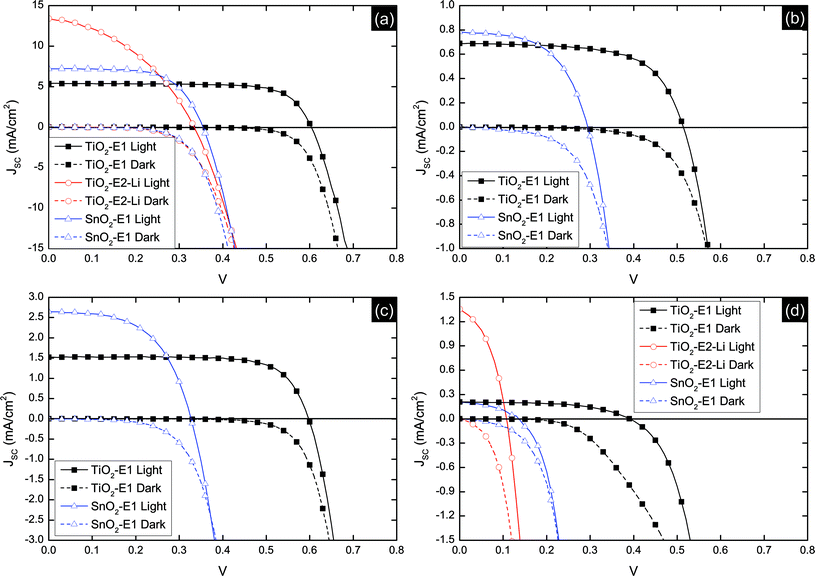 | ||
| Fig. 5 J–V characteristics for all device architectures studied. (a) D102 (dTiO2 = 7.0 μm, dSnO2 = 7.0 μm), (b) P1 (dTiO2 = 7.0 μm, dSnO2 = 3.5 μm), (c) P2 (dTiO2 = 7.0 μm, dSnO2 = 3.5 μm) and (d) P3 (dTiO2 = 7.0 μm, dSnO2 = 7.0 μm). | ||
| Device Structure | d oxide (μm) | J sc (mA cm−2) | V oc | FF | η (%) |
|---|---|---|---|---|---|
| TiO2–D102–E1 | 7.0 | 5.39 | 0.60 | 0.74 | 2.35 |
| TiO2–D102–E2–Li | 7.0 | 13.39 | 0.33 | 0.41 | 1.76 |
| SnO2–D102–E1 | 7.0 | 7.21 | 0.35 | 0.64 | 1.58 |
| TiO2–P1–E1 | 7.0 | 0.69 | 0.51 | 0.64 | 0.22 |
| SnO2–P1–E1 | 3.5 | 0.78 | 0.29 | 0.56 | 0.13 |
| TiO2–P2–E1 | 7.0 | 1.52 | 0.60 | 0.73 | 0.66 |
| SnO2–P2–E1 | 3.5 | 2.65 | 0.33 | 0.55 | 0.47 |
| TiO2–P3–E1 | 7.0 | 0.21 | 0.39 | 0.55 | 0.045 |
| TiO2–P3–E2–Li | 7.0 | 1.36 | 0.11 | 0.40 | 0.059 |
| SnO2–P3–E1 | 7.0 | 0.21 | 0.14 | 0.39 | 0.011 |
The porphyrin dye P2 exhibited a higher Jsc and Voc than P1. The difference in photocurrent generation is an expected result of the difference in light absorption in the Q-band of the respective dyes. The steric consequence of the twisted anthracene unit in P2 may also make it less likely to aggregate compared to P1. The relatively higher density of free charge in devices based on P2 will lead to a wider splitting of the electron and hole quasi-Fermi levels therefore increasing Voc. The orientation and packing of the dye on the surface may also be influenced by the additional anthracene moiety which may result in relative shift in potential at the dye-sensitized interface due to either a change in the dipole nature of the dye layer, or influencing the ion concentration on the surface of the TiO2.
Interestingly, although P3 absorbs a broader spectrum of incident light than P1 and P2, devices based on this dye exhibited lower Jsc and Voc. The normalized IPCE as a function of incident wavelength for a TiO2–P3–E1 device is shown in Fig. 6. This indicates that despite the dye having an absorption onset at ∼1000 nm in solution, charge collection only occurs for wavelengths shorter than ∼700 nm, i.e. photon absorption in the Q-band, corresponding to the first excited state, contributes no current. This may be a consequence of poor electron injection from the dye LUMO into the conduction band-edge of TiO2 resulting from an insufficient energetic driving force between these levels. This is consistent with the electrochemical and absorption data.
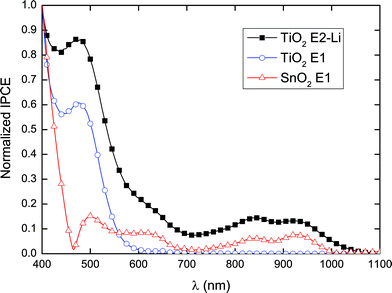 | ||
| Fig. 6 Normalized incident photon-to-current conversion efficiency (IPCE) for P3 with various oxide-electrolyte configurations. | ||
To investigate the origin of the poor performance of devices based on P3 in more detail, devices using SnO2 as the electron transporting layer were fabricated. Exchanging TiO2 for SnO2 allows the assessment of device performance using an oxide with a deeper conduction band-edge,35,38 which, in principle, should enhance the rate of electron injection from the lowest excited states on the dye. The conduction band of SnO2 is significantly below the LUMO of dye P3, as shown in Fig. 3. For all dyes studied using SnO2, an increase in Jsc and a decrease in Voc was observed. This is consistent with a deeper conduction band in the oxide enhancing the favorability of electron injection but lowering the potential difference between the electrons and holes collected at their respective electrodes.
The normalized IPCE spectrum for a SnO2–P3–E1 device shown in Fig. 6 indicates a peak in the NIR, revealing that lowering the conduction band-edge of the oxide enables electron injection from the first excited state. Similarly, introducing lithium ions into the electrolyte has been observed to lower the conduction band-edge of the oxide with respect to vacuum,39 increasing the energetic favorability for electron injection but at the cost of lowering the open-circuit voltage. This was achieved here using an iodide/triiodide electrolyte with a high concentration of lithium iodide, E2–Li. When using this electrolyte, devices based on the control dye, D102, exhibited a dramatically increased Jsc, with a concurrent decrease in Voc and FF, consistent with a lowering of the oxide conduction band-edge. Using E2–Li in devices based on P3 yielded an order of magnitude increase in Jsc in comparison to devices employing E1. As in the case of SnO2, lowering the conduction band-edge by introducing Li+ results in an increase in IPCE in the Q-band with an onset of ∼1100 nm (see Fig. 6) suggesting that electron injection from the first excited state is achievable once the density of states in the oxide conduction band is large enough at the LUMO energy of the dye. To expand upon this, it is important to note that with similar Soret- and Q-band peak extinction coefficients, as shown in Fig. 2b, observing a difference in IPCE peak heights from devices on TiO2 suggests that the electron injection efficiency is higher in the Soret band, both with and without the addition of LiI. This could be due to “hot” electron-transfer into TiO2 where it is unnecessary for the electron to relax to the dye LUMO level following higher energy excitation.40–42
The absolute values for the IPCE spectra for the P3 dye reached up to ∼6% and ∼1% for the Soret and Q-bands respectively (see Fig. S33, ESI†). These values suggest that the low IPCE of devices based on P3 are associated with the kinetics of electron injection. The calculated LUMO distribution of P3 indicates there is no intramolecular charge transfer nature to the first excited state following fusion of the anthracene moiety. Extension of the conjugated core in P3 shifts the LUMO away from the ethynylphenyl group on the anchor which is likely to decrease the rate for electron injection from the dye into the metal oxide. This highlights an important design consideration for panchromatic dyes where the absorption is controlled by changing the size of the conjugated core. Although extending the core will narrow the optical gap, enhancing absorption at longer wavelengths, choosing appropriate chromophore substituents is crucial to ensuring there is a sufficient electronic coupling at the interface between the dye and the oxide for charge transfer whilst maintaining a large Voc. Additionally, careful tailoring of the orbital localization is required to enhance the competitiveness of electron injection from the excited state of the dye into the metal oxide versus natural decay.
Transient absorbance spectroscopy
Although inefficient electron injection is likely to be responsible for the low photocurrents of P3, slow regeneration of the dye cation by the electrolyte may also limit current generation by allowing time for the injected electron to recombine with the hole on the dye. To further investigate the charge transfer dynamics at the dye interface, transient absorbance spectra were obtained from porphyrin-loaded TiO2 films in an environment of MPN or of electrolyte E1. Hence measuring the lifetime of the cation when it can or cannot be regenerated by the electrolyte provides information about the rate limiting step during cell operation. Fig. 7 shows the transient absorbance decay probed at 700 nm, where the oxidized dye strongly absorbs (see Fig. S31 in the ESI† for the spectrum), following excitation at 510 nm. The lifetime for both decays are significantly longer than typical S1–S0 relaxation of a porphyrin43 hence we conclude that the observed processes originate from the decay of the cation following charge transfer from the dye to the oxide. TAS does not provide accurate information about the efficiency of electron transfer however, so it is not possible to correlate the result directly with the IPCE spectrum.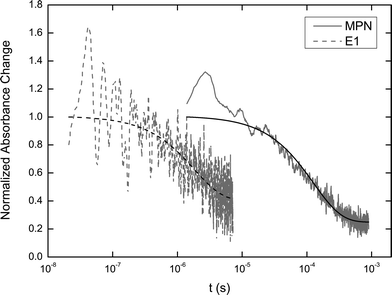 | ||
| Fig. 7 Normalized transient absorbance decay of the P3 cation on TiO2 probed at 700 nm in MPN or in electrolyte E1, following excitation at 510 nm. Data are shown in grey and exponential fits are in black. | ||
When the P3-dyed TiO2 film is contained in an environment of MPN, i.e. when the only pathway to recover the ground state after cation formation is recombination with the conduction band electrons in the oxide, the lifetime for the decay (derived from fitting a monoexponential decay) is 126 μs. In contrast, when electrolyte is present the oxidized dye is “intercepted” by the iodide and therefore the lifetime of the cation, 1.8 μs, is almost two orders of magnitude shorter. The same trend was observed for both P1 and P2 as shown in the ESI (Fig. S32).† The dye regeneration yields44 are calculated to be 99.7%, 99.2%, and 98.6% for P1, P2 and P3 respectively. These regeneration yields suggest that interception of the cation by electrons in the oxide conduction band rarely occurs because the dye is regenerated by the electrolyte on a much shorter timescale at low charge density. We therefore conclude that electron injection into the oxide from P3 is the limiting step for efficient photocurrent generation under these conditions (we note that at higher light intensities near open-circuit, where charge densities in the metal oxide are higher, the rate of cation interception increases, lowering the regeneration efficiency therefore potentially resulting in a loss in fill factor45,46). This supports the device data and dye characterization, suggesting that the energetic driving force and orbital localization require further optimization.
Conclusion
Three new porphyrin-based dyes were synthesized and characterized as sensitizers in DSSCs, using both TiO2 and SnO2 as the as the electron-transport material. Control of the dye properties by molecular tailoring was manifest in the DSSC performance. Moving from an unsubstituted porphyrin to an anthracene-substituted porphyrin resulted in enhanced photocurrent through enhanced light absorption in the Q-band. Fusing the anthracene to the porphyrin core narrowed the HOMO–LUMO gap enabling a photon-to-current conversion onset of ∼1100 nm, when using SnO2 or lithium-doped TiO2 as the metal oxide. This is the longest wavelength photocurrent generation reported for a porphyrin dye. However, narrowing the gap was found to reduce the favorability for photoinduced electron transfer into the metal oxide conduction band by lowering the energetic driving force and shifting the location of the LUMO away from the anchor to the metal oxide surface. This may be a generic problem for porphyrin-based panchromatic sensitizers with enlarged chromophores. However, based on the observations in the present work this may be addressed through (i) anchoring the dye through carboxyl groups directly attached to the chromophore to enhance coupling of the molecular orbitals with the oxide conduction band and (ii) further controlling the molecular orbital energies to achieve a more ideal absorption onset of around 940 nm through linking alternative appropriate substituents.Acknowledgements
For financial support JMB and HJS acknowledge the European Commission, under the SANS project, grant agreement number 246124; NKSD, JDW, HLA, HJS and JT acknowledge the Engineering and Physical Sciences Research Council; NKSD, JDW and HLA acknowledge the Defence Science and Technology Laboratory; and JK is a member of the Oxford Centre of Collaborative Applied Mathematics (OCCAM) where his work is supported by award number KUK-C1-013-04, made by King Abdullah University of Science and Technology.References
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Grätzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624–3629 CrossRef CAS.
- Y. Tachibana, S. A. Haque, I. P. Mercer, J. R. Durrant and D. R. Klug, J. Phys. Chem. B, 2000, 104, 1198–1205 CrossRef CAS.
- T. Ma, K. Inoue, K. Yao, H. Noma, T. Shuji, E. Abe, J. Yu, X. Wang and B. Zhang, J. Electroanal. Chem., 2002, 537, 31–38 CrossRef CAS.
- F. Odobel, E. Blart, M. Lagree, M. Villieras, H. Boujtita, N. El Murr, S. Caramori and C. A. Bignozzi, J. Mater. Chem., 2003, 13, 502–510 RSC.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514–6517 CrossRef CAS.
- S. Eu, S. Hayashi, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 4396–4405 CAS.
- Y. Liu, N. Xiang, X. Feng, P. Shen, W. Zhou, C. Weng, B. Zhao and S. Tan, Chem. Commun., 2009, 2499–2501 RSC.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CAS.
- M. V. Martinez-Diaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS.
- I. Radivojevic, A. Varotto, C. Farley and C. M. Drain, Energy Environ. Sci., 2010, 3, 1897–1909 CAS.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS.
- C.-L. Wang, Y.-C. Chang, C.-M. Lan, C.-F. Lo, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2011, 4, 1788–1795 CAS.
- S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846–847 CrossRef CAS.
- C. Jiao, N. Zu, K.-W. Huang, P. Wang and J. Wu, Org. Lett., 2011, 13, 3652–3655 CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- H. J. Snaith, Adv. Funct. Mater., 2010, 20, 13–19 CrossRef CAS.
- N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945–3947 CrossRef CAS.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124–2127 CrossRef CAS.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2010, 133, 30–31 CrossRef.
- K. A. Nguyen, P. N. Day and R. Pachter, J. Chem. Phys., 1999, 110, 9135–9144 CrossRef CAS.
- K. A. Nguyen and R. Pachter, J. Chem. Phys., 2001, 114, 10757–10767 CrossRef CAS.
- K. A. Nguyen, P. N. Day, R. Pachter, S. Tretiak, V. Chernyak and S. Mukamel, J. Phys. Chem. A, 2002, 106, 10285–10293 CrossRef CAS.
- P. Docampo and H. J. Snaith, Nanotechnology, 2011, 22, 225403 CrossRef.
- S. Ito, P. Chen, P. Comte, M. K. Nazeeruddin, P. Liska, P. Péchy and M. Grätzel, Progr. Photovolt.: Res. Appl., 2007, 15, 603–612 CrossRef CAS.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fré, P. Rubino, C. Choné, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036–3037 RSC.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–815 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS.
- S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3480–3487 CrossRef CAS.
- J. B. Asbury, E. Hao, Y. Wang and T. Lian, J. Phys. Chem. B, 2000, 104, 11957–11964 CrossRef CAS.
- F. Lenzmann, J. Krueger, S. Burnside, K. Brooks, M. Grätzel, D. Gal, S. Rühle and D. Cahen, J. Phys. Chem. B, 2001, 105, 6347–6352 CrossRef CAS.
- J. B. Asbury, N. A. Anderson, E. Hao, X. Ai and T. Lian, J. Phys. Chem. B, 2003, 107, 7376–7386 CrossRef CAS.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- Z. Zhang, S. Ito, J.-E. Moser, S. M. Zakeeruddin and M. Grätzel, ChemPhysChem, 2009, 10, 1834–1838 CrossRef CAS.
- K. Zhu, S.-R. Jang and A. J. Frank, J. Phys. Chem. Lett., 2011, 2, 1070–1076 CrossRef CAS.
- J. R. Jennings, Y. Liu and Q. Wang, J. Phys. Chem. C, 2011, 115, 15109–15120 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20952g |
| This journal is © The Royal Society of Chemistry 2012 |