Electron detachment dynamics of Cu−(H2O)n (n = 1–3): a direct ab initio MD study†
Hiroto
Tachikawa
*
Division of Materials Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan
First published on 18th October 2012
Abstract
Direct ab initio molecular dynamics (MD) has been applied to the electron detachment dynamics of Cu−(H2O)n (n = 1–3). The initial structures of Cu(H2O)n were generated randomly around the equilibrium point of anionic complex Cu−(H2O)n and then trajectories were run from the vertical electron detachment points of Cu−(H2O)n. It was found that three reaction channels compete with each other when n = 1: dissociation channel (the product is Cu + H2O) and complex formation channel (the product is neutral CuH2O complex). The complex channel is further classified to two kind of complexes: a strongly bound (I) complex and a weakly bound (II) complex. The dissociation channel occurred from an inner Franck–Condon (FC) region where the distance Cu−–H2O is shorter than the equilibrium point. On the other hand, the complex formation channel occurred from the wide FC region. In case of n = 2 and 3, dissociation channels were the main products. The mechanism of the electron detachment dynamics was discussed on the basis of the theoretical results.
1. Introduction
Determining the influence of solvation on reaction dynamics is one of the scientifically challenging problems in the field of chemical physics. There is particular interest in contrasting differences in the behavior and reactivity of ions in the gaseous phase compared to the condensed phase. The studying of clusters offers the opportunity to explore the change caused by solvation.1–8 In particular, microsolvation of a metal ion by water molecules is clearly a key issue to determine the initial solvation process of metal ions from the metal surface in an aqueous environment.Lineberger and co-workers investigated that microsolvation dynamics of metal–water systems of Cu−(H2O)n (n = 1 and 2) using femtosecond photodetachment-photoionization experiments.9–11 Electron photodetachment of Cu−(H2O) produces neutral Cu(H2O) initially with the anion geometry and with an internal energy that is near the Cu–H2O dissociation energy. Immediately after the birth of neutral Cu(H2O), the water molecule will begin to rotate and vibrate with respect to Cu, and a fraction of the complexes will dissociate.
From a theoretical point of view, they calculated the structures of Cu(H2O) and Cu−(H2O) systems using high-level ab initio and wave-packet dynamics calculations and showed that Cu−(H2O) is planar with a hydrogen atom oriented toward the copper anion. The neutral complex Cu(H2O) has an oxygen atom oriented toward the copper atom and a shorter Cu–OH2 separation compared to the anion.9 The calculated probability amplitude was in good agreement with the experiment. Thus, the decay process of Cu–H2O following the electron detachment of Cu−(H2O) and static electronic properties of Cu–H2O system are well understood by their experiment and theoretical calculations. However, the time profile of molecular geometry was not clearly understood.
In the present study, the direct ab initio molecular dynamics (MD) method is applied to the electron detachment dynamics of Cu−(H2O)n (n = 1–3) to elucidate the dynamics features of the Cu–H2O system. In particular, the dominant factor of the reaction channels was determined in the dynamical features. Also, the structural change of Cu–H2O is visualized using the direct ab initio MD results. In previous papers,12–15 we investigated the electron detachment and attachment dynamics of Na−(H2O), (H2O)n and CF2Cl2 using the direct ab initio MD method. The mechanism and time scale of the reaction were determined. In this work, a similar technique was applied to the Cu−(H2O)n system.
2. Computational methods
The structures of anionic and neutral complexes, Cu−(H2O)n and Cu(H2O)n, were fully optimized at the MP2/6-311++G(2d,2p) level of theory. The electron detachment dynamics of Cu−(H2O)n (n = 1 and 2) were calculated by means of direct ab initio MD method at the MP2/6-311++G(2d,2p) level. The initial geometries were randomly generated around the equilibrium point of Cu−(H2O)n, and then the geometries having the energy lower than 1.0 kcal mol−1 relative to the equilibrium point were selected. The trajectories for neutral system Cu(H2O)n were run from these selected points on the assumption of vertical electron detachment. The electronic state of the system was monitored during the simulation. We confirmed carefully that the electronic state is kept during the reaction. For Cu(H2O)n (n = 3), direct ab initio MD calculation was carried out at the MP2/6-311++G(d,p) level.The equations of motion were numerically solved by the velocity Verlet algorithm method. No symmetry restriction was applied to the calculation of the energy gradients. The time step size was chosen as 0.10 fs, and a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 or 20000 steps were calculated for each dynamics calculation. The drift of the total energy is confirmed to be less than 10−3% throughout at all steps in the trajectory. The momentum of the center of mass and the angular momentum were assumed to zero. Details of the direct MD calculations are also described elsewhere.12–15
000 or 20000 steps were calculated for each dynamics calculation. The drift of the total energy is confirmed to be less than 10−3% throughout at all steps in the trajectory. The momentum of the center of mass and the angular momentum were assumed to zero. Details of the direct MD calculations are also described elsewhere.12–15
Static ab initio and DFT calculations were carried out using Gaussian 03.16 To confirm the stability of the complexes at all stationary points, harmonic vibrational frequencies were calculated at the MP2/6-311++G(2d,2p) level of theory. All vibrational frequencies obtained were positive, indicating that all stationary points were located at local minima on the potential energy surface. The relative energies were also calculated at the MP4SDQ, CCSD, and QCISD levels of theory for comparison. The excited states of the neutral states were 2.0–3.0 eV higher than the anionic state (TD-DFT calculation). Therefore, we considered only the ground state potential energy surface of the Cu(H2O) systems. The excitation energies of Cu(H2O) after the electron detachment of anion system were calculated by means of symmetry adapted cluster/configuration interaction (SAC-CI) method with a 6-311++G(d,p) basis set.
3. Results
A. Structures of Cu−(H2O) and CuH2O
The structures of anionic and neutral complexes, Cu−(H2O) and CuH2O, were optimized at the MP2/6-311++G(2d,2p), MP4SDQ/6-311++G(2d,2p), CCSD/6-311++G(2d,2p), QCISD/6-311++G(2d,2p) and QCISD/6-311++G(3df,3pd) levels of theory. The geometry optimizations were started from several initial configurations of H2O around Cu− (or Cu atom). All levels of theory gave the similar geometrical parameters (all geometrical parameters are given in Table S1, ESI†). The optimized structures obtained at the MP2/6-311++G(2d,2p) level are given in Fig. 1.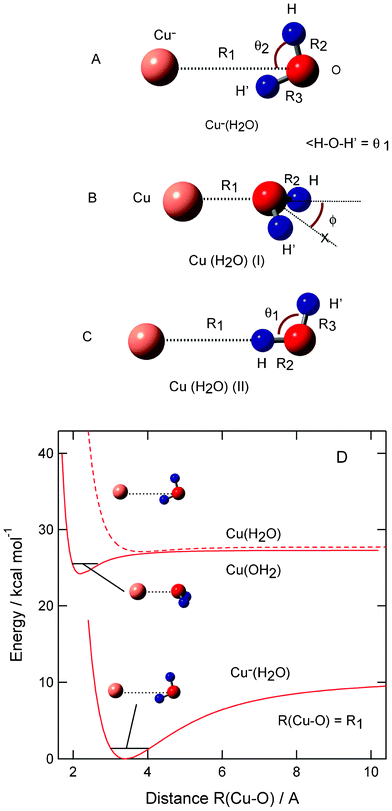 | ||
| Fig. 1 Optimized structures of (A) anion complex Cu−(H2O), (B) neutral complex CuH2O (I) and (C) CuH2O (II) calculated at the MP2/6-311++G(2d,2p) level. (D) Potential energy curves for Cu–H2O systems plotted as a function of R(Cu–O) distance. | ||
In the case of Cu−(H2O), one of the hydrogen atoms of H2O orients to the copper anion with a hydrogen bond of H2O. The structure is planar with a Cs symmetry. The Cu–O distance and angles (θ1 and θ2) in Cu−(H2O) are calculated to be R1 = R(Cu–O) = 3.508 Å, θ1 = 100.1°, and θ2 = 74.4°, respectively, at the QCISD/6-311++G(3df,3pd) level. This geometry is slightly distorted from a C2v dipole orientation structure. This feature is similar to those of halogen ion–water systems, (X−·H2O; X = F, Cl, and Br).17
In the case of neutral system Cu(OH2), two complexes were found: a strongly bound complex (I) and a weak bound complex (II). In complex (I), the oxygen atom of H2O binds directly to the copper atom. The structure of complex (I) is non-planar with a bending angle of H2O. The Cu–OH2 distances in complexes (I) and (II) are calculated to be R1 = R(Cu–O) = 2.158 and 3.908 Å, respectively. It should be noted that MP2 calculation gives reasonable geometrical parameters for the Cu–H2O system. These results are also in reasonable agreement with previous theoretical studies.9,10
Harmonic vibrational frequencies of Cu−(H2O) and CuH2O (I and II) are calculated at the MP2/6-311++G(2d,2p) level. All frequencies are given in Table S2, ESI†. All frequencies are positive, indicating that the complexes are located on the local minima of the ground state potential energy surface. The frequencies of Cu−(H2O) are calculated to be 3874 cm−1 (O–H asym.), 3601 cm−1 (O–H sym.), 1670 cm−1 (H–O–H scission), and 538 cm−1 (out of plane H–O–H bending). The neutral complex (I) showed 3917 cm−1 (O–H asym.), 3794 cm−1 (O–H sym.), 1649 cm−1 (H–O–H scission), and 392 cm−1 (out of plane H–O–H bending). The symmetric and asymmetric O–H stretching modes of H2O are red-shifted by the interaction with both Cu− and Cu. The intermolecular stretching modes for Cu−(H2O) and CuH2O (I) and (II) are calculated to be 169, 337, and 61 cm−1, respectively, indicating that the curvature of intermolecular potential energy curve of complex (I) is larger than those of anion complex and complex (II). These features are in good accord with those in previous works.9–11
The binding energies of H2O to Cu and Cu− are given in Table 1, and potential energy curves for Cu–H2O systems are plotted in Fig. 1. For example, the binding energy (Eb) for Cu− + H2O is defined by
| −Eb = E(Cu−(H2O)) − [E(Cu−) + E(H2O)] | (1) |
| Cu−(H2O) | Cu(H2O) (I) | Cu(H2O) (II) | |
|---|---|---|---|
| MP2/6-311++G(2d,2p) | 10.3 | 3.0 | 0.6 |
| MP2+ΔZPE(MP2) | 9.7 | 2.0 | 0.4 |
| QCISD/6-311++G(3df,3pd) | 8.8 | 5.0 | 0.6 |
| QCISD+ΔZPE(MP2) | 8.2 | 4.0 | 0.4 |
| Taylor et al. (ref. 10) | 10.6 | 5.1 | 1.2 |
After inclusion of zero point energies (ZPEs), the binding energies of H2O are slightly changed to be 9.7 kcal mol−1 in Cu−(H2O), 2.0 kcal mol−1 in complex (I), and 0.4 kcal mol−1 in complex (II). These energies are in reasonable agreement with the values of QCISD/6-311++G(3df,3pd): namely, 8.2 kcal mol−1 in Cu−(H2O), 4.0 kcal mol−1 in Cu(H2O) (I), and 0.4 kcal mol−1 in Cu(H2O) (II).
B. Electron detachment dynamics of Cu−(H2O)
The structure of the anion complex Cu−(H2O) fluctuates around the equilibrium point due to thermal activation. The MP2 calculation of Cu−(H2O) indicates that the bending angle of H2O in Cu−(H2O) vibrates in the range of ϕ = 0 ± 20° at the vibrational ground state (v = 0). Also, the intermolecular distance R(Cu–O) is distributed in the range 3.2–3.6 Å. Therefore, several initial points should be examined in the trajectory calculations. To elucidate the effects of the initial geometrical configuration on the reaction dynamics, 30 geometries were randomly selected and then the trajectories were run from the selected points, R(Cu–O) = 3.2–3.6 Å and ϕ = 0 ± 20°. The calculations showed that three reaction channels, one dissociation and two complex formation channels, were obtained as product channels.In a direct dissociation channel, the water molecule is dissociated from Cu(H2O) after the electron detachment of Cu−(H2O). The complex channels can be further classified as strong and weak complex formations. In the strong complex formation, complex (I), expressed as Cu(OH2) (I), is formed as a product. The other complex formation channel is a weakly bound complex formation (complex II). The reaction scheme is expressed by:
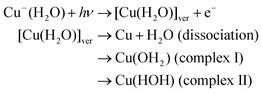
In the next section, the reaction dynamics for the dissociation channel is discussed.
C. Dissociation channel
The result for the dissociation channel is given in Fig. 2. Potential energy of Cu(H2O) formed from the vertical electron detachment of Cu−(H2O) decreases suddenly due to the structural change (points a → b). The potential energy reaches a minimum at time = 152 fs (−1.2 kcal mol−1 at point b). This minimum corresponding to complex II. The Cu–O distance of Cu(H2O) is gradually elongated from 3.200 Å (0.0 fs) to 3.978 Å (152 fs). After the minimum, the energy increases again and saturated to a limiting value (−0.75 kcal at point d). The vibrational structure appearing in the potential energy curve is caused by the H–O–H bending mode and O–H stretching modes. Time profile of R1 = R(Cu–O) indicates strongly that Cu and H2O separate smoothly from each other. The amplitude of the H–O–H bending mode (points c and d) is estimated to be 10°, which corresponds to a slight vibrational excited state of H2O.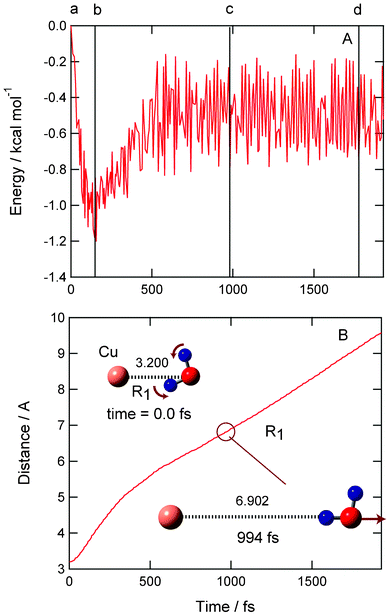 | ||
| Fig. 2 Time propagations of (A) potential energy of the system and (B) Cu–O distance (R1) after the electron detachment of Cu−(H2O) in the equilibrium point. Snapshots are illustrated as insert figures. Dissociation channel was obtained as product channel. No symmetry restriction was applied to the calculation. The distances are in Å. | ||
Snapshots of Cu(H2O) are illustrated in Fig. 2 as insert figures (detailed snapshots at all points are given in the ESI†). At time zero (point a), the Cu–O bond distance is R(Cu–O) = 3.200 Å (point a). This distance approximately corresponds to an inner edge of the Franck–Condon (FC) region of Cu−(H2O). After 152 fs, the Cu–OH2 bond is elongated to 3.978 Å. The potential energy reaches a minimum at this point (point b). The H2O molecule further leaves from Cu and the distance is 6.902 Å at 994 fs (point c). Finally, both H2O and Cu are fully separated from each other. The distance is 9.189 Å at 1790 fs (point d). In the case of this trajectory, the rotational excitation of H2O was not found. The relative translational energy between H2O and Cu is calculated to be 0.1 kcal mol−1, which is about 30% of the available energy for the reaction. Thus, the electron detachment of Cu−(H2O) in the inner edge of FC gives a dissociation channel.
D. Complex formation channel (I)
A sample trajectory for the complex formation channel (I) is given in Fig. 3. After the electron detachment of Cu−(H2O), the potential energy decreases slightly and is first minimized at point b (−0.6 kcal mol−1 at time = 125 fs). The energy increases slightly and decreases again around point c (606 fs). At 756 fs, the energy reaches the lowest energy point (point d). The energy at the lowest point is −2.7 kcal mol−1 relative to that of the starting point. After that, the energy increases suddenly at 780 fs due to the collision between Cu and H2O. At 1063 fs, the potential energy shows −0.8 kcal mol−1 (point e), and complex (I) is formed again at 1400 fs.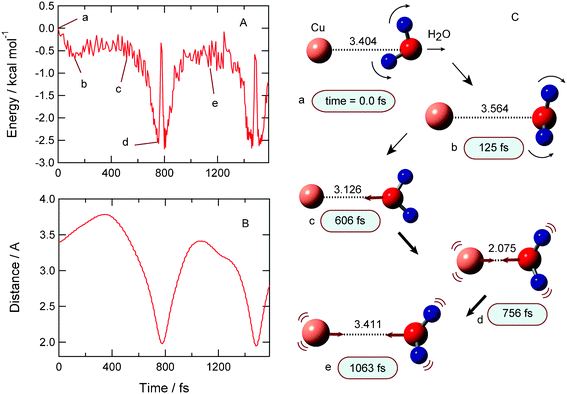 | ||
| Fig. 3 Result of complex formation channel (I). (A) Time propagations of potential energy of the system, (B) Cu–O distance (R1) after the electron detachment of Cu−(H2O), and (C) snapshots. | ||
Time evolution of the distance between Cu and H2O (R1) is plotted in Fig. 3B. The distance is 3.404 Å at the starting point (time zero). Once the distance shows a maximum at 380 fs with R1 = 3.75 Å, and it is shortened gradually and reaches a shortest distance at 756 fs with R1 = 2.075 Å. This result indicates that the water molecule leaves Cu after the electron detachment, but the water molecule returns and collides with the Cu atom. The collision occurs at 790 fs with R1 = 1.98 Å. After that, the water molecule leaves from Cu, but the dissociation does not occur. Instead, the large amplitude stretching vibration between H2O and Cu occurs continuously within complex (I).
Snapshots of Cu(H2O) following the electron detachment Cu−(H2O) are illustrated in Fig. 3 (right). At time zero, the Cu–O bond is R(Cu–O) = 3.404 Å (point a) and the dipole moment is oriented toward the Cu atom. This structure is close to that of the equilibrium point. In case of this sample trajectory, the initial bending angle is ϕ = 175°, meaning that the structure is a non-planar at time zero. After the electron detachment, the water molecule rotates a large amount. This structural change causes a decrease of potential energy. When the oxygen of H2O orients toward the Cu atom, the potential energy reaches a minimum point. On the other hand, the energy increases if the dipole moment of H2O orients to the Cu atom. At points c and d, the water closely approaches the Cu atom. After that, both rotation and stretching modes of H2O occur around the Cu atom. Thus, it is clearly shown that the complex Cu(H2O) has large amplitude motions after the electron detachment of Cu−(H2O). This dynamical feature is common in all trajectories in the complex formation channel (I).
E. Complex formation channel (II)
The result for the complex formation channel (II) is given in Fig. 4. After the electron detachment of Cu−(H2O), the potential energy decreases slightly and is minimized at point b (−0.65 kcal mol−1 at time = 209 fs). The energy increases and decreases again around point d (1139 fs). One of the hydrogen atoms of H2O is oriented to the Cu atom. The large amplitude complex was formed as shown in snapshots. The complex (II) is an intermediate to the dissociation product. If the excess energy is enough for dissociation and is concentrated into the Cu–O intermolecular mode, this complex can be easily dissociated to Cu + H2O.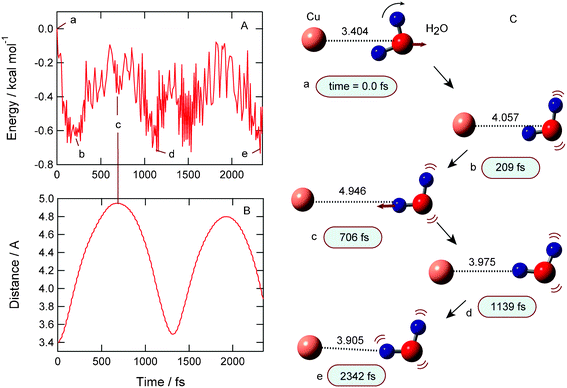 | ||
| Fig. 4 Result of complex formation channel (II). (A) Time propagations of potential energy of the system, (B) Cu–O distance (R1) after the electron detachment of Cu−(H2O), and (C) snapshots. | ||
F. The origin of the reaction channels
As shown in previous sections, the three reaction channels are competitive as product states. In this section, the dominant factor on the reaction channels is considered on the basis of the calculated results. The relation between initial bending angle (ϕ) and product channel is analyzed in all trajectories. The angle ϕ is defined as shown in Fig. 5, i.e. a measure of the deviation from planarity of Cu−(H2O). From this analysis, it was found that the initial bending angle affects the product channel. If the complex Cu−(H2O) has a near planar structure (namely, ϕ ≃ 180.0°), the trajectory calculations give the dissociation channel or complex (II). On the other hand, the trajectories with smaller bending angles (ϕ < ca. 177°) give complex channel (I). This feature is clearly shown in all trajectories.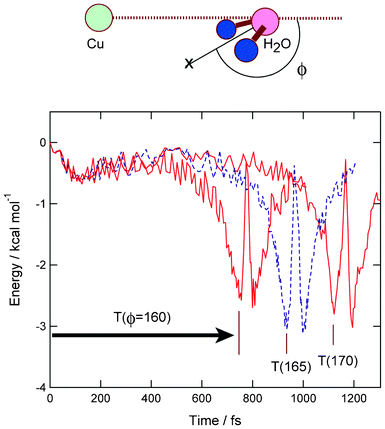 | ||
| Fig. 5 Potential energies for the complex formation channel (I). Three typical cases (ϕ = 160, 165, and 170°) are plotted as a function of time. T(ϕ) means elapsed time required for the complex formation from time zero. | ||
Next, the elapsed time for the complex formation is analyzed in the trajectory leading to the complex channel (I). The results of trajectory calculations for three typical cases are given in Fig. 5. The initial angles are chosen as ϕ = 160, 165 and 170° for example. The elapsed time, expressed by T(ϕ), is defined as a time from the starting point (time zero) to minimum energy point. As clearly seen in Fig. 5, T(ϕ) is strongly dependent on the initial angle α: namely, T(170) = 1125 fs, T(165) = 936 fs and T(160) = 756 fs. This result strongly suggests that the increase of bending angle enhances the complex formation. Thus, it is found that the initial configuration of Cu−(H2O) strongly affects the reaction channels.
G. Time evolution of excitation energies
The present calculations showed that the three reaction channels compete after the electron detachment of the anion system. Transient absorption spectra of Cu(H2O) for complex (I) formation channel are given in Fig. 6. The first band with strong intensity corresponds to the 2S → 2P electronic transition of the Cu atom in CuH2O complex. The directions of spectrum shift are slightly different from each other. In the case of isolated Cu atom, the electronic transition for the 2S → 2P band is observed at 3.81 eV. The present calculation gives 4.02 eV, which is in reasonable agreement with the experiment. Fig. 6 shows that the peak appears at Eex = 4.03 eV at time zero. This band is gradually red-shifted as a function of time. At final stage, the peak is appeared at Eex = 3.10 eV (756 fs), which corresponds to the absorption spectrum of Cu(OH2) (I) complex. Thus, the 2S → 2P band is largely red-shifted in complex formation channel (I). On the other hand, the spectrum shifts were negligibly small in both the dissociation channel and complex formation channel (II).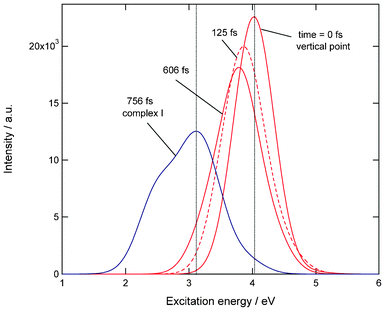 | ||
| Fig. 6 Transient absorption spectra of Cu(H2O) (complex formation channel (I)). Excitation energies and intensities were calculated at the TDDFT-B3LYP/6-311++G(d,p) level. | ||
Fig. 7 shows absorption spectra of Cu atom, Cu(H2O) complex (I), and complex (II). Also, the absorption spectrum of Cu(H2O) at the vertical electron detachment point is given. The peak of complex (II) is close to that of free Cu atom. Also, the vertical point has the peak at a similar position. The peak of complex (I) is significantly red-shifted from that of the Cu atom. This result indicates that the complex formation channel (I) is only observed as a large spectrum shift.
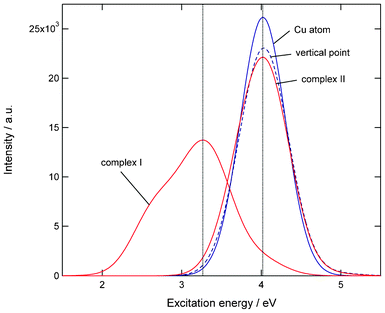 | ||
| Fig. 7 Absorption spectra of Cu atom, Cu(H2O) (complex (I)), Cu(H2O) (complex (II)), and Cu(H2O) at teh vertical excitation point. Excitation energies and intensities were calculated at the TDDFT-B3LYP/6-311++G(d,p) level. | ||
Thus, the present study implies that measurement of spectrum shift gives an assignment of reaction channels. Muntean et al. observed the spectrum shift by the hydration: Cu(2S → 2P transition) is remarkably shifted by only one water molecule. This result is in accord with previous experiments.9–11
H. Potential energy surfaces of Cu–H2O system and reaction model
As shown in section 3F, it is found that the bending angle of H2O (α and ϕ) relative to Cu strongly dominates the reaction channels. Two dimensional potential energy surfaces (PESs) plotted as functions of R1 and α are given in Fig. 8. Fig. 8A and B show PESs for Cu−(H2O) and CuH2O systems, respectively. On the PES of Cu−(H2O), the energy minimum dotted corresponds to the equilibrium point of Cu−(H2O). The shaded region around the minimum point indicates the FC region projected onto the 2D PES at the vibrational ground state of Cu−(H2O). These two modes are flexible and have a wide FC region. Therefore, several geometrical configurations are involved in the electron detachment dynamics.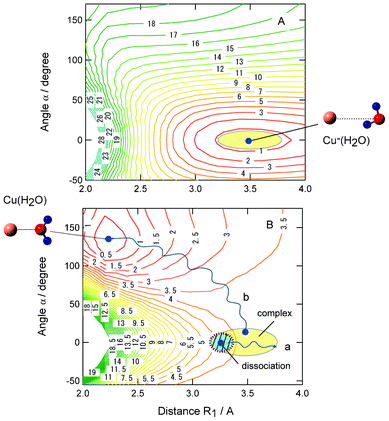 | ||
| Fig. 8 Potential energy surfaces for (A) anionic complex Cu−(H2O) and (B) neutral complex Cu(H2O) plotted as functions of Cu–O distance (R1) and bending angle α. The values are calculated at the MP2/6-311++G(d,p) level. Shaded region schematically indicates Franck–Condon region of Cu−(H2O). Paths a and b schematically indicate reaction paths for dissociation and complex formation channels, respectively. The values are drawn in kcal mol−1. | ||
The energy minimum corresponding to the equilibrium point of Cu(OH2) is located at R1 = 2.25 Å and α = 130° in Fig. 8B. However, the minimum is very shallow. The shaded region indicates the FC region from the electron detachment of Cu−(H2O). The reaction paths a and b show schematic illustrations of typical trajectories leading to dissociation and complex formation channels, respectively. The trajectory for path a starts from the inner FC region (R < R1) and near planar structure (ϕ ≈ 0°), and path b starts from a point with a large bending angle (α). The path a leads to a dissociation channel, whereas the path b reaches the Cu(OH2) complex (I) after the electron detachment of Cu−(H2O). Thus, the structural configuration before electron detachment correlates strongly with product reaction channels.
I. Electron detachment dynamics of Cu−(H2O)2
In this section, the direct ab initio MD method is extended to Cu−(H2O)n (n = 2). In the optimized structure of Cu−(H2O)2, two protons of water dimer orient to the Cu− ion with bond distances of 3.29 and 3.68 Å. The bond distance of the hydrogen bond in the water dimer is 2.05 Å as an O–OH distance.Rathbone et al. suggested from the photoelectron image and spectrum of Cu−(H2O)2 that two water molecules make a hydrogen bond around Cu−, and water molecules are not separated each other. The present calculation strongly supports their experiment.11
In the direct ab initio MD calculation, a total of 20 trajectories were run around the equilibrium structure of Cu(H2O)2. Four channels were found as product channels (i.e., dissociation, partial dissociation, and complex channels I and II). The reaction channels are summarized in Table 2. The main product is a direct dissociation channel: [Cu(H2O)2]ver → Cu + (H2O)2. The next channel is a partial dissociation channel where one of the water molecules (W1) is preferentially dissociated: [Cu(H2O)2]ver → Cu(H2O) + H2O. The other channels are minor. The time evolution of potential energy and snapshots of Cu(H2O)2 for the direct dissociation channel are given in Fig. 9. After the detachment, the potential energy largely decreases and is minimized at 50 fs (−4.5 kcal mol−1). The magnitude of energy change with n = 2 is larger than that of n = 1 due to the large solvation energy when n = 2. The water molecule (W1) is rapidly rotated and a proton of W1 orients to the opposite direction from the Cu atom. At 280 fs, the water dimer is located in 4.30 Å (W1) and 3.86 Å (W2), indicating that the water dimer leaves gradually from the Cu atom. At 500 fs, the water dimer is significantly far from the Cu atom (5.04 and 4.47 Å). This result indicates that the water dimer leaves directly from the Cu atom after the electron detachment in n = 2.
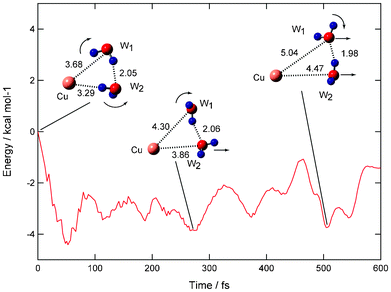 | ||
| Fig. 9 Time evolution of potential energy of the Cu(H2O)n (n = 2) system (direct dissociation channel) and snapshots for electron detachment of Cu−(H2O)2 obtained by direct ab initio MD calculation at the MP2/6-311++G(2d,2p) level. Values indicate bond distances between molecules in Å. Dissociation channel is obtained as product channel. | ||
| Channel | Product | Number of trajectory |
|---|---|---|
| Dissociation | Cu + (H2O)2 | 12 |
| Partial dissociation | Cu(H2O) + H2O | 4 |
| Complex (I) | Cu(H2O)2 | 2 |
| Complex (II) | Cu(HOH)(H2O) | 2 |
The time evolution of potential energy and snapshots of Cu(H2O)2 for the partial dissociation channel are given in Fig. S3, ESI.† In this channel, one of the water molecules (W2) is preferentially dissociated. Atomic forces on the oxygen atoms of W2 and W1 at the vertical electron detachment point [Cu(H2O)2]ver are calculated to be 19.7 and 12.2 mhartree bohr−1, respectively. The atomic force on W2 is significantly larger than that on W1. Therefore, the water molecule (W2) is preferentially dissociated from the [Cu(H2O)2]ver.
At time zero, the water dimer orients to the Cu atom. After the electron detachment, the water molecule leaves rapidly from the Cu atom and the water molecule (W1) is rotated. At 700 fs, the oxygen atom of W1 orients toward the Cu atom and Cu–O strong bond is formed. The potential energy reaches a minimum point corresponding to the Cu–OH2 (W1) complex. The water dimer is still remained at this time. The excess energy is dissipated into the translational mode to W2, so that the water molecule (W2) can leave from the Cu–OH2 system. The final product is [Cu(H2O)2]ver → Cu–OH2 + H2O in the partial dissociation channel.
J. Electron detachment dynamics of Cu−(H2O)3
In the optimized structure of n = 3, the water trimer interacts with the Cu− ion (Fig. 10). Three protons of the water trimer orient to the Cu− ion. The distances of proton–Cu− are ca. 2.60 Å, and three water molecules interact with the Cu− ion.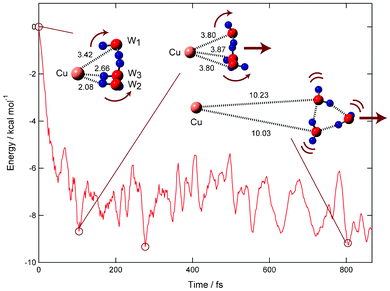 | ||
| Fig. 10 Time evolution of potential energy of the Cu(H2O)n (n = 3) system (direct dissociation channel) and snapshots for electron detachment of Cu−(H2O)2 obtained by direct ab initio MD calculation at the MP2/6-311++G(d,p) level. Values indicate bond distances between molecules in Å. The dissociation channel is obtained as product channel. | ||
At time zero, the protons of water trimer (W1–W3) orient to the Cu atom. After the electron detachment, the protons rotate in the opposite direction to the Cu atom due to a repulsive interaction. The distance of water trimer to the Cu atom is slightly elongated from 3.42 Å (time = 0.0 fs) to 3.80 Å (104 fs). After the rotation of protons, the excess energy is mainly transferred into the translational mode between Cu atom and the water trimer, so that the water trimer leaves rapidly from the Cu atom. The Cu–O distances at 104 and 277 fs are 3.80 and 5.17 Å, respectively. At 806 fs, the water trimer is fully separated from the Cu atom (10.23 Å). Thus, the electron detachment of Cu−(H2O)3 gives a dissociation channel.
4. Discussion
A. Comparison with previous studies
In the present study, the electron detachment dynamics of Cu−(H2O)n has been investigated by means of direct ab initio MD method. The dynamics calculation for n = 1 showed that three reaction channels (dissociation and two complex formation channels) are competitive with each other after the electron detachment of Cu−(H2O). It was also found that the initial geometrical configuration of Cu−(H2O) affects strongly to the reaction channels. In particular, the bending angle of H2O relative to the Cu− ion in Cu−H2O complex strongly couples with the formation of complex channel (complex I). If the electron detachment occurs from near the equilibrium point (Cs and near Cs, and α = 0°), the dissociation channel via region of channel (II) is dominant. The time scale of complex (II) formation is about 100–300 fs. The dissociation channel occurs via the region of complex (II). On the other hand, a deformed structure (α > ca. 3°) causes complex formation channel (I). Time scales for the complex formation are roughly estimated to be 500–1000 fs, and it decreases with increasing bending angle (α). Thus, the present calculation clearly visualizes the electron detachment dynamics of Cu−(H2O) and selectivity of reaction channels.The electron detachment dynamics of Cu−(H2O) was experimentally investigated by Lineberger and co-workers using femtosecond photodetachment-photoionization spectroscopy.9–11 They observed both complex and dissociation channels after the electron detachment of Cu−(H2O). The present calculation strongly supports the experiments.9–11
For n = 2, Taylor et al. investigated electron photodetachment dynamics of Cu−(H2O)2.18 It was found that the parent molecule from the initial ion Cu−(H2O)2 is not detected above a back-ground signal. On the other hand, Cu and Cu(H2O) were found as products. The Cu atom is formed from direct dissociation of Cu(H2O)2 and via partial dissociation of Cu(H2O). The partial dissociation occurs as [Cu(H2O)2]ver → Cu–OH2 (complex I) + H2O, and then the complex I dissociates into Cu + H2O. The present calculations found that the partial dissociation takes place after the detachment. This result is in good agreement with the experiments.
For n = 3, unfortunately, there is no experiment. The present calculation predicts that the main reaction channel is direct dissociation to form Cu + water trimer. As a result of the present calculations, it is predicted for n > 3 that the branching ratio of the complex formation channels becomes smaller because the repulsive interaction between Cu and (H2O)n at the vertical point becomes stronger in a larger system. This is due to the fact that the number of protons increases in the larger system.
For n = 6, we calculated the optimized structure of Cu−(H2O)n (n = 6), and the structure is given in the ESI.† In Cu−(H2O)6, two “cyclic water trimer”s interact with Cu− from up and down positions. On the basis of present results, this form will give the product Cu + (water trimer) + (water trimer) after the electron photodetachment from Cu−(H2O)6.
In our previous paper,12 we investigated the reaction dynamics of Na(H2O) following electron detachment from Na−(H2O) in a zero-point vibrational state. The photodetachment-photoionization spectra using 4.3–5.3 eV photons were theoretically predicted by the calculation. In the previous work, we considered only one solvent and calculated photodetachment photoionization spectra of Na(H2O). On the other hand, in the present study, the reaction mechanism of Cu(H2O)n (n = 1–3) following the electron detachment was discussed in detail. The purpose of this study is much different from that of previous work.
The electronic non-adiabatic effect including spin–orbit coupling is frequently important in heavy atom system.19,20 In the present study, we did not consider electronic non-adiabatic effects in the dynamics calculation. The Cu atom has a low-lying electronic excited state (2D) near ground state (2S). Therefore, more accurate dynamics including non-adiabatic effects and spin–orbit coupling may be needed to obtain a more detailed dynamic feature.
B. Temperature effect on the reaction channels
Muntean et al. reported temperature effects on the reaction channels in the electron detachment dynamics of Cu−(H2O).9 A large temperature effect on the product channel was found. They showed that increase of temperature enhances the complex formation. The present calculation strongly supports their observation. Here, we propose a model of the temperature effect on the product channels. At low temperature, the initial complex Cu−(H2O) vibrates slightly around the equilibrium point. The deformation from the planar structure is significantly small. The electron detachment from the structure leads to the dissociation channel. On the other hand, the structure of Cu−(H2O) is deformed largely from the planar structure at high temperature. The bending mode (deformation from the planar structure) is excited due to the low frequency mode. This excitation enhances the complex (I) formation in the electron detachment of Cu−(H2O). This is an origin of enhancement of complex (I) and temperature dependence. Thus, the present calculations qualitatively support their observation.Acknowledgements
The author acknowledges a partial support from a Grant-in-Aid from JSPS (Project No. 2455000102).References
- O. H. Kwon, T. H. Yoo, C. M. Othona, J. A. Van Deventer, D. A. Tirrell and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17101–17106 CrossRef CAS.
- H. Ishikawa, T. Nakano, T. Eguchi, T. Shibukawa and K. Fuke, Chem. Phys. Lett., 2011, 514, 234–238 CrossRef CAS.
- D. Pentlehner, R. Riechers, A. Vdovin, G. M. Potzl and A. Slenczka, J. Phys. Chem. A, 2011, 115, 7034–7043 CrossRef CAS.
- L. Sheps, E. M. Miller, S. Horvath, M. A. Thompson, R. Parson, A. B. McCoy and W.C. J. Lineberger, Chem. Phys., 2011, 134, 184311 Search PubMed.
- G. Matisz, A. M. Kelterer, W. M. F. Fabian and S. Kunsagi-Mate, J. Phys. Chem. A, 2011, 115, 10556–10564 CrossRef CAS.
- K. Pathak-Arup, ChemPhysChem, 2011, 12, 2641–2645 CrossRef CAS.
- H. J. Ireta, J. Chem. Theory Comput., 2011, 7, 2630–2637 CrossRef.
- F. M. V. Leavens, C. D. M. Churchill, S. Y. Wang and S. D. Wetmore, J. Phys. Chem. B, 2011, 115, 10990–11003 CrossRef CAS.
- F. Muntean, M. S. Taylor, A. B. McCoy and W. C. Lineberger, J. Chem. Phys., 2004, 121, 5676–5687 CrossRef CAS.
- M. S. Taylor, F. Muntean, W. C. Lineberger and A. B. McCoy, J. Chem. Phys., 2004, 121, 5688–5699 CrossRef CAS.
- G. J. Rathbone, T. Sanford, D. Andrews and W. C. Lineberger, Chem. Phys. Lett., 2005, 401, 570–574 CrossRef CAS.
- S. K. Kondo, K. Hashimoto and H. Tachikawa, Chem. Phys. Lett., 2006, 431, 45 CrossRef CAS.
- H. Tachikawa and A. J. Orr-Ewing, J. Phys. Chem. A, 2008, 112, 11575–11581 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2011, 115, 9091–9096 CrossRef CAS.
- H. Tachikawa, Phys. Chem. Chem. Phys., 2011, 13, 11206–11212 RSC.
- Gaussian 03, Revision B.04,M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong and C. GonzalezJ. A. PopleGaussian, Inc., Pittsburgh PA, 2003.
- D. Y. Wu, S. Duan, X. M. Liu, Y. C. Xu, Y. X. Jiang, B. Ren, X. Xu, S. H. Lin and Z. Q. Tian, J. Phys. Chem. A, 2008, 112, 1313–1321 CrossRef CAS.
- M. S. Taylor, J. Barbera, C. P. Schulz, F. Muntean, A. B. McCoy and W. C. Lineberger, J. Chem. Phys., 2005, 122, 054310 CrossRef.
- S. Mahapatra, Acc. Chem. Res., 2009, 42, 1004–1015 CrossRef CAS.
- S. Mahapatra, Int. Rev. Phys. Chem., 2004, 23, 483 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20907a |
| This journal is © The Royal Society of Chemistry 2012 |