What is your actual catalyst? TMS cleavage rates of diarylprolinol silyl ethers studied by in situ NMR†
Michael H.
Haindl
,
Markus B.
Schmid
,
Kirsten
Zeitler
* and
Ruth M.
Gschwind
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstraße 31, D-93053, Regensburg, Germany. E-mail: ruth.gschwind@chemie.uni-regensburg.de; kirsten.zeitler@chemie.uni-regensburg.de; Fax: +49 941 943 4617
First published on 12th June 2012
Abstract
Diarylprolinol silyl ethers are excellent and broadly applicable organocatalysts for various enamine and iminium-type synthetic transformations. However, their undesired degradation to the corresponding diarylprolinols and the subsequent formation of oxazolidines during reaction with aldehydes may significantly affect their catalytic performance. Therefore, in situ NMR was used to examine the TMS cleavage rate of diarylprolinol silyl ethers as a function of solvent properties, acidic/basic additives and the presence of water. Highly polar solvents with strong hydrogen bond acceptor properties and especially moderate acidic additives with pKa (DMSO) values around 10 accelerate the deprotection significantly, whereas basic and highly acidic additives are not detrimental. Additional mechanistic studies reveal that the substitution reaction takes places at the silicon atom.
Jørgensen–Hayashi-type diarylprolinol silyl ethers have demonstrated an excellent performance in a variety of asymmetric organocatalytic transformations.1–7 Even multi-component domino reactions have been developed allowing for one pot syntheses of complex organic molecules with many stereocenters.8–12 Recently, different additives have been reported to be highly beneficial in organocatalysis.13–17 For diarylprolinol silyl ethers, especially, benzoic acid has successfully been used to increase both yields and the catalytic activity.13–15 However, a critical shortcoming of diarylprolinol silyl ethers is the potential loss of the TMS group leading to the formation of diarylprolinol catalysts. Although both catalyst classes, diarylprolinol silyl ethers and their deprotected analogues, share excellent stereocontrolling properties leading to high ee values, diarylprolinols are known to be less active.18–21 Recently, we have been able to monitor for the first time the deprotection of a diarylprolinol silyl ether by in situ NMR.20 Further extensive spectroscopic studies of the enamine intermediates revealed different preferred conformations of the diarylprolinol silyl ether enamines and diarylprolinol enamines.22 In addition, the lower reactivity of diarylprolinols could be correlated to the hardly reversible formation of oxazolidines with carbonyl species and strongly reduced amounts of enamine intermediates being present during the reaction with aldehydes.20 Since typical reaction times in diarylprolinol silyl ether catalysis vary from hours to days, the stability of the TMS protecting group under particular reaction conditions (solvents and additives) is of utmost importance. Herein we present a systematic study on the cleavage conditions for the TMS protecting group of diarylprolinol silyl ethers 1 and 3 (Fig. 1) in solution by means of modern in situ NMR experiments.
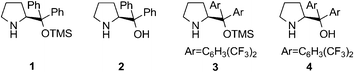 | ||
| Fig. 1 The diarylprolinol silyl ethers 1 and 3 used as model systems and their desilylated analogues 2 and 4. | ||
The NMR spectroscopic analysis of the mixtures of 1 and its desilylated analogue 2 as well as of 3 and its free-OH analogue 4 in solution reveals different chemical shifts for the pyrrolidine protons of 1 and 2, and of 3 and 4, respectively (for the 1H NMR chemical shift assignments see the ESI†). Based on this the identification of the silylated and desilylated species can be accomplished either via their different diffusion coefficients in DOSY spectra or via NOE contacts between the pyrrolidine protons and the TMS protons or the OH proton (the latter being detectable in DMSO, data not shown). In this study, the 1H NMR signals originating from the α-protons of species 1–4, which are clearly separated from other 1H NMR resonances (Fig. 2A), are used as a probe to monitor the kinetics of the TMS cleavage reaction.
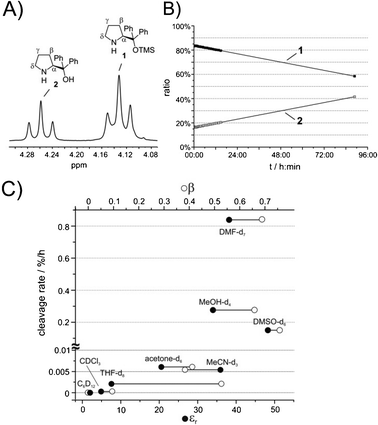 | ||
| Fig. 2 (A) 1H NMR resonances of the α protons of 1 and 2, respectively, in DMSO-d6 at room temperature at 600 MHz. (B) Reaction profile of 1 and 2 during the TMS cleavage reaction in MeOH-d4 at room temperature. (C) Experimental cleavage rate (c.r.) of 1 in different solvents correlated with the dielectric constants εr (•) and hydrogen bond acceptor properties β (○) (Kamlet–Taft parameter) of these solvents. | ||
Here, our first surprising observation was that the commercially purchased catalysts 1 and 3 contained at least 10–15% of their deprotected analogues 2 and 4 (Fig. 1). Since typically the activity of diarylprolinol catalysts is low in comparison with diarylprolinol silyl ethers6 and stereocontrol might follow different modes with possible opposite stereoinduction, the observed purity grade of the purchased catalysts may not satisfy the needs of synthetic organic chemists. A simple 1H NMR experiment and integration of the well separated α-proton resonances of 1 and 2 (Fig. 2A) and of 3 and 4, respectively, may be used to decide if purification of the catalyst is necessary prior to its application (for chemical shift assignments of 1 and 2 in various solvents and full proton spectra see the ESI†).
In order to examine the influence of solvent properties (i.e. polarity, as expressed by the dielectric constant εr27 and hydrogen bond acceptor properties as expressed by the Kamlet–Taft parameter β27) on the TMS cleavage of 1, we performed a solvent screening at room temperature using cyclohexane-d12, chloroform-d1, THF-d8, acetone-d6, MeOH-d4, acetonitrile-d3, DMF-d7 and DMSO-d6. For example, the concentration–time curves of 1 and 2 in MeOH-d4 are presented in Fig. 2B. The negative slope of the deprotection of 1 represents the cleavage rate per hour (c.r.) in Fig. 2C. The fastest cleavage rates were observed in the polar solvents DMF-d7 (c.r. = 0.84% h−1), MeOH-d4 (c.r. = 0.27% h−1) and DMSO-d6 (c.r. = 0.15% h−1). Virtually no cleavage was monitored in the non-polar solvents cyclohexane-d12 (c.r. < 0.0001% h−1) and chloroform-d1 (c.r. = 0.0003% h−1) and only very slow deprotection was observed in acetone-d6 (c.r. = 0.006% h−1) and acetonitrile-d3 (c.r. = 0.005% h−1). Considering that the typical reaction times of 1 in DMF at room temperature range from hours to a few days28–31 the deprotected analogue 2 should be present in high amounts and should compete with 1 as the active organocatalyst. In fact, this could explain some of the reported drawbacks of such solvent–catalyst combinations.28–31 Interestingly, the cleavage rate of 1 only correlates roughly with the solvent parameters εr and β (Fig. 2C). However, clearly a combination of high polarity and strong hydrogen bond acceptor properties (DMF-d7, MeOH-d4 and DMSO-d6) leads to a relevant cleavage rate with an exceptionally high rate in DMF. Solvents lacking one of these properties (THF-d8: large β, small εr; MeCN-d3: small β, large εr) show significantly slower cleavage tendencies.
We continued our study with an investigation of the influence of various acidic and basic additives on the cleavage reaction and selected DMSO-d6 as the model solvent for that purpose (Fig. 3A). Benzoic acid as a typical additive for diarylprolinol silyl ethers was tested with all the solvents used in the additive free study (see above). To cover a broad pKa range, acids with pKa values in DMSO varying from 0 to 11.1 were selected (Fig. 3A). Along with the acidic additives picric acid (I), HCl aq. (II), TFA (trifluoroacetic acid) (III), tetrazole (IV), succinic acid (V), p-nitrophenol (VI) and benzoic acid (VII) we also applied the basic additives DABCO (1,4-diazabicyclo[2.2.2]octane), DMAP (4-dimethylaminopyridine) and DBU (1,8-diazabicycloundec-7-ene).
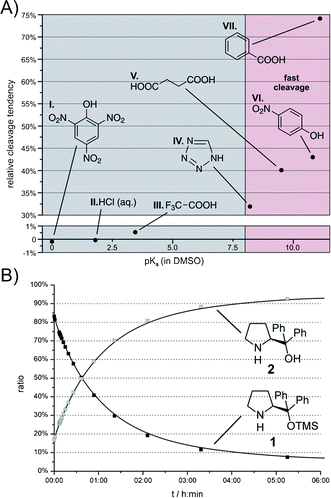 | ||
| Fig. 3 (A) Relative cleavage tendency (the amount of the TMS group cleaved after 5 h minus the corresponding amount cleaved in pure DMSO-d6) of 1 with acidic additives (100 mol%, 50 mM) in DMSO-d6 at room temperature as a function of their pKa(DMSO) values.23–26 (B) Concentration–time curve of 1 and 2 in DMSO-d6 with PhCOOH as additive at room temperature. | ||
As expected, the basic additives such as DBU pKaH (acetonitrile) = 23.9,33 DMAP pKaH (acetonitrile) = 18.1834 or DABCO pKaH (DMSO) = 8.9323 only show a minor effect. Most strikingly, all the tested acidic additives with pKa values above 7.5 strongly accelerated the cleavage reaction (Fig. 3A). The fastest deprotection was observed with benzoic acid. In this case, the amount of 1 decreased exponentially from about 84% at the beginning to less than 10% after 6 h (Fig. 3B). Under these “fastest cleavage conditions”, the progress of deprotection can easily be detected by the crystallisation of the benzoate salt of 2 after a few hours.32 Surprisingly, Enders et al.9 described an enantioselective Michael addition–α-alkylation cascade reaction in which a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of catalyst 1 and benzoic acid in DMSO was successfully applied during reaction times from one to four days. However, our experimental findings suggest that after a few hours 1 should not be the predominant catalytic species in the described reaction any longer. Interestingly, strong acids such as picric acid pKa(DMSO) = 0,24 hydrochloric acid pKa(DMSO) = 1.824 and TFA pKa(DMSO) = 3.4524 also have only a negligible effect on the desilylation rate (Fig. 3A).
:1 mixture of catalyst 1 and benzoic acid in DMSO was successfully applied during reaction times from one to four days. However, our experimental findings suggest that after a few hours 1 should not be the predominant catalytic species in the described reaction any longer. Interestingly, strong acids such as picric acid pKa(DMSO) = 0,24 hydrochloric acid pKa(DMSO) = 1.824 and TFA pKa(DMSO) = 3.4524 also have only a negligible effect on the desilylation rate (Fig. 3A).
Since benzoic acid shows the highest acceleration rate and is broadly applied as an additive in combination with diarylprolinol silyl ethers we examined the deprotection rates with this additive also in other solvents (cyclohexane, chloroform, THF (tetrahydrofuran), acetone, MeOH, acetonitrile, DMF (dimethyl formamide) and DMSO (dimethyl sulfoxide)). A strong acceleration effect of benzoic acid was only observed in DMSO and DMF.
We then investigated the cleavage mechanism of the TMS deprotection in an attempt to better understand the difference between benzoic acid and strong acids. Apart from the expected hydrolysis product TMS–OH 5 and its condensation product TMS–O–TMS 6 (Fig. 4) we observed the formation of silyl benzoate 7 after an induction period as a result of the nucleophilic attack of PhCOO− at the Si atom. This product could be fully suppressed if additional water (up to 16 vol%) was used. Unlike using dry conditions, the TMS cleavage is decelerated (stopped at ca. 35%) and only silyl ester 7 is formed (for the reaction profile and the chemical shift assignments, see the ESI†). Further experiments using methanol as the competing nucleophile and the resulting exclusive formation of methoxytrimethylsilane during the TMS cleavage also revealed that the nucleophilic attack occurred at the electrophilic silicon atom.
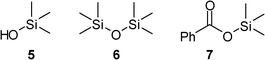 | ||
| Fig. 4 The detected silyl products 5, 6 and 7 during the TMS cleavage of silylether 1 in the presence of benzoic acid. | ||
Unlike weak acids (pKa(DMSO) > 7.5, see Fig. 3A), which tend to greatly accelerate the TMS deprotection in DMSO with increasing pKa, strong acids (pKa(DMSO) < 5, see Fig. 3A: TFA, HCl etc.), featuring a full protonation of the catalyst's pyrrolidine nitrogen as detected by NMR (for spectra see the ESI†),35 are ineffective for the TMS cleavage. For the ammonium species of 2 two separated NH signals (δΔ ≥ 0.38 ppm) were detected indicating an intramolecular hydrogen bond between one NH proton and the ether oxygen atom;36 for the ammonium form of 1 only one broad signal could be observed. Hence shifting the equilibrium from 50%:50%37 amine R2NH vs. protonated analogue R2NH2+ to ≈ 100% of the corresponding ammonium species R2NH2+ of 1 (respectively 3) with decreasing pKa mirrors the diminishing cleavage tendency. Cleavage “protection” by full protonation may arise from both a steric and an electrostatic shielding of the silicon atom against nucleophilic attack by the closely attached counterion of the acid.
Our extensive NMR spectroscopic studies on the TMS cleavage rates of diarylprolinol silyl ethers in various solvents and with different additives reveal that the experimental conditions for this important and widely applied catalyst have to be chosen with care as significant catalyst degradation might occur during the reaction. In polar solvents with strong H-bond acceptor properties cleavage rates up to 0.84% h−1 are observed and weak acidic additives (pKa(DMSO) ≈ 10) even result in more than 90% of the desilylated catalyst within 5 h. In contrast, non-polar solvents and basic or strongly acidic additives as well as benzoic acid in non-polar solvents are not detrimental. We expect our findings to aid chemists working with diarylprolinol silyl ethers in choosing optimal reaction conditions and designing new reactions.
Acknowledgements
This work was supported by the DFG (SPP 1179).References
- M. Marigo, D. Fielenbach, A. Braunton, A. Kjaersgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703–3706 CrossRef CAS.
- M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS.
- M. Marigo, J. Franzén, T. B. Poulsen, W. Zhuang and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 6964–6965 CrossRef CAS.
- A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922–948 CrossRef CAS.
- C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876–7880 CrossRef CAS.
- L. W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243–279 CrossRef CAS.
- D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS.
- D. Enders, C. Wang and J. W. Bats, Angew. Chem., Int. Ed., 2008, 47, 7539–7542 CrossRef CAS.
- M. Marigo, S. Bertelsen, A. Landa and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 5475–5479 CrossRef CAS.
- M. Marigo, T. Schulte, J. Franzén and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 15710–15711 CrossRef CAS.
- W. Wang, H. Li, J. Wang and L. Zu, J. Am. Chem. Soc., 2006, 128, 10354–10355 CrossRef CAS.
- J.-L. Li, T.-R. Kang, S.-L. Zhou, R. Li, L. Wu and Y.-C. Chen, Angew. Chem., Int. Ed., 2010, 49, 6418–6420 CrossRef CAS.
- S. Brandau, E. Maerten and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 14986–14991 CrossRef CAS.
- S. Cabrera, J. Alemán, P. Bolze, S. Bertelsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2008, 47, 121–125 CrossRef CAS.
- J. E. Hein, J. Burés, Y.-h. Lam, M. Hughes, K. N. Houk, A. Armstrong and D. G. Blackmond, Org. Lett., 2011, 13, 5644–5647 CrossRef CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem.–Eur. J., 2012, 18, 3362–3370 Search PubMed.
- E. Alza and M. A. Pericàs, Adv. Synth. Catal., 2009, 351, 3051–3056 CrossRef CAS.
- U. Grošelj, D. Seebach, M. Badine, B. Schweizer, A. K. Beck, I. Krossing, P. Klose, Y. Hayashi and T. Uchimaru, Helv. Chim. Acta, 2009, 92, 1225–1259 CrossRef CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, J. Am. Chem. Soc., 2011, 133, 7065–7074 CrossRef CAS.
- M. C. Varela, S. M. Dixon, K. S. Lam and N. E. Schore, Tetrahedron, 2008, 64, 10087–10090 CrossRef CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem. Sci., 2011, 2, 1793 RSC.
- R. L. Benoit, D. Lefebvre and M. Fréchette, Can. J. Chem., 1987, 65, 996 CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- I. M. Kolthoff and M. K. Chantooni, J. Am. Chem. Soc., 1976, 98, 5063–5068 Search PubMed.
- I. M. Kolthoff, M. K. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23–28 CrossRef CAS.
- P. D. D. Adams and S. Tavener, Chemistry in Alternative Reaction Media, Wiley & Sons, Ltd., 2004 Search PubMed.
- P. García-García, A. Ladépêche, R. Halder and B. List, Angew. Chem., Int. Ed., 2008, 47, 4719–4721 CrossRef CAS.
- J. Franzén and A. Fisher, Angew. Chem., Int. Ed., 2009, 48, 787–791 CrossRef CAS.
- T. Govender, L. Hojabri, F. M. Moghaddam and P. I. Arvidsson, Tetrahedron: Asymmetry, 2006, 17, 1763–1767 CrossRef CAS.
- I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sundén, P. Dziedzic and A. Córdova, Chem.–Eur. J., 2008, 14, 7867–7879 CrossRef CAS.
- For the crystal structure see the ESI.† CCDC 869742 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
- K. T. Leffek, P. Pruszynski and K. Thanapaalasingham, Can. J. Chem., 1989, 67, 590–595 CAS.
- D. Augustin-Nowacka, M. Makowski and L. Chmurzynski, Anal. Chim. Acta, 2000, 418, 233–240 CrossRef CAS.
- The corresponding trifluoroacetic acid and perchlorate salts of 3 have been isolated by Hayashi et al.: Y. Hayashi, S. Samanta, H. Gotoh and H. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 6634–6637 Search PubMed.
- In experiments with diphenylprolinol methyl ether a similar signal pattern (δΔ = 2.94 ppm) was detected in DMSO-d6 with 100 mol% TFA where NOESY data show that the proton in proximity to the methoxy group has the higher chemical shift (downfield).
- As an estimation for the pKa values of the ammonium species R2NH2+ the pKa value of pyrrolidinium pKa(DMSO) = 11.1 was used: M. R. Crampton and I. A. Robotham, J. Chem. Res. (S), 1997, 22–23 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: spectra and tables. CCDC reference number 869742. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20860a |
| This journal is © The Royal Society of Chemistry 2012 |