DOI:
10.1039/C2RA20825C
(Paper)
RSC Adv., 2012,
2, 8110-8115
Ultralong monoclinic ZnV2O6 nanowires: their shape-controlled synthesis, new growth mechanism, and highly reversible lithium storage in lithium-ion batteries†
Received
1st May 2012
, Accepted 19th June 2012
First published on 21st June 2012
Abstract
In this paper, we report on the shape-controlled synthesis of monoclinic (m-) ZnV2O6 micro/nanostructures through a simple hydrothermal approach and their highly reversible lithium storage for anode materials in lithium-ion batteries. m-ZnV2O6 structures with different diameters were selectively explored by changing the critical experimental parameters of dwell time and reaction temperatures. A novel “dissolution recrystalizaion–Ostwald ripening–splitting” combination mechanism for uniform nanowires is proposed by further monitoring the time-dependent evolution of morphologies and phases. Furthermore, these m-ZnV2O6 nanowires with high aspect ratio exhibit a better reversible capacity and a much excellent cyclic retention than that of as-obtained mesostructures and bulk counterparts because of better contact behavior and a shorter diffusion length for Li+, implying a promising candidate for the application in high-energy batteries.
I. Introduction
One dimensional (1D) electrode materials applied in lithium-ion batteries have triggered widespread attention in recent years because these novel structures can satisfy the demand for high-energy density performances.1–3 Metal vanadium oxides nanomaterials,4–8 including lithium vanadates and transition metal (Zn, Ag, Cu, Fe, Co, Ni, Mn, Y, In, etc.) vanadium oxides, have fascinating properties in electrochemical energy storage and conversion for reversible Li insertion/extraction.4,9–34 For zinc vanadium oxide, Zn0.84V4O10 with a microscale square plate shape also has promising applications for lithium ion batteries due to their active electrochemical properties and good reversibility for their crystal structure,34 although increased polarization appears during the smooth potential-composition behavior.5–7 Recently, zinc vanadium oxide hydroxide hydrate (Zn3V2O7(OH)2(H2O)2 and Zn3(OH)2V2O7·nH2O) 2D and 3D structures have been fabricated.11,13–15 Nevertheless, few studies in the literature on the morphology and electrochemical performance of zinc vanadium oxide 1D nanomaterials have been reported. The fundamental scientific issues are how to control the complex reaction process containing the vanadium element, and modify the preferential growth along certain crystal direction to enhance the properties of reversible lithium storage. To solve these central problems, the hydrothermal method is a good choice due to the following advantages: Firstly, it provides thermal/kinetic behavior to make it possible to synthesize functional vanadium based materials. Secondly, homogeneous nucleation can be successfully achieved to monitor the shape of the final products.
In this paper, we focus on the facile and highly efficient hydrothermal technology for the preparation of m-ZnV2O6 nanowires of a highly crystalline with a diameter of approximately 70–120 nm and a length of about 80–100 μm. The close relationship between crucial synthesizing conditions and prevailing microstructures of samples is systematically ascertained. A novel growth mechanism of one-dimensional nanomaterials is proposed by continuously changing the time-dependent evolution of phases and topologies. In addition, the electrochemical measurements of as-prepared m-ZnV2O6 nanowires have a highly reversible capacity and excellent high-rate stability for the lithium insertion/extraction, which make it a potential anode material in lithium ion battery.
II. Experimental section
A. Sample preparation
All chemical reagents of zinc nitrate (Zn(NO3)2·6H2O), and ammonium metavanadate (NH4VO3) were analytical grade and used as purchased without further purification from No.3 Factory of Tianjin Chemical Regent Company (China). Distilled water was applied in the whole process.
Zinc vanadium oxide materials were successfully fabricated through the hydrothermal method. Important parameters, including the reaction time and temperature, were altered in a series of parallel experiments. In a typical preparation, the synthesizing process can be described as follows: Firstly, 0.3635 g of Zn(NO3)2·6H2O crystals were dispensed into 5.0 mL of distilled water. NH4VO3 powders (0.0024 mole) were transferred into 18.5 mL of hot distilled water (75 °C) and then mixed for 15 min. Secondly, the NH4VO3 solution was added dropwise to the Zn(NO3)2 solution with continuous stirring for 5 min. Finally, the above mixture was sealed in 30 mL Teflon-lined stainless steel autoclaves, and a constant temperature of 100–200 °C was maintained for 3–168 h. After the dwell time, the autoclaves were allowed to naturally cool to room temperature. A fluffy wool-like product emerged near the inner wall of the Teflon vessel was collected, centrifuged, washed with water and absolute ethanol several times to remove the byproduct, and then dried at 80 °C for 12 h for further analysis.35,36 For comparison, microscale bulk counterparts with layered structures were fabricated through the solid-state reaction method via calcined ZnO and V2O5 powders at 600 °C for 12 h in air atmosphere (Fig. S1 in the ESI†). The overall experiments and corresponding morphologies were summarized in Table S1. The chemical reactions during the process are listed in equations as follows:
| | 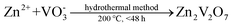 | (1) |
| | 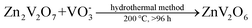 | (2) |
B. Characterization
The powder X-ray diffraction (XRD) pattern was performed on a Rigaku D/Max2500PC and MiniFlex II (Japan) diffractometer with Cu-Kα radiation (λ = 1.54056 Å). Field-emission scanning electron microscope (FE-SEM) images were determined by a Hitachi S-4800 using accelerating voltages of 10 kV to measure the size and morphology of as-synthesized samples. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were measured on a Philips Tecnai G2 F20 transmission electron microscope with a 200 kV acceleration voltage. The Brunauer–Emmett–Teller (BET) specific surface areas of typical products were preformed on a Quantachrome Instruments Nova 4200 e apparatus.
C. Electrochemical measurement
Electrochemical analyses were carried out using a two-electrode system containing a working electrode, a porous polypropylene separator and a high-pure lithium counter electrode in CR2032 button cells. The working electrode was prepared by the mixture of zinc vanadium oxide materials![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbon black:polyvinylidene fluoride (PVDF) in a weight ratio of 45:50:5. The electrolyte solution was 1M LiPF6 in a 50:50 vol.% mixture of ethylene carbonate (EC) and diethylene carbonate (DEC). The button cells were sealed at a pressure of 100 kg cm−2 for 15 s in an argon-filled glovebox (Mikrouna China Universal 2240/750). The electrochemical performances of electrochemical impedance spectroscopy (EIS) were measured on a Parstat 2273 potentiostat/galvanostat analyzer (Princeton Applied Research & AMETEK Company).17 Discharge-charge characteristics at different current densities were tested galvanostatically between 0.025 and 3.0 V (vs. Li+/Li) using an Neware BTS-5V10mA Battery Test System (Newware Technology Co., Ltd., China) at 25 °C.
:carbon black:polyvinylidene fluoride (PVDF) in a weight ratio of 45:50:5. The electrolyte solution was 1M LiPF6 in a 50:50 vol.% mixture of ethylene carbonate (EC) and diethylene carbonate (DEC). The button cells were sealed at a pressure of 100 kg cm−2 for 15 s in an argon-filled glovebox (Mikrouna China Universal 2240/750). The electrochemical performances of electrochemical impedance spectroscopy (EIS) were measured on a Parstat 2273 potentiostat/galvanostat analyzer (Princeton Applied Research & AMETEK Company).17 Discharge-charge characteristics at different current densities were tested galvanostatically between 0.025 and 3.0 V (vs. Li+/Li) using an Neware BTS-5V10mA Battery Test System (Newware Technology Co., Ltd., China) at 25 °C.
III. Results and discussion
A. Characterization of the as-obtained m-ZnV2O6
Fig. 1 characterizes the microstructure of m-ZnV2O6 nanowires for sample 1 in detail by the XRD, SEM, TEM and HRTEM images. Fig. 1a shows the typical XRD pattern of zinc vanadium oxide synthesized via the facile hydrothermal method with the concentration of 0.25 mol L−1 Zn(NO3)2 at 200 °C for 168 h. The experimental detail is summarized in Table S1 in the ESI.† From the image, all the diffraction peaks are assigned to a monoclinic symmetry with a space group of C2 (No. 5), and lattice constants of a = 9.242 Å, b = 3.526 Å, c = 6.574 Å, and β = 111.55°. The result can be easily indexed to the standard values of m-ZnV2O6 (ICDD-JCPDS card No. 74-1262). No peaks from impurities are detected, confirming the high purity of the product. The shape of m-ZnV2O6 nanomaterial is further examined by SEM with different magnifications as shown in Fig. 1b–d. From Fig. 1b, wire-like ZnV2O6 structures with 80–100 μm in length are distributed homogeneously over a wide area. Further magnifications in Fig. 1c,d elucidate that these nanowires have very smooth surface and a diameter in the range of approximately 70–120 nm. Until now, there has been little research concerning the shape-controlled fabrication of zinc vanadium oxide with nanoscale structures. In this regard, it is necessary to emphasize the characterization of the representative m-ZnV2O6 nanomaterials, and subsequently analyze the kinetic and dynamic effects at a series of experimental parameters, such as reaction time and the heating temperatures. In addition, a large quantity of ultralong nanowires with uniform diameter is also observed in the TEM image of Fig. 1e. Careful observation from HRTEM reveals that the m-ZnV2O6 nanowire recorded near the upper part (Fig. 1f) confirms apparent lattice fringes, suggesting the good single-crystalline nature of the wire. Interestingly, clear fringes with an interplanar distance calculated from the line profile (Fig. S-2 in the ESI†) are 0.326 nm, which is in good agreement with the (110) crystal planes. Thus, the wire-like products grow preferentially along the [100] direction as marked by a white arrow in Fig. 1e. In the crystal diagram of m-ZnV2O6 (Fig. 1g, and Fig. S-3 in the ESI†), the double layered structure presents stacks of chains of corner-sharing [VO4] tetrahedra along the b-axis. While, the [ZnO6] units occupied by a Zn center through octahedral coordination are also arranged along the b-axis. These [ZnO6] chains are connected by oxygen-sharing with [VO4] tetrahedra double chains along the vertical direction of the b-axis. The alternation of double and single layered structure paves a good way for reversible lithium insertion/extraction, favoring highly efficient energy storage and good capacity retention. In particular, the spacing (0.326 nm) of the neighboring (110) planes in the schematic diagram is equal to the interplanar distance along the b-axis in Fig. 1f. Therefore, the microstructure analysis plays an important role in designing the oriented growth to fabricate ultralong 1D nanoscale morphology.
![Characterization of the as-synthesized products: (a) XRD pattern of sample 1 prepared through hydrothermal method; (b–d) low and high magnification SEM images of ultralong m-ZnV2O6 nanowires; (e) overview TEM and (f) HRTEM images of an individual nanowire (sample 1); (g) schematic diagram of a single nanowires denoted the preferential growth along [100] by the black arrow, which is in good agreement with the experimental results from HRTEM.](/image/article/2012/RA/c2ra20825c/c2ra20825c-f1.gif) |
| | Fig. 1 Characterization of the as-synthesized products: (a) XRD pattern of sample 1 prepared through hydrothermal method; (b–d) low and high magnification SEM images of ultralong m-ZnV2O6 nanowires; (e) overview TEM and (f) HRTEM images of an individual nanowire (sample 1); (g) schematic diagram of a single nanowires denoted the preferential growth along [100] by the black arrow, which is in good agreement with the experimental results from HRTEM. | |
To monitor the formation process of these nanowires, morphological and phase evolution of zinc vanadium oxide as a function of different synthetic periods permits in-depth understanding. Fig. 2 shows the SEM images of shape-changes for the as-synthesized m-ZnV2O6 micro/nanostructure, obtained at the hydrothermal temperature of 200 °C with a Zn(NO3)2 concentration of 0.25 mol L−1 by adjusting dwell time to 3, 12, and 48 h for sample 2, 3 and 4, respectively. Fig. S-4 in the ESI† provides the phase variations for these samples varying with reaction time. In the initial stage, the main phase of the Zn2V2O7 product consists of abundant microflowers assembled by several microsheets with an average diameter of 3 μm (Fig. 2a). The high-magnification SEM image shown in Fig. 2b clearly demonstrates that the ordered microsheets only 30 nm thick assemble into three-dimensional flower shaped superstructures. In addition, the angle between these two groups of crosslinked sheets is around 120°, which is attributed to 2D oriented growth of individual microsheet. After extending the hydrothermal time to 12 h (Fig. 2c,d), the coexistence of ZnV2O6 and Zn2V2O7 microsheets (1–2 μm in size) and nanowires (80 nm in diameter and 5–20 μm in length) began to appear. The formation of radial nanowires on the outer surface of microsheets is achieved, leading to the decreased size of microscale sheets. When the reaction time reaches 48 h (sample 4), two obvious phenomena can be illustrated from Fig. 2e,f: (i) the formation of pure-phase m-ZnV2O6 with a dramatic decrease of Zn2V2O7 (Fig. 2e and Fig. S-4 in the ESI†); and (ii) the fabrication of meso/nanowires with the gradual consumption of microsheets during the transformation period of 12–48 h. As we knew, the dwell time and reaction temperatures are key factors for controlling the shape evolution and crystal phase of the nanostructures. Therefore, reaction temperatures as a sole variable from 160–180 °C were changed in the growth process by fixing other experimental parameters (i.e., dwell time for 168 h, and  of 0.25 mol L−1) as given in the ESI of Fig. S-5.† However, no pure phase was gained (Fig. S-6 in the ESI†). Therefore, the optimum conditions for the highly pure m-ZnV2O6 nanowire are a reaction temperature of 200 °C for 168 h, and a Zn(NO3)2 concentration of 0.25 mol L−1.
of 0.25 mol L−1) as given in the ESI of Fig. S-5.† However, no pure phase was gained (Fig. S-6 in the ESI†). Therefore, the optimum conditions for the highly pure m-ZnV2O6 nanowire are a reaction temperature of 200 °C for 168 h, and a Zn(NO3)2 concentration of 0.25 mol L−1.
 |
| | Fig. 2 Typical SEM images of zinc vanadium oxides fabricated at selected reaction time with a constant heating temperature of 200 °C and Zn(NO3)2 concentration of 0.25 mol L−1: (a,b) 3 h, Zn2V2O7 and ZnV2O6 microflowers for sample 2; (c,d) 12 h, the coexistence of ZnV2O6 and Zn2V2O7 microsheets and nanowires for sample 3; and (e,f) 48 h, m-ZnV2O6 meso/nanowires with diameter of 100–400 nm for sample 4. | |
According to the above corresponding shape-evolution, the present experimental exploration gains strong evidence for the growth process. A novel combination of “dissolution recrystallization” and the “Ostwald ripening–splitting” mechanisms is quite plausible for the rationally controlled synthesis of 1D m-ZnV2O6 nanowires (Fig. 3). Two steps can mainly be observed to design 1D material from 3D structures, which achieves a different formation mechanism of zinc vanadates. In the early stage, 3D superstructure microflowers were assembled and then aggregated by numerous nuclei of vanadium compounds (Fig. 3a,b). With the prolonged time, the microflowers were split into dispersed microsheets with nanoscale thickness. At the same time, the yield of wire-like product was increased with the decrease of the diameter of sheets (Fig. 3b,c). The high-energy sites of nanostructures on the surface of microsheets are helpful for the growth of single crystallized nanowires. This interesting changes can be explained by the kinetically controlled “dissolution recrystallization” mechanism.35–38 Secondly, the observations of ultralong nanowires with uniform morphology (Fig. 3e) suggests that these wires are split from mesowires with larger diameters (Fig. 3d), which corresponds with the well-known “Ostwald ripening–splitting” mechanism17,23,25,39–41 in this step. Hence, it is worth mentioning that a precise growth mechanism addressed in the present work is very different from that in the reported references, and the growth process of the 1D nanowire along the preferential growth direction at a large scale can depend on the dissolution of 3D superstructures.
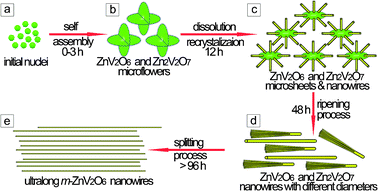 |
| | Fig. 3 Proposed formation process of the zinc vanadium oxide nanostructures: (a) nanoparticles, the mixture of Zn2V2O7 and ZnV2O6; (b) micro/nanoflowers, the coexistence of Zn2V2O7 and ZnV2O6; (c) microsheets and nanowires, the mixture of Zn2V2O7 and ZnV2O6; (d) dominating meso/nanowires, the coexistence of Zn2V2O7 and ZnV2O6; and (e) ultralong nanowires, pure m-ZnV2O6. | |
B. Electrochemical measurements
The electrochemical properties of the representative m-ZnV2O6 nano/mesomaterials and bulk powders electrodes over the potential range between 3.0–0.025 V were systematically measured as shown in Fig. 4 and 5. Fig. 4 discusses the close-relationship between the microstructures and charge–discharge behaviors at a current density of 100 mA g−1 at 25 °C. From the figure, the discharge curves for the initial cycle of these samples identify a pronounced plateau at approximately 1.00–0. 45 V (vs. Li/Li+) and have relatively small specific capacities at a lower voltage of 0.45–0.10 V (vs. Li/Li+) without distinct potential plateau. The appearance of the first plateau at a higher voltage might be assigned to a faradic capacitance of the material.42 Meanwhile, the capacity of a potential window lower than 0.45 V (vs. Li/Li+) is prone to be associated with a gradual lithium storage process into the layered structure for monoclinic-type ZnV2O6. Obviously, the first discharge capacities are 1555.5, 1377.3, and 1299.6 mAh g−1 for m-ZnV2O6 nanowires (sample 1), meso/nanowires (sample 4), and bulk specimen (sample 7), respectively. Obviously, the discharge capacities for the second cycle are 1035.4, 822.8, and 818.2 mAh g−1 for these samples. The reason for the remarkable irreversible capacities between the first two cycles is mainly straightforward and due to the irreversible electrode reaction leading to the phase transformation of active materials.6,7 Furthermore, the reversible capacity of the products after cycling for 10 cycles, indicating an excellent reversible retention capacity, are well maintained 972.9, 807.4, and 647.3 mAh g−1 corresponding to the curves in Fig. 4a–c. Surprisingly, the battery made from ultralong nanowires notably exhibit the highest initial capacity, and the greatest stability during lithium insertion/extraction among the three batteries (Fig. 4). The electrochemical performance at different current densities of these samples was characterized as shown in Fig. 5. The specific capacity decreased with the gradually increasing current densities. The values in the sequence are sample 1 > sample 4 > sample 7, coinciding with the order of the specific surface areas and the particle sizes. It is worth noticing that the ultralong m-ZnV2O6 nanowires with uniform diameter exhibit the highest capacity 688.2 mAh g−1 even at a large current density of 800 mA g−1. After altering discharge current density from 100 to 800 mA g−1 and then returning to 100 mA g−1, only 5.3% was lost for the mean capacity of m-ZnV2O6 nanowires. Meanwhile, primary 1D nanowires (Fig. S-7 in the ESI†) are preserved after disassembling the anode electrode. It proves that no significant morphology change was found during lithium insertion/extraction even at different current densities. The 1D nanostructures in the paper show superior properties than that of bulk, 2D and 3D zinc vanadium-based materials in the published literatures.11,12,14a The improvements of the electrochemical performance are induced by the following factors: (1) a suitable contact behavior among nanowires as active material, carbon and binder in the electrode is provided, decreasing the charge-transfer resistance (8.2 Ω) as analyzed from EIS in Fig. 4d. (2) the ultralong wire-like morphology with a large aspect ratio possess more active sites for the electrochemical reactions on the outer surface of layered structure, and the nanoscale diameter is also beneficial to shorten the diffusion distance and hasten the kinetics for Li+, resulting in the dramatic decrease of electrode polarization. Therefore, ultralong m-ZnV2O6 nanowires are promising anode electrode materials for future lithium ion batteries with fast reaction kinetics, high capacity, and excellent cyclic retention.
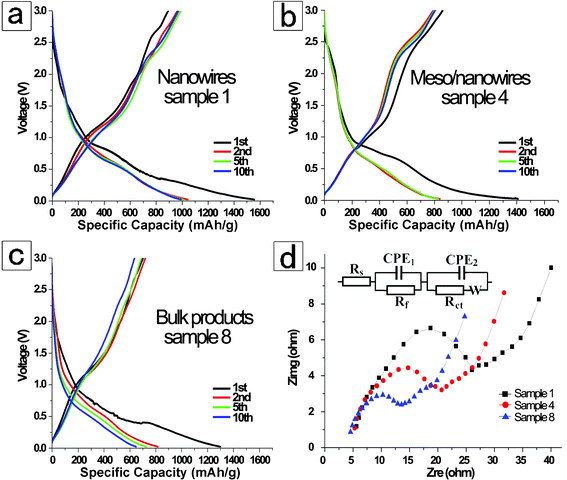 |
| | Fig. 4 Typical charge–discharge performance for as-prepared m-ZnV2O6 materials in coin-type half cells during the 1st, 2nd, 5th, and 10th cycles over a current density of 100 mA g−1 at 25 °C: (a) ultralong nanowires with diameter of 70–120 nm, sample 1; (b) meso/nanowires with 100–400 nm in diameter, sample 4; (c) bulk counterparts via solid-state method, sample 7. (d) EIS plots of the three products over the voltage of 0.8 V (vs. Li/Li+) in the frequency ranging from 100 kHz to 10 mHz. By fitting the circuit model in the inset, the charge-transfer resistance for sample 1, 4, and 8 are 8.2, 73.7, and 99.9 Ω, respectively. | |
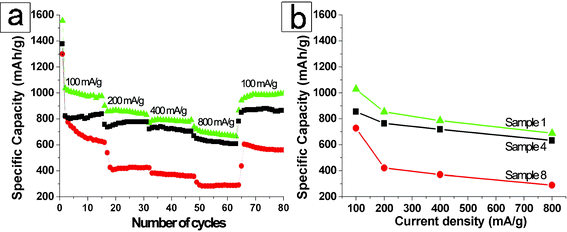 |
| | Fig. 5 (a) discharge capacities of m-ZnV2O6 nanowires for sample 1 (blue triangles), meso/nanowire for sample 4 (black squares), and bulk counterparts for sample 7 (red circles) between 3.0 and 0.025 V vs. Li/Li+ at a constant temperature of 25 °C; (b) cycle life of the electrodes made from these three m-ZnV2O6 product at altering discharge current density from 100 to 800 mA g−1 and then return to 100 mA g−1 at 25 °C. | |
IV. Conclusions
In summary, ultralong m-ZnV2O6 nanowires with an aspect ratio as high as 1100 have been controllably fabricated via a facile hydrothermal technology at a synthesis temperature of 200 °C for 168 h with a Zn(NO3)2 concentration of 0.25 mol L−1. The shape evolution and crystal phases for the micro/nanostructures were monitored by changing the kinetic and dynamic factors, including the heating temperatures, reaction time, and concentrations of precursors. Furthermore, a novel mechanism of “dissolution recrystallization” following the “Ostwald ripening–splitting” process is a quite plausible alternate to facilitate the rationally designed preparation of 1D m-ZnV2O6 wires. In addition, the electrochemical properties of m-ZnV2O6 nanowires exhibit a high initial discharge capacity of 1555.5 mAh g−1, 972.9 mAh g−1 after the 10th cycle at a current density of 100 mA g−1, and excellent cyclic retention, which is due to the decrease of electrode polarization, increase of active sites and reaction kinetics for Li+ insertion/extraction. The present investigation reveals that the ultralong m-ZnV2O6 nanomaterials with uniform morphology are promising anode electrode materials for future lithium ion batteries.
Acknowledgements
This work was supported by the Open Project of Key Laboratory of Advanced Energy Materials Chemistry (Nankai University) (KLAEMC-OP201201), the Science and Research Program of Hebei Province (No.11213939, and No.11276744), the Doctor Foundation of Hebei United University (No. 35393901 and No. 35393801), and the Student Innovational Plans of Hebei United University (X2011086).
References
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. V. Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef.
-
(a) Y. S. He, P. F. Gao, J. Chen, X. W. Yang, X. Z. Liao, J. Yang and Z. F. Ma, RSC Adv., 2011, 1, 958–960 RSC;
(b) Z. Z. Lin, W. B. Yue, D. Z. Huang, J. Y. Hu, X. Y. Zhang, Z. Y. Yuan and X. J. Yang, RSC Adv., 2012, 2, 1794–1797 RSC;
(c) X. L. Zhang, F. Y. Cheng, K. Zhang, Y. L. Liang, S. Q. Yang, J. Liang and J. Chen, RSC Adv., 2012, 2, 5669–5675 RSC.
- R. S. Diggikar, V. M. Dhavale, D. B. Shinde, N. S. Kanbargi, M. V. Kulkarni and B. B. Kale, RSC Adv., 2012, 2, 3231–3233 RSC.
-
(a) W. Xu, X. Bai, S. Xu, Y. S. Zhu, L. Xia and H. W. Song, RSC Adv., 2012, 2, 2047–2054 RSC;
(b) S. W. Cao, J. Fang, M. M. Shahjamali, F. Y. C. Boey, J. Barber, S. C. J. Loo and C. Xue, RSC Adv., 2012, 2, 5513–5515 RSC.
- M. S. Whittingham, Y. Song, S. Lutta, P. Y. Zavalij and N. A. Chernova, J. Mater. Chem., 2005, 15, 3362–3379 RSC.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526–2552 RSC.
- F. Y. Cheng and J. Chen, J. Mater. Chem., 2011, 21, 9841–9848 RSC.
- E. M. Sorensen, H. K. Izumi, J. T. Vaughey, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2005, 127, 6347–6352 CrossRef CAS.
- J. M. Song, Y. Z. Lin, H. B. Yao, F. J. Fan, X. G. Li and S. H. Yu, ACS Nano, 2009, 3, 653–660 CrossRef CAS.
- A. F. Fuentes, L. Treviño, A. Martínez-de la Cruz and L. M. Torres-Martínez, J. Power Sources, 1999, 81–82, 264–267 CrossRef CAS.
- L. F. Xiao, Y. Q. Zhao, J. Yin and L. Z. Zhang, Chem.–Eur. J., 2009, 15, 9442–9450 CrossRef CAS.
- R. Shi, Y. J. Wang, F. Zhou and Y. F. Zhu, J. Mater. Chem., 2011, 21, 6313–6320 RSC.
-
(a) S. B. Ni, G. Zhou, S. M. Lin, X. H. Wang, Q. T. Pan, F. Yang and D. Y. He, Mater. Lett., 2009, 63, 2459–2461 CrossRef CAS;
(b) S. B. Ni, S. M. Lin, Q. T. Pan, K. Huang, F. Yang and D. Y. He, J. Alloys Compd., 2009, 477, L1–L3 CrossRef CAS;
(c) S. B. Ni, X. H. Wang, X. L. Sun, F. Yang, Y. Q. Liu and D. Y. He, Mater. Chem. Phys., 2010, 124, 803–808 CrossRef CAS.
- M. Wang, Y. J. Shi and G. Q. Jiang, Mater. Res. Bull., 2012, 47, 18–23 CrossRef CAS.
- K. J. Takeuchi, A. C. Marschilok, S. M. Davis, R. A. Leising and E. S. Takeuchi, Coord. Chem. Rev., 2001, 219–221, 283–310 CrossRef CAS.
- S. Y. Zhang, W. Y. Li, C. S. Li and J. Chen, J. Phys. Chem. B, 2006, 110, 24855–24863 CrossRef CAS.
- T. A. Albrecht, F. Sauvage, V. Bodenez, J. M. Tarascon and K. R. Poeppelmeier, Chem. Mater., 2009, 21, 3017–3020 CrossRef CAS.
- F. Sauvage, V. Bodenez, J. M. Tarascon and K. R. Poeppelmeier, J. Am. Chem. Soc., 2010, 132, 6778–6782 CrossRef CAS.
- M. Morcrette, P. Rozier, L. Dupont, E. Mugnier, L. Sannier, J. Galy and J. M. Tarascon, Nat. Mater., 2003, 2, 755–761 CrossRef CAS.
- M. Morcrette, P. Martin, P. Rozier, H. Vezin, F. Chevallier, L. Laffont, P. Poizot and J. M. Tarascon, Chem. Mater., 2005, 17, 418–426 CrossRef CAS.
- X. Y. Cao, J. G. Xie, H. Zhan and Y. H. Zhou, Mater. Chem. Phys., 2006, 98, 71–75 CrossRef CAS.
- H. Ma, S. Y. Zhang, W. Q. Ji, Z. L. Tao and J. Chen, J. Am. Chem. Soc., 2008, 130, 5361–5367 CrossRef CAS.
- S. Y. Zhang, L. J. Ci and H. R. Liu, J. Phys. Chem. C, 2009, 113, 8624–8629 CAS.
- C. J. Patridge, C. Jaye, H. S. Zhang, A. C. Marschilok, D. A. Fischer, E. S. Takeuchi and S. Banerjee, Inorg. Chem., 2009, 48, 3145–3152 CrossRef CAS.
- S. Denis, R. Dedryvère, E. Baudrin, S. Laruelle, M. Touboul, J. Olivier-Fourcade, J. C. Jumas and J. M. Tarascon, Chem. Mater., 2000, 12, 3733–3739 CrossRef CAS.
- P. Poizot, E. Baudrin, S. Laruelle, L. Dupont, M. Touboul and J. M. Tarascon, Solid State Ionics, 2000, 138, 31–40 CrossRef CAS.
- M. Hibino, N. Ozawa, T. Murakami, M. Nakamura and T. Yao, Electrochem. Solid-State Lett., 2005, 8, A500–A503 CrossRef CAS.
- S. N. Ichikawa, M. Hibino and T. Yao, J. Electrochem. Soc., 2007, 154, A1156–A1159 CrossRef CAS.
- S. J. Lei, K. B. Tang, Y. Jin and C. H. Chen, Nanotechnology, 2007, 18, 175605 CrossRef.
- T. Morishita, K. Nomura, T. Inamasu and M. Inagaki, Solid State Ionics, 2005, 176, 2235–2241 CrossRef CAS.
- T. Morishita, H. Konno, Y. Izumi and M. Inagaki, Solid State Ionics, 2006, 177, 1347–1353 CrossRef CAS.
- S. L. Chou, J. Z. Wang, J. Z. Sun, D. Wexler, M. Forsyth, H. K. Liu, D. R. MacFarlane and S. X. Dou, Chem. Mater., 2008, 20, 7044–7051 CrossRef CAS.
- F. Zhang, P. Y. Zavalij and M. S. Whittingham, Mater. Res. Bull., 1997, 32, 701–707 CrossRef CAS.
- C. S. Li, S. Y. Zhang, F. Y. Cheng, W. Q. Ji and J. Chen, Nano Res., 2008, 1, 242–248 CrossRef CAS.
- C. S. Li, F. Y. Cheng, W. Q. Ji, Z. L. Tao and J. Chen, Nano Res., 2009, 2, 713–721 CrossRef CAS.
- X. L. Hu, Y. Masuda, T. Ohji and K. Kato, Cryst. Growth Des., 2010, 10, 626–631 CAS.
- G. C. Xi, K. Xiong, Q. B. Zhao, R. Zhang, H. B. Zhang and Y. T. Qian, Cryst. Growth Des., 2006, 6, 577–582 CAS.
- Z. Y. Tang, Y. Wang, S. Shanbhag, M. Giersig and N. A. Kotov, J. Am. Chem. Soc., 2006, 128, 6730–6736 CrossRef CAS.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 1389–1395 CrossRef CAS.
-
W. Ostwaldin Lehrbuch der Allgemeinen Chemie, Leipzig, Germany, 1896, vol. 2, part 1 Search PubMed.
- E. J. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: (1) Table S1: Summary of the experimental parameters and the corresponding structures of zinc vanadium oxide micro/nanomaterials. (2) Fig. S-1: SEM and XRD of typical sample 8 synthesized through solid-state reaction at 600 °C for 12 h. (3) Fig. S-2: Line profile from the HRTEM image. (4) Fig. S-3: Crystallographic view of m-ZnV2O6 along the b-axis. (5) Fig. S-4: XRD of zinc vanadium oxide synthesized at various reaction time. (6) Fig. S-5: SEM images of products obtained at variety of hydrothermal temperatures. (7) Fig. S-6: XRD patterns of the as-synthesized products at different reaction temperatures. (8) Fig. S-7: SEM images of cross-section for the m-ZnV2O6 electrode after 80 charge/discharge cycles. See DOI: 10.1039/c2ra20825c/ |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.