DOI:
10.1039/C2RA20747H
(Paper)
RSC Adv., 2012,
2, 7439-7448
Copolymers from naphtho[2,3-c]thiophene-4,9-dione derivatives and benzodithiophene: synthesis and photovoltaic applications†
Received
22nd April 2012
, Accepted 10th June 2012
First published on 13th June 2012
Introduction
Recently, polymer solar cells (PSCs) have attracted broad attention due to their unique advantages of light weight, flexibility, low cost and roll to roll production. The bulk-heterojunction (BHJ) PSCs are the most widely used device architecture, which can ensure maximum internal donor–acceptor (D–A) interfacial area for efficient charge separation.1 In the past several years, great progress in BHJ PSCs has been witnessed, the power conversion efficiency (PCE) of organic photovoltaic (OPV) devices has been increased up to 6–9%.2–8 The present studies indicate that PSCs have a bright future, but their efficiency and stability still need to be improved compared to their inorganic counterparts towards commercialization. To achieve high efficiency PSCs, the greatest challenge is to develop low band-gap polymers with broad absorption and high hole mobility.9 The D–A approach is the most effective strategy for obtaining low band-gap polymers and modulating their electronic properties. Coplanar geometries and rigid structures can usually lead to high charge mobility.10 However, the performance of these copolymers is still lower than the theoretical PCE value deduced from the model. The deviation indicates that the photovoltaic properties are not only determined by the band-gap and energy level; some other characteristics such as mobility, molecular weight, solubility and morphology etc. are also very important for high efficiency photovoltaics.11 Much attention has been paid to the effects of morphology on the performance of PSCs. Usually, bicontinuous nanoscale morphology can lead to a relatively high short circuit current (Jsc), a better fill factor (FF), and therefore a higher PCE.
Last year, Li and co-workers reported a new strong electron withdrawing unit—naphtho[2,3-c]thiophene-4,9-dione (NTDO). The copolymer with NTDO as the acceptor unit and dithienosilole as the donor unit exhibited a high PCE of 5.21%.12 The NTDO unit possesses a relatively simple, planar structure and has strong electron withdrawing ability, resulting in lower HOMO and LUMO energy levels of the D–A copolymers. Usually a lower HOMO level is beneficial in order to get a higher open circuit voltage (Voc) of the PSCs. Besides, benzodithiophene (BDT) is used as a donor unit, and has demonstrated great potential in efficient polymer photovoltaics, due to its coplanar structure and a more electron-rich building block than such units as fluorene or carbazole, etc.3,5 For instance, Yang, Yu and co-workers have synthesized a family of BDT based copolymers with optimized electronic and optical properties, which led to PCEs as high as 7%.3 Furthermore, side chains also play a very important role in the electronic level, morphology, mobility and therefore PCE.13 However, the polymers using NTDO derivatives with different side chains as the acceptor units have scarcely been explored for photovoltaic applications. Just recently, Li and co-workers reported two copolymers using NTDO derivatives as the acceptor and benzodithiophene as the donor; the polymers exhibited different photovoltaic properties with different side alkyl chains of NTDO derivatives. However, the best PCE only reached up to 1.52% due to a low FF of 0.28–0.30.14 Along these lines, the copolymers from the BDT and NTDO derivatives with different side chains probably lead to some interesting photovoltaic properties.
In this work, a series of new D–A copolymers from a BDT donor unit and NTDO derivative acceptor units with different side chains, PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 (as shown in Scheme 1) were designed and synthesized towards photovoltaic applications. The relationship between the structure and properties has been investigated in detail. The copolymers show good solubility in common organic solvents and a broad absorption from 300–700 nm. The absorption edges of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 are located at 686 nm, 689 nm and 692 nm, respectively, from their absorption spectra. In addition, the blend films suggested a hole mobility of up to 2.04 × 10−5 cm2 V−1 s−1, 2.50 × 10−6 cm2 V−1 s−1 and 3.04 × 10−5 cm2 V−1 s−1, respectively, measured by the SCLC method. The PSCs based on PBDTNTDO-C3 as the donor and PC71BM as the acceptor demonstrated a PCE of up to 2.2% with a Voc of 0.81 V, a Jsc of 4.93 mA cm−2 and a FF of 55.2%, under the illumination of AM1.5, 100 mW cm−2.
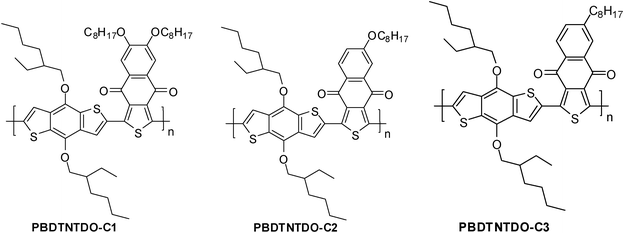 |
| | Scheme 1 Molecular structures of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3. | |
Experimental
Materials
Tetrakis(triphenylphosphine) palladium (Pd(PPh3)4), thiophene and octylbenzene (compound 9) were obtained from Pacific Chem Source, and they were used as received. Toluene was dried over Na/benzophenone ketyl and freshly distilled prior to use. Other reagents and solvents were purchased commercially as analytical-grade quality and used without further purification. Column chromatography was carried out on silica gel (size: 200–300 mesh).
Characterization
1H NMR spectra were recorded using a Bruker AV-400 spectrometer in deuterated chloroform solution at 298 K, unless specified otherwise. Chemical shifts were reported as δ values (ppm) relative to an internal tetramethylsilane (TMS) standard. The molecular weight of the polymers was measured by gel permeation chromatography (GPC) and polystyrene was used as a standard. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TGA-7 with a heating rate of 20 K min−1 under a nitrogen atmosphere. The temperature of degradation (Td) corresponds to a 5% weight loss. Mass spectra were obtained with a Shimadzu QP2010 spectrometer. The UV-vis absorption spectra were recorded on a JASCO V-570 spectrophotometer. For solid state measurements, the polymer solution in chloroform was spin-coated onto quartz plates. The cyclic voltammetry was performed on a Zahner IM6e electrochemical workstation with a three-electrode system in a solution of 0.1 M Bu4NPF6 in acetonitrile at a scan rate of 50 mV s−1. The polymer films were coated on a glassy carbon electrode (1.0 cm2) by dipping the electrode into the corresponding solutions and then drying. A Pt wire was used as the counter electrode, and Ag/Ag+ was used as the reference electrode. The morphology of the polymer/PC71BM blend films was investigated by a SPI 3800N atomic force microscope (AFM) in contacting mode with a 5 μm scanner.
Device fabrication and characterization
The PSCs were fabricated in the configuration of the common sandwich structure with an indium tin oxide (ITO) glass anode and a Ca/Al cathode. Patterned ITO glass with a sheet resistance of 15–20 Ω □−1 was purchased from CSG HOLDING Co., Ltd. (China). ITO glass was pre-cleaned and modified by a thin layer of PEDOT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PSS, which was spin-cast from a PEDOT:PSS aqueous solution (Baytron PVP Al 4083, Germany) on the ITO substrate and dried at 150 °C for 30 min in air, and the thickness of the PEDOT:PSS layer is about 40 nm. The polymers and [6,6]-phenyl-C71butyricacidmethylester (C70-PCBM) (Nano-C, USA) were solubilized in ortho-dichlorobenzene (ODCB) overnight, and then spin-coated on the ITO/PEDOT:PSS electrode. Then the Ca/Al cathode was deposited on the active layer by vacuum evaporation under 5 × 10−5 Pa. The effective area of one cell is 4 mm2. The current–voltage (I–V) measurement of the devices was conducted on a computer-controlled Keithley 236 Source Measure Unit. A xenon lamp with an AM1.5 filter was used as the white light source and the optical power was 100 mW cm−2. All fabrications and characterizations of devices were performed in a glove box.
:PSS, which was spin-cast from a PEDOT:PSS aqueous solution (Baytron PVP Al 4083, Germany) on the ITO substrate and dried at 150 °C for 30 min in air, and the thickness of the PEDOT:PSS layer is about 40 nm. The polymers and [6,6]-phenyl-C71butyricacidmethylester (C70-PCBM) (Nano-C, USA) were solubilized in ortho-dichlorobenzene (ODCB) overnight, and then spin-coated on the ITO/PEDOT:PSS electrode. Then the Ca/Al cathode was deposited on the active layer by vacuum evaporation under 5 × 10−5 Pa. The effective area of one cell is 4 mm2. The current–voltage (I–V) measurement of the devices was conducted on a computer-controlled Keithley 236 Source Measure Unit. A xenon lamp with an AM1.5 filter was used as the white light source and the optical power was 100 mW cm−2. All fabrications and characterizations of devices were performed in a glove box.
Synthesis of monomers and polymers
Synthetic routes of the monomers and polymers are shown in Scheme 2. 2,6-bis(trimethyltin)-4,8-di(2-ethylhexyloxyl)benzo[1,2-b:3,4-b]dithiophene 10 was prepared according to the literature.10a All of the other compounds were synthesized according to the following procedures.
 |
| | Scheme 2 Synthetic routes of the monomer and the corresponding polymers. | |
Tetrabromothiophene 1.
To a chloroform solution (20 mL) of thiophene (50.0 g, 0.6 mol), a chloroform solution (40 mL) of bromine (180 mL, 3.5 mol) was added dropwise within 4 h at 0 °C. The reaction mixture was warmed to room temperature and an additional amount of bromine (35 mL, 0.7 mol) was added, and the reaction mixture was subsequently stirred under reflux for 3 h. A saturated aqueous solution of NaOH was added and the mixture was stirred under reflux for one hour to remove the bromine. The mixture was extracted with dichloromethane (DCM) three times and the combined organic portions were dried over magnesium sulfate. After filtration, the organic solution was condensed under reduced pressure. The crude solid was recrystallized from a 1:1(v/v) solution of chloroform and ethanol to give pure 1 as colorless crystals (223 g, 93% yield). MS: 400 (M+).
3,4-Dibromothiophene 2.
In a 500 mL flask with 180 mL glacial acetic acid–water (1:2, v/v) the compound 1 (113 g, 283 mmol) and zinc powder (60 g, 918 mmol) were added alternately in several portions. The mixture was stirred for one hour at room temperature, and then stirred under reflux for another three hours. The mixture was cooled to room temperature and filtered to remove the excess zinc powder; the filtrate was extracted with ether. The combined organic portions were washed several times with water and then dried over magnesium sulfate. The crude product, which remained after the solvent had evaporated, was distilled under reduced pressure and a colorless oil was obtained (58.6 g, 86% yield). Bp: 104–108 °C /10–12 mm Hg. 1H NMR (400 MHz, CDCl3): δ = 7.27 ppm (s, 2H); MS: 242 (M+).
3,4-Dicyanothiophene 3.
A stirred solution of 3,4-dibromothiophene 2 (24.9 g, 102 mmol) and cuprous cyanide (55.3 g, 618 mmol) in dimethylformamide (50 mL) was maintained at reflux overnight. The dark mixture was cooled to room temperature and then poured into a solution of hydrated ferric chloride (200.0 g, 740 mmol) in hydrochloric acid (300 mL, 2.0 M) and maintained at 60–70 °C for one hour with vigorous stirring. After filtration, the mixture was extracted with a large amount of dichloromethane, the organic extraction was washed successively with 6 M hydrochloric acid, water and saturated sodium bicarbonate solution twice and the combined organic phases were dried over magnesium sulfate. After filtration, the solvents were evaporated under vacuum and the crude product was purified on a silica gel column, eluting with CH2Cl2–hexane (1:1, v/v). White crystals were obtained (5.7 g, 42% yield). Mp: 170–172 °C. 1H NMR (400 MHz, CDCl3): δ = 8.13 ppm (s, 2H) MS: 134 (M+).
2,5-Dibromothiophene-3,4-dicarboxylic acid 5.
A solution of thiophene-3,4-dicarboxylic acid 4 (1.83 g, 10.6 mmol) and glacial acetic acid (20 mL) was stirred in a 50 mL round-bottomed flask. Bromine (3.22 mL, 62.8 mmol) was added dropwise and the mixture was stirred overnight. Aqueous sodium bisulfate solution was added until the reddish color disappeared. The mixture was filtered and washed with water and a grey solid was obtained (2.76 g, 79% yield). 1H NMR (400 MHz, CDCl3): δ = 13.6 ppm (s, 2H). MS: 330 (M+)
1, 2-Bis(octyloxy)benzene 7.
Bromooctane (38.6 g, 202.2 mmol) was added to a vigorously stirred solution of catechol (10.0 g, 91.0 mmol) and K2CO3 (38.0 g, 270.0 mmol) in 50 mL dimethylformamide under nitrogen. The mixture was stirred for 40 h at 100 °C, then poured into water and extracted with dichloromethane. The combined organic portions were dried over magnesium sulfate and evaporated to afford the crude product. The crude product was purified on a silica gel column, eluting with hexane, and a pale yellow oil was obtained (25.2 g, 83% yield). 1H NMR (400 MHz, CDCl3): δ = 6.90 (s, 4H), 4.05 (t, 4H), 1.86–1.76 (m, 4H), 1.60–1.33 (m, 20H), 0.90 (t, 6H).
Octyloxybenzene 8.
This compound was synthesized in the same manner as described for compound 7. Bromooctane (11.1 g, 57.4 mmol) and phenol (4.5 g, 47.8 mmol) were used as the starting materials. Pale yellow oil was obtained (9.2 g, 93% yield). 1H NMR (400 MHz, CDCl3): δ = 6.83–7.22 (s, 5H), 4.05 (t, 2H), 1.76–1.88 (m, 4H), 1.28–1.32 (m, 8H), 0.88 (t, 3H).
Compound M1.
A solution of 2,5-dibromothiophene-3,4-dicarboxylic acid chloride 6 (1.1 g, 3 mmol) in dichloromethane was slowly added to a suspension of aluminum chloride (1.77 g, 13.2 mmol) in 10 mL dichloromethane at 0 °C. The mixture was stirred at 0 °C for 10 min. Then 1,2-bis(octyloxy)benzene 7 (1.2 g, 3.6 mmol) was added dropwise, the mixture was stirred for one hour at room temperature and then poured into ice. Dichloromethane was added and the mixture was shaken vigorously. The mixture was extracted with dichloromethane. The combined organic phases were washed with a saturated sodium bicarbonate aqueous solution and dried over magnesium sulfate. After filtration, the crude product was obtained by evaporating the solvents, then the crude product was purified on a silica gel column eluting with hexane–ethyl acetate (10:1, v/v). Finally a pale yellow solid was obtained (1.18 g, 63% yield). 1H NMR (400 MHz, CDCl3): δ = 7.65 (s, 2H), 4.15–4.18 (t, 4H), 1.86–1.89 (m, 4H), 1.29–1.57 (m, 20H), 0.90 (t, 6H). MS: 628 (M+).
Compound M2.
This compound was synthesized in the same manner as described for compound M1. 2,5-Dibromothiophene-3,4-dicarboxylic acid chloride 6 (1.1 g, 3 mmol) and octyloxybenzene 8 (0.74 g, 3.6 mmol) were used as the starting materials. A yellow solid was obtained (0.82 g, 55% yield). 1H NMR (400 MHz, CDCl3): δ = 7.62 (s, 2H), 7.50 (s, 1H), 4.16 (t, 2H), 1.76–1.84 (m, 4H), 1.29–1.32 (m, 8H), 0.89 (t, 3H). MS: 500 (M+).
Compound M3.
This compound was synthesized in the same manner as described for compound M1. 2,5-Dibromothiophene-3,4-dicarboxylic acid chloride 6 (1.1 g, 3 mmol) and n-octylbenzene 9 (0.68 g, 3.6 mmol) were used as the starting materials. A pale yellow solid was obtained (0.74 g, 51% yield). 1H NMR (400 MHz, CDCl3): δ = 8.09–8.19 (d, 2H), 7.60 (s, 1H), 2.76 (s, 2H), 1.26–1.56 (m, 13H), 0.87 (s, 2H) MS: 500 (M+).
Synthesis of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3.
Polymerization was performed by a typical Stille coupling reaction.
PBDTNTDO-C1.
Compound 10 (193.1 mg, 0.25 mmol) and M1 (157.1 mg, 0.25 mmol) were dissolved in 15 mL toluene. The mixture was flushed with argon for 20 min, and then Pd(PPh3)4 (11.6 mg, 0.01 mmol) was added into the flask. The flask was purged three times with successive vacuum and argon filling cycles. The polymerization reaction was heated to 110 °C, and the mixture was stirred for 24 h under an argon atmosphere. The mixture was cooled to room temperature and poured slowly into methanol (50 mL), the reactant was filtered through a Soxhlet thimble. Then the crude polymer was washed with methanol and hexane to remove the oligomers and catalyst residue. Finally the polymer was extracted with chloroform. The polymer solution was condensed and slowly poured into methanol. The precipitate was collected by filtration and dried under high vacuum to afford PBDTNTDO-C1 as a dark purple solid (170 mg, 72% yield).
PBDTNTDO-C2.
PBDTNTDO-C2 was synthesized with a similar procedure to PBDTNTDO-C1 from monomer 10 (200.8 mg, 0.26 mmol) and M2 (130 mg, 0.26 mmol), giving a dark purple solid (140 mg, 66% yield).
PBDTNTDO-C3.
PBDTNTDO-C3 was synthesized with a similar procedure to PBDTNTDO-C1 from monomer 10 (232 mg, 0.3 mmol) and M3 (145 mg, 0.3 mmol), giving a purple solid (120 mg, 50% yield).
Results and discussion
Synthesis and characterization
The general syntheses of the monomers and the copolymers are outlined in Scheme 2. Compound 10 copolymerized with compound M1, compound M2 or compound M3 through Stille coupling reactions to afford the target polymers, PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3, respectively. The polymers were purified by sequential Soxhlet extraction with methanol, hexane and CHCl3. The CHCl3 fraction was then reduced in volume, precipitated into methanol and collected by filtration, yielding a purple solid. The chemical structures of the polymers were verified by 1H NMR. The molecular weight of the polymers was measured by gel-permeation chromatography (GPC) in tetrahydrofuran (THF) solution relative to polystyrene standards. The weight-average molecular weight (Mw) is 11 KDa for PBDTNTDO-C1, 17 KDa for PBDTNTDO-C2 and 53 KDa for PBDTNTDO-C3. PDI values for DTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 were 1.7, 4.3 and 2.4, respectively, and the related data were listed in Table 1. PBDTNTDO-C2 has a relatively low molecular weight and a high PDI, which may be related to its relatively low solubility; improvement in molecular weight is in progress. PBDTNTDO-C1 has the highest polymerization yield. However, its molecular weight is relatively low according to the GPC measurement; a small section with high molecular weight cannot be dissolved in THF as its symmetrical structure leads to very low solubility, resulting in crystallization. Of all the polymers, the solubility of PBDTNTDO-C3 is the best, and PBDTNTDO-C1 has the worst solubility; the difference in solubilities results in some inversions between the reaction yield and the molecular weight. However, when heated, all of them can be dissolved in CHCl3 and ortho-dichlorobenzene. The thermal properties of the copolymers were investigated with thermogravimetric analysis (TGA) as shown in Fig. 1. TGA measurement shows that the 5% weight-loss temperatures (Td) of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 were 314 °C, 313 °C and 329 °C under inert atmosphere, respectively. The thermal curves of the three copolymers are almost similar, which indicates that the thermal stability of the copolymers is enough for optoelectronic applications.
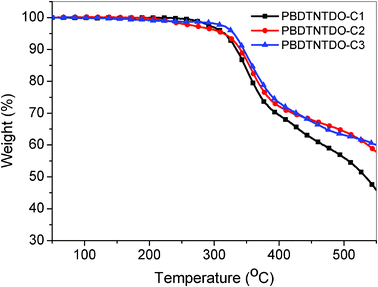 |
| | Fig. 1 TGA curves of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 with a heating rate of 20 K min−1. | |
Table 1 Molecular weights and thermal properties of the copolymers
| Polymers |
M
w
a
|
M
n
a
|
PDIa |
T
d(°C)b |
|
M
w, Mn, and PDI of the polymers were determined by GPC using polystyrene standards in THF.
T
d is the 5% weight-loss temperature of the polymers under inert atmosphere.
|
| PBDTNTDO-C1 |
11069 |
6348 |
1.7 |
314 |
| PBDTNTDO-C2 |
17157 |
3900 |
4.3 |
313 |
| PBDTNTDO-C3 |
53365 |
21887 |
2.4 |
329 |
Optical properties
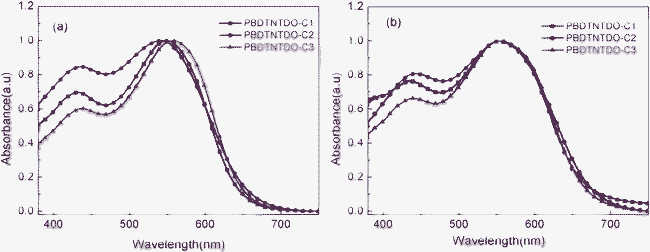
UV-vis absorption spectra could provide much information on the electronic structure of the conjugated polymers. The photophysical characteristics of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 were investigated by UV-vis absorption spectra in dilute chloroform solution and in solid films spin-coated on the quartz substrate. Fig. 2 shows the UV-vis absorption spectra of the polymer solutions and films; the corresponding optical data are summarized in Table 2. The absorption spectra of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 in solutions showed absorption maxima at 548 nm, 541 nm and 557 nm, respectively, whereas in the film states the absorption peaks shifted to 550 nm, 554 nm and 556 nm, respectively. Compared to the solution absorption, the UV-vis absorption of the PBDTNTDO-C2 film gets broader and shows a red shift of 13 nm, indicating good intermolecular interactions in the solid state, which is probably due to the asymmetric electron-donating alkoxy group resulting in decreased vibration thermal loss. All the copolymers have the absorption range 400 nm to 680 nm. The optical bandgap (Eg) was calculated from the onset wavelength of the absorption spectra to be 1.81 eV, 1.80 eV and 1.79 eV, respectively. The results show that the side chains do not obviously affect the optical energy gap, they only change the relative absorption intensity from 400 nm to 550 nm.
Table 2 Optical and electrochemical properties of the copolymers
| Copolymers |
Absorption spectra |
Cyclic voltammetry |
| Solutiona |
Filmb |
Energy levels |
|
λ
max (nm) |
λ
max (nm) |
λ
onset (nm) |
Eoptgc (eV) |
HOMO (eV) |
LUMO (eV) |
|
Measured in chloroform solution.
Spin-coated from chloroform solution.
Band gap estimated from the onset wavelength of the optical absorption.
|
| PBDTNTDO-C1 |
548 |
550 |
685 |
1.81 |
−5.19 |
−3.38 |
| PBDTNTDO-C2 |
541 |
554 |
689 |
1.80 |
−5.23 |
−3.33 |
| PBDTNTDO-C3 |
557 |
556 |
693 |
1.79 |
−5.27 |
−3.39 |
Electrochemical properties
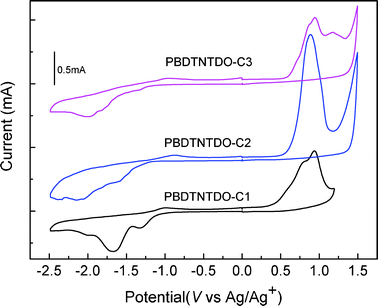
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of conjugated polymers are important for PSCs, which are closely concerned with charge separation. Energy levels can be estimated by electrochemical cyclic voltammetry (CV). Fig. 3 shows cyclic voltammograms of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3, which were carried out in a 0.1 M solution of Bu4NPF6 in acetonitrile at room temperature under argon with a scanning rate of 50 mV s−1. All the potentials are reported vs. Ag/Ag+ with the ferrocene/ferrocenium couple as an internal standard. The onset reduction potentials (Ered) are −1.33 V vs. Ag/Ag+ for PBDTNTDO-C1, −1.38 V vs. Ag/Ag+ for PBDTNTDO-C2 and −1.32 V vs. Ag/Ag+ for PBDTNTDO-C3, while the onset oxidation potentials (Eox) are 0.48 V vs. Ag/Ag+ for PBDTNTDO-C1, 0.52 V vs. Ag/Ag+ for PBDTNTDO-C2 and 0.56 V vs. Ag/Ag+ for PBDTNTDO-C3. From the Eox and Ered of the polymers, we calculated the HOMO and LUMO energy levels of the polymer according to the equations.15
| HOMO = −e (Eox + 4.71) (eV) |
| LUMO = −e (Ered + 4.71) (eV) |
 |
| | Fig. 3 Cyclic voltammograms of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 films on a glassy carbon electrode in 0.1 M Bu4NPF6, CH3CN solution. | |
The HOMO energy levels of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 are −5.19 eV, −5.23 eV and −5.27 eV, respectively, which indicates that the introduced alkoxyl group can increase the HOMO level to some extent compared to the corresponding alkyl group; the LUMO energy levels are −3.38 eV, −3.33 eV and −3.28 eV, respectively. The related electrochemical data are summarized in Table 2 for comparison.
Calculation
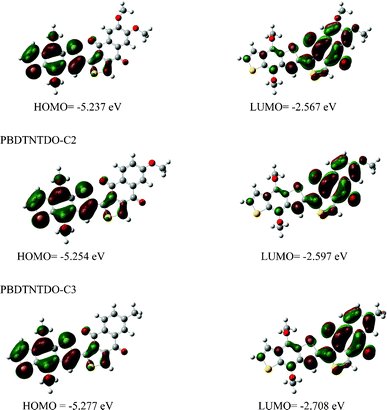
To provide an insight into the molecular architecture of the polymers, molecular simulation was carried out for the three copolymers with a chain length of n = 1 using density functional theory (DFT) at the B3LYP/6-31G level with the Gaussian 09 program package. To simplify the calculations, all of the alkyl chains were replaced by methyl groups. DFT/B3LYP/6-31G* has been found to be an accurate method for calculating the optimal geometry and electronic structures of many molecular systems. Fig. 4 shows the calculated molecular orbital geometry and energy levels on the model compound of the polymers. The HOMOs of the copolymers are mainly delocalized over the polymer backbones. Because the HOMOs can be partially delocalized over the oxygen atom of the alkoxy side substituents of PBDTNTDO-C1 and PBDTNTDO-C2, PBDTNTDO-C1 and PBDTNTDO-C2 have higher HOMO levels compared to that of PBDTNTDO-C3. From the DFT B3LYP/6-31G* level calculations combined with the equations provided by the Leclerc group, considering our saturated calomel electrode (SCE) level here of 4.4 eV with the Ag/Ag+ reference electrode at −4.71 eV, not at −4.7 eV as in that publication,16 the HOMO and LUMO levels of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 are calculated to be −5.27 and −3.28 eV, −5.27 and −3.30 eV, and −5.29 and −3.39 eV, respectively, which are in good agreement with the experimental values for the energy gap and the HOMO and LUMO levels from the electrochemical measurement. From the theoretical calculations and experimental data, the side chains affect the electronic and optical properties of the copolymers a little, which may originate from the side chain separated from the polymer backbone by a naphthdione unit. Thus, it is clear that the calculation can be used as a good tool to predict the electronic and optical properties of the polymers.
 |
| | Fig. 4 Molecular orbital geometry and energy levels obtained from DFT calculations on the copolymers with chain length n = 1 at the B3LYP/6-31G* level. | |
Hole mobility
Besides absorption and energy level, hole mobility is another important parameter of the conjugated polymers for photovoltaic applications, due to its direct effect on charge transport. We investigated the hole mobility of PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 blends with PC71BM by the space-charge-limited current (SCLC) model which is based on Poole–Frenkel Law17 with a device structure of ITO/PEDOT:PSS/polymer: PC71BM (1:2, w/w)/Au. The SCLC model can be described by the equation:| | 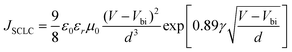 | (1) |
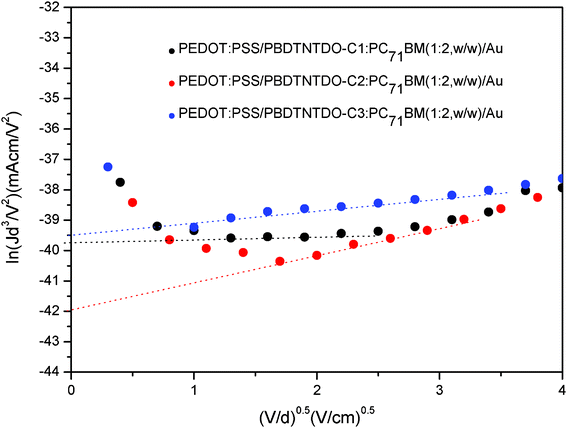
The results are plotted as ln(Jd3/V2) vs. (V/d)0.5, as shown in Fig. 5. Herein, J stands for current density, d is the thickness of the device and V = Vappl − Vbi, where Vappl is the applied potential and Vbi is the built-in potential. According to eqn (1) and Fig. 5, the hole mobilities of the polymer blends are evaluated to be 2.04 × 10−5 cm2 V−1 s−1, 2.50 × 10−6 cm2 V−1 s−1 and 3.04 × 10−5 cm2 V−1 s−1 for PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3, respectively. Obviously, the PBDTNTDO-C3 blend possesses the highest hole mobility among the three copolymers, which may originate high molecular weight and the orientation of the alkyl group.13f The high hole mobility of the polymer is expected to facilitate charge transport and reduce recombination loss in the PSCs, therefore higher hole mobility can usually lead to an improved device performance.
 |
| | Fig. 5 ln(Jd3/V2) vs. (V/d)0.5 plots of the polymers for measurement of hole mobility by the SCLC method. | |
Photovoltaic properties
To explore the photovoltaic properties of the three copolymers, BHJ PSC devices with a structure of ITO/PEDOT:PSS/polymer: PC71BM (1:2, w/w)/Ca/Al were fabricated. The polymer active layers were spin-coated from an ortho-dichlorobenzene blend solution. Fig. 6 shows typical current density–voltage (J–V) curves and the external quantum efficiency (EQE) of the PSCs. As is well known, the PCE is proportional to the open circuit voltage (Voc), short circuit current (Jsc) and fill factor (FF) of the PSCs. The corresponding Voc, Jsc, FF and PCEs of the devices are summarized in Table 3. From the photovoltaic results, PBDTNTDO-C1 exhibits the highest Voc, which is not consistent with its HOMO level and related to the blend morphology and interface resistance.4 PBDTNTDO-C1 and PBDTNTDO-C3 have a much higher FF than that of PBDTNTDO-C2, which is probably a result of higher hole mobility, balanced charge transport, relatively high molecular weight and better blend morphologies. Among these copolymers, PBDTNTDO-C3 has the highest Jsc combined with a high Voc of 0.81 V and a high FF of 0.55, which led to the highest PCE of up to 2.2%. The complementary absorbance of PC71BM and the copolymers leads to a relatively high photoconversion efficiency over the wavelength range 350–600 nm, with monochromatic EQE values above 25%. The Jsc calculated by integrating the EQE curve with an AM1.5 G reference spectrum agrees well with the Jsc value obtained from the J–V measurements. Although PBDTNTDO-C1 and PBDTNTDO-C3 have a good FF, the Jsc value of 4–5 mA cm−2 for the devices of the three polymers is relatively low compared to those of the high efficiency polymers, which may be related to the moderate hole mobilities of 10−5–10−6 cm2 V−1 s−1 and absorption bands below 700 nm with some sunlight loss. From the EQE spectra, no photoresponse above 700 nm combined with low EQE values from 300–700 nm with a maximum EQE of 40% to some extent limited the increase of Jsc. However, the initial result of PCE up to 2.2% is promising, and we are currently aiming at trying new modifications of NTDO and different device conditions for improving the photovoltaic properties.
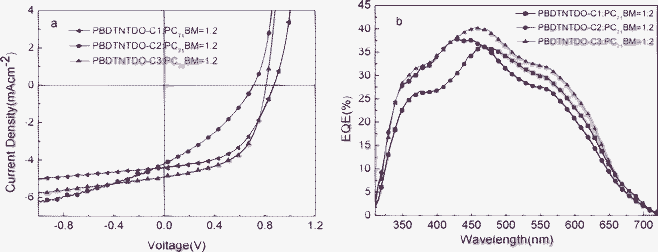 |
| | Fig. 6 (a) J–V curves of the PSCs based on PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3:PC71BM (1:2, w/w), under illumination of AM1.5, 100 mW cm−2. (b) EQE spectra of PSCs based on PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3:PC71BM (1:2, w/w). | |
Table 3 Photovoltaic performances of the polymer solar cells based on PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3
| Polymer/PC71BM (1:2, w/w) |
V
oc (V) |
J
sc (mA cm−2) |
FF (%) |
PCE (%) |
| PBDTNTDO-C1 |
0.87 |
4.42 |
50.8 |
1.96 |
| PBDTNTDO-C2 |
0.71 |
4.21 |
34.0 |
1.01 |
| PBDTNTDO-C3 |
0.81 |
4.93 |
55.2 |
2.21 |
Morphology
The morphology of the photoactive layer is very important for PSCs, and in some cases the performance of the device strongly depends on its morphological features.18 To explain in more detail the reasons for the different PCEs of the polymers, we used atomic force microscopy (AFM) to investigate the morphological characteristics of the copolymer: PC71BM (1:2, w/w) blend films by spin coating the o-dichlorobenzene solution of the blends on an ITO glass in a similar manner to the preparation of the active layer in photovoltaic devices. Five measurements for every polymer were investigated here. The mobilities of the blend films correspond to the hole mobility part in this manuscript. The AFM topography and phase images are shown in Fig. 7. The surface rms (root-mean-square) roughness of the polymer: PC71BM films (1:2, w/w) are 1.76 nm for PBDTNTDO-C1, 3.22 nm for PBDTNTDO-C2 and 2.90 nm for PBDTNTDO-C3, respectively. For the PBDTNTDO-C2/PC71BM blend, Fig. 7b,e reveal no detectable phase separation of the two components in the film, which probably affects the separation of the charges; therefore, an unbalanced charge transport was produced. For PBDTNTDO-C1/PC71BM, Fig. 7a,d, the blend films show some aggregation and nanophase separation occurred. For the PBDTNTDO-C3/PC71BM blend, the blend films are relatively smooth compared to that of PBDTNTDO-C2/PC71BM. The domains are well distributed throughout the surface, indicating a significant aggregation as shown in Fig. 7c, which may be compact chain packing and crystallization in the aggregation form, beneficial for obtaining higher hole mobility. As shown in the corresponding phase image of Fig. 7f, pronounced nanophase separation was formed, which to some extent explains the higher photovoltaic properties of PBDTNTDO-C3/PC71BM among the three copolymers.19 The good nanophase separation morphology can in part explain the high hole mobility of the copolymer and the relatively high PCE.
 |
| | Fig. 7 AFM height images (a, b and c) and phase images (d, e and f) for the polymer: PC71BM blend films (1:2, w/w): (a and d) PBDTNTDO-C1; (b and e) PBDTNTDO-C2; (c and f) PBDTNTDO-C3, spin-coated with o-dichlorobenzene solution. | |
Conclusions
In summary, we synthesized three new D–A conjugated polymers containing a BDT donor unit and a NTDO acceptor unit, PBDTNTDO-C1, PBDTNTDO-C2 and PBDTNTDO-C3 by Stille cross-coupling polymerization reactions. The synthesized copolymers exhibited broad absorption in the visible region. Preliminary studies on photovoltaic cells using the blends of PBDTNTDO-C3 and PC71BM (1:2, w/w) as the active layer afforded devices with a Voc of 0.81 V, Jsc of 4.93 mA cm−2, FF of 55.2% and PCE of 2.2%. This indicates that the NTDO derivatives are probably promising electron accepting building blocks for construction of D–A copolymers for printed electronics. A higher PCE can be anticipated by increasing the molecular weight, and by careful device engineering such as additives and thermal annealing, etc.
Acknowledgements
This work was supported by NSFC (Nos. 51173206, 21161160443), National High Technology Research and Development Program (NO. 2011AA050523), the Natural Science Foundation of Hunan Province, China (No. 11JJ4010), China Postdoctoral Science Foundation (No.20110490150), the Fundamental Research Funds for the Central Universities (No.2010QZZD0112).
References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062 CrossRef CAS.
- H. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS.
- Z. He, C. Zhong, X. Huang, W. Y. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636 CrossRef CAS.
-
(a) Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS;
(b) L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697 CrossRef CAS;
(c) Z. Tan, W. Zhang, Z. Zhang, D. Qian, Y. Huang, J. Hou and Y. Li, Adv. Mater., 2012, 24, 1476 CrossRef CAS.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupr, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS.
-
(a) Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952 CrossRef CAS;
(b) Y. Cheng, S. Yang and C. Hsu, Chem. Rev., 2009, 11, 5868 CrossRef;
(c) B. Liu, W. Wu, B. Peng, Y. Liu, Y. He, C. Pan and Y. Zou, Polym. Chem., 2010, 1, 678 RSC;
(d) P. Ding, C. C. Chu, B. Liu, B. Peng, Y. Zou, Y. He, K. Zhou and C. Hsu, Macromol. Chem. Phys., 2010, 211, 2555 CrossRef CAS;
(e) C. Y. Chang, Y. J. Cheng, S. H. Hung, J. S. Wu, W. S. Kao, C. H. Lee and C. S. Hsu, Adv. Mater., 2012, 24, 549 CrossRef CAS.
-
(a) Z. Zhang, B. Peng, B. Liu, C. Pan, Y. Li, Y. He, K. Zhou and Y. Zou, Polym. Chem., 2010, 1, 1441 RSC;
(b) W. Yue, Y. Zhao, S. Shao, H. Tian, Z. Xie, Y. Geng and F. Wang, J. Mater. Chem., 2009, 19, 2199 RSC;
(c) Y. Zou, D. Gendron, R. B. Aıch, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891 CrossRef CAS;
(d) E. Ahmed, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2009, 42, 8615 CrossRef CAS.
-
(a) I. Osaka, M. Saito, H. Mori, T. Koganezawa and K. Takimiya, Adv. Mater., 2012, 24, 425 CrossRef CAS;
(b) J. Chen and C. Hsu, Polym. Chem., 2011, 2, 2707 RSC.
- C. Cui, X. Fan, M. Zhang, J. Zhang, J. Min and Y. Li, Chem. Commun., 2011, 47, 11345 RSC.
-
(a) C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595 CrossRef CAS;
(b) Q. Peng, X. Liu, D. Su, G. W. Fu, J. Xu and L. M. Dai, Adv. Mater., 2011, 23, 4554 CrossRef CAS;
(c) Z. Gu, P. Tang, B. Zhao, H. Luo, X. Guo, H. Chen, G. Yu, X. Liu, P. Shen and S. Tan, Macromolecules, 2012, 45, 2359 CrossRef CAS;
(d) S. Ko, E. Hoke, L. Pandey, S. Hong, R. Mondal, C. Risko, Y. Yi, R. Noriega, M. McGehee, J. L. Brédas, A. Salleo and Z. N. Bao, J. Am. Chem. Soc., 2012, 134 DOI:10.1021/ja210954r;
(e) A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Frechet, J. Am. Chem. Soc., 2012, 134, 2180 CrossRef CAS;
(f) J. Lu, T. Y. Chu, S. Beaupré, Y. Zhang, J. R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345 CrossRef.
- C. Cui, H. Fan, X. Guo, M. Zhang, Y. He, X. Zhan and Y. Li, Polym. Chem., 2012, 3, 99 RSC.
- M. Yang, B. Peng, B. Liu, Y. Zou, K. Zhou, Y. He, C. Pan and Y. Li, J. Phys. Chem. C, 2010, 114, 17989 CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. N. Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS.
-
(a) G. G. Malliaras, J. R. Salem, P. J. Brock and C. Scott, Phys. Rev. B: Condens. Matter, 1998, 58, 13411 CrossRef;
(b) H. C. F. Martens, H. B. Brom and P. W. M. Blom, Phys. Rev. B: Condens. Matter, 1999, 60, 8489 CrossRef.
-
(a) C. V. Hoven, X. D. Dang, R. C. Coffin, J. Peet, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2010, 22, E63 CrossRef CAS;
(b) J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef CAS.
-
(a) W. Li, Y. Zhou, B. V. Andersson, L. M. Andersson, Y. Thomann, C. Veit, K. Tvingstedt, R. Qin, Z. Bo, O. Inganas, U. Würfel and F. Zhang, Org. Electron., 2011, 12, 1544 CrossRef CAS;
(b) W. Wang, H. Wu, C. Yang, C. Luo, Y. Zhang, J. Chen and Y. Cao, Appl. Phys. Lett., 2007, 90, 183512 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Figures giving the 1H NMR spectra of the monomers. See DOI: 10.1039/c2ra20747h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.