Particulate transepithelial drug carriers: barriers and functional polymers
Krzysztof
Babiuch
ab,
Michael
Gottschaldt
ab,
Oliver
Werz
bc and
Ulrich S.
Schubert
*abd
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: Ulrich.Schubert@uni-jena.de; Fax: 49 3641 948202; Tel: 49 3641 948201
bJena Center for Soft Matter (JCSM), Friedrich Schiller University, Humboldtstr, 10, 07743, Jena, Germany
cChair of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich Schiller University Jena, Philosophenweg, 14, D-07743, Jena, Germany
dDutch Polymer Institute (DPI), John F. Kennedylaan, 2, 5612, AB Eindhoven, The Netherlands
First published on 3rd August 2012
Abstract
This review may serve as a compilation of helpful guidelines for design, synthesis, formulation, and in vitro, ex vivo as well as in vivo evaluation of particulate drug delivery systems that cross epithelial barriers. It provides both chemist and biologist with a comprehensive overview of complex anatomorphological features encountered by intranasally, pulmonary and orally administered particles. In addition, the materials (structures, modifications, formulations, and activity) used in transepithelial delivery are discussed. It covers the biological as well as physicochemical aspects crucial for the successful development of particulate drug carriers and methods for their application.
1. Introduction
Polymeric nano- and microparticles have found a wide range of applications in drug and antigen delivery via transepithelial (mucosal) routes. They include intranasal, intrapulmonary and oral ways of administration among others and offer ubiquitous advantages over parenteral routes, e.g. no injection requirement and, consequently, no need for trained personnel, better patient compliance and lower risk of infection. However, introduction of drugs by mucosal routes poses one of the greatest challenges of the 21st century in the pharmaceutical field. In order to avoid drug/antigen degradation in the gut, to enhance its retention in mucus and to improve the overall permeation through epithelia, thus, augmenting the effectiveness of mucosally administered drugs, different delivery systems have been developed and are reviewed in the scientific literature. They include: microemulsions, ISCOMs (immunostimulating complexes), liposomes, virosomes, virus-like particles as well as polymeric nano- and microparticles.1–6 There are plenty of factors that influence the uptake of particulates across the epithelia. They can be divided into two main groups; the ones concerning the material and formulation: polymer composition, particle size, hydrophobicity and surface charge, use of surfactants and penetration enhancers as well as additional targeting agents. And the ones related to the method of introduction to the body: dose of particles, administration vehicle, means of delivery, animal species and age as well as fed state of the patients. All of these factors are caused by the differences in cellular as well as anatomorphological features of the respiratory and gastrointestinal tracts described within this review. Therefore, each of the particulate systems or drug carriers has to be optimized for an appropriate drug administration route. The administration route of antigen-carrying particles affects the quality of immune responses.7 The application of polymeric particles as drug carriers opens an avenue for the modification of traits that enable the deposition of particulate material at the required site of action. By adjustment of macromolecular composition and processing parameters ideal properties of a transmucosal drug delivery system can be obtained. They are listed in Table 1. Additionally, for transmucosal vaccination systems the ideal particles should retain antigens at the mucosal surface for targeting antigen-presenting cells (APCs), stimulate both innate and adaptive immunity at the epithelium, induce homing of effector cells to the site of infection and cause long-term immunity. In order to produce drug carriers fulfilling these requirements, a multitude of materials and formulations has been investigated, out of which selected examples are presented within this review. Since the mucosal surfaces are the primary site of pathogen infection, and most of the particulate drug delivery systems are in the size range of viruses and bacteria, their interaction with, as well as transport via, mucosa-associated lymphoid tissues (MALT) is widely exploited for vaccination purposes. It has been established that protection against pathogens invading the organism through epithelia correlates better with the presence of specific antibodies in local secretions than with serum antibodies, thus, the induction of immune response by antigen-carrying particles at the site of infection is crucial for effective defense.8 Particulated material acts not only as a delivery system but also as an immunologic adjuvant, i.e., a substance that accelerates, prolongs, or enhances antigen-specific immune responses.9 The adjuvancy of particles is moderated by different factors, such as: specific uptake by APCs, prolonged release of antigen in MALT due to slow biodegradation, direct stimulation of the response by particles themselves, as well as the route of uptake (direct to cytoplasm or via endosomal compartment).10,11 As a consequence, the adjuvant action of the particulate delivery system is extremely complex but may be adjusted by formulation processes and the material itself. Furthermore, particles can act as an all-in-one vaccination package by integration of targeting groups, permeation enhancers, antigens, and adjuvants for transmucosal delivery to the same group of APCs. Application of particulates for delivery of pharmacologically active substances other than vaccines offers some unique advantages such as reduction of required dose of the drug by the increase of its bioavailability through protection from the acidic environment in the gut and encapsulation of poorly water-soluble drugs within carriers. By tailoring of the polymer's solubility/degradability, changes in formulation, e.g. introduction of mucoadhesive coatings and surfactants, drug-loaded particles with different sizes, porosities and surface charges, enable organ and cell targeted delivery trough mucus-associated tissues. Additionally, adjustment of aerodynamic properties of particulates, administration volumes, as well as vehicles allows the design of advanced systems for inhalation therapies.| • Safe, no side effects in adult and newborn animals/humans |
| • Biodegradable, does not accumulate in the body |
| • Does not agglomerate in suspensions before administration |
| • Crosses epithelial barrier |
| • Does not induce toxic or inflammatory reactions |
| • Entraps drug/antigen while maintaining its activity |
| • Contains targeting moieties for specific cellular uptake |
This review aims to provide practical guidelines for researchers tackling the problem of transmucosal drug carriers. Within, the reader can find a comprehensive description of the anatomorphological features of the pulmonary, nasal and gastrointestinal epithelia (Section 2) crucial for the understanding of the barriers' location and function. Furthermore, Section 3 provides an overview of the materials and their particulate formulations as well as administration methods which have been employed in the development of transepithelial drug delivery systems.
2. Structure of intestinal, lung, and nasal epithelial barriers
The intestinal, lung, and nasal epithelial barriers belong to mucosal surfaces, also called mucous membranes, which form the lining of digestive, respiratory, and urogenital tracts with a combined surface area of about 400 m2.12 Since they are the most common route for the entry of foreign pathogens, environmental particulates, and toxins, nature has developed an appropriate system of anatomical and physiological defences (Fig. 1).13,14 The primary protection is achieved by the formation of a tight boundary layer of intestinal, lung, and nasal epithelial cells amplified by the properties of mucus covering these epithelia. The secondary protection is offered by region-specific mucosa-associated lymphoid tissue (MALT), namely, in the herein discussed barriers, the gastrointestinal-associated lymphoid tissue (GALT) and the nasopharynx-associated lymphoid tissue (NALT).15–17 Structural features of the mucus as well as epithelial cells, GALT, and NALT, which are crucial for the passage of particulate matter through the barriers, are summarized below as well as in Table 2.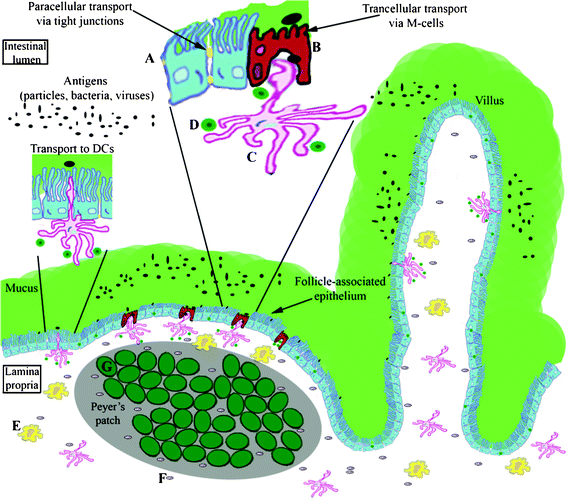 | ||
| Fig. 1 Schematic representation of the intestinal epithelial barrier. A –epithelial cell, B —M-cell, C—dendritic cell (DC), D—lymph cell (B/T-lymphocyte), E—macrophage, F—plasma cell, G—lymphoid follicle. | ||
| Type of epithelium | Surface area | Thickness of epithelium | Mucus layer thickness | Special surface properties |
|---|---|---|---|---|
| Intestinal | 400 m2 (small intestine 200 m2),62 | Varies with different regions and cell types | Ileum (10 μm), cecum (36.7 ± 7.2 μm), colon (average 100 μm),47–51 | Leaky tight junctions in jejunum and ileum cause enhanced paracellular transport. Transport of antigen-functionalized particles through follicle-associated epithelium and dendritic cells in GALT.103 |
| Nasal | 160 cm2,41,70 | 40 to 100 μm,68,69 | Approx. 10 μm,41,42 | Nasal mucus is considered highly permeable. Possibility of nasal immunization by targeting to NALT.223 |
| Pulmonary | Approx. 120 m2 during inhalation,72 | Thickness decreases in the direction of alveoli, less than 1 μm in the alveolar epithelium,74 | 5 to 20 μm,44–46 | Direct access to the blood capillaries wrapped around the alveoli. Lung surfactant present instead of mucus in deep lung.39,40 Targeting of particles to alveolar macrophages for vaccination.181,182 |
2.1 The structure of mucus
The mucus is the first obstacle encountered by all drug carriers delivered to the respiratory, gastrointestinal (GI), or cervicovaginal tracts.18,19 To reach the underlying epithelium the particles must be able to overcome the mucus barrier.20,21 The mucus is a complex, viscoelastic hydrogel, composed of highly branched glycoproteins (mucins), lipids, cellular and serum macromolecules, electrolytes, proteolytic enzymes, bile salts, cells, and other cellular debris.18,22 Mucins are the key component of mucus. They are large, amphiphilic macromolecules with a molar mass in the range of 200 kDa to 20–40 MDa.23,24 Their structure is complex and highly segregated into heavily glycosylated hydrophilic regions and polypeptide regions with little or no glycosylation.25 Mucins are present in a cell-associated, membrane bound shape (in glycocalyx) and in a secreted form in the mucus gel. The glycocalyx is composed of integral membrane proteins with projecting glycosaminoglycan side chains and is thought to prohibit microorganisms from attaching and invading enterocytes. The carbohydrate content in mucins ranges from 40 to 80% and consists mainly of N-acetylgalactosamine, N-acetylglucosamine, fucose, galactose, N-acetylneuraminic acid and derivatives thereof (sialic acids), traces of mannose, and sulphated sugars. The mucins of mucus gel form fibers densely coated with short glycans, usually end-terminated with a negatively charged group (carboxyl or sulfate). These hydrophilic domains are separated by relatively hydrophobic regions of the protein and fold into disulfide-bond stabilized globules that tend to accumulate at interfaces with their glycosylated parts oriented toward polar/aqueous phases. This amphiphilic structure enables efficient entrapment of foreign particulates by polyvalent adhesive interactions of mucin fibers. Hydrophilic particles adhere strongly to the glycosylated and negatively charged regions, whereas hydrophobic ones are captured by multiple low-affinity bonds between the protein part and the particle surface.26 More detailed information about mucins can be found in several reviews.23,25,27–34 The pH value of the mucus varies greatly depending on the nature of surface covered. Furthermore, highly acidic environments cause aggregation of mucin fibers, thus, increasing mucus viscoelasticity.35,36 Lung and nasal mucus are in general neutral in pH. From luminal to basal surface within the cross-section of gastric mucus, a rising pH gradient is observed from around 1–2 up to approximately 7.37,38 Within the deep lung, the mucus layer is not present; instead, the alveolar epithelium is lined by a complex surfactant that consists of approximately 90% lipids and 10% proteins.39,40 The thickness of the mucin gel layer is constantly regulated by its constant secretion, as well as clearance, and varies greatly at different mucosal surfaces. Furthermore, toxic and irritating substances cause an increase in the production of mucus and its thickness.19 The nasal mucosa is covered by a limited, approximately 10 μm thick coating and is considered highly permeable in comparison to other mucosal surfaces.41,42 It consists of two layers; the outer (luminal) forms a gel blanket layer and the inner an aqueous periciliary sol phase (Fig. 2).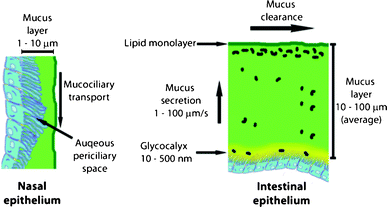 | ||
| Fig. 2 General features of nasal and intestinal mucus. | ||
The pulmonary mucus has a similar double-layered structure.43 The luminal gel part is considered to be approximately 5 to 10 μm thick and the inner, less viscoelastic sol, which is occupied by cilia, forms the second layer of 5 to 10 μm.44–46 However, the relative thickness of the human GI tract mucus layer still requires proper characterization.20 The reported average values for the intestinal surfaces are dependent on the covered region: ileum (10 μm), cecum (36.7 ± 7.2 μm), and colon (different values are published, average 100 μm, see the references for details).47–51 Intestinal mucus also exhibits a layered structure illustrated in Fig. 2. Due to its multi-constituent structure and the variety of intermolecular interactions, like hydrogen bonding, electrostatic and hydrophobic interactions, together with physical entanglement of the macromolecular components, the mucus has certain unique physical properties. The most important ones are high viscoelasticity and effective porosity of the gel network.52 The viscoelasticity of the mucus increases together with the concentration of the mucins and decreases with the level of hydration.53 Hydration is regulated by the underlying mucosal epithelium by adjustment of the ionic environment.54 The estimated average pore size within the mucin fiber matrix is assumed to be 100 nm and is in good agreement with the dimensions of mucosa-infecting viruses.55,56 Another significant feature of mucus is the shear-thinning behavior.55 The bulk viscosity of healthy human mucus is normally (at low shear rates) 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher than the viscosity of water, however, under the shear stress present in GI and nasal tracts, a lubricating slippage plane is formed and the upper, luminal layers are removed much faster than the lower ones. At maximum physiological shear rates the viscosity of mucus approaches that of water making it an excellent low viscosity lubricant. Due to this shear-thinning behavior, a stable, unstirred layer is formed on the epithelial surfaces. The constant secretion, degradation, and shedding of the mucus makes it necessary for pathogens and drug-delivery agents to migrate “upstream”, markedly faster than mucus renewal and clearance takes place, in order to reach the epithelium.19,34 The different techniques to determine materials with mucoadhesive and/or mucopenetration properties have been recently reviewed.57 Nearly 10 liters of mucus are secreted into the gastrointestinal tract (GIT) each day.58 The continuous production and clearance by peristaltic forces and ciliary motion leads to quick turnover times in the range of 4–6 h in the rat GIT and approximately 10–20 min in the human nasal tract, respectively.20,59,60 Mucus is not only a barrier for foreign microorganisms but also provides the environment for commensal bacteria, native to the intestinal and nasal flora, which compete with microbes for attachment sites and nutrients.61 The normal commensal flora may exert an anti-infectious and anti-inflammatory influence and protect epithelial cells from toxins.62 Most of the native intestinal bacteria do not penetrate the mucus layer and occupy only the luminal surface (Fig. 2).63
000 times higher than the viscosity of water, however, under the shear stress present in GI and nasal tracts, a lubricating slippage plane is formed and the upper, luminal layers are removed much faster than the lower ones. At maximum physiological shear rates the viscosity of mucus approaches that of water making it an excellent low viscosity lubricant. Due to this shear-thinning behavior, a stable, unstirred layer is formed on the epithelial surfaces. The constant secretion, degradation, and shedding of the mucus makes it necessary for pathogens and drug-delivery agents to migrate “upstream”, markedly faster than mucus renewal and clearance takes place, in order to reach the epithelium.19,34 The different techniques to determine materials with mucoadhesive and/or mucopenetration properties have been recently reviewed.57 Nearly 10 liters of mucus are secreted into the gastrointestinal tract (GIT) each day.58 The continuous production and clearance by peristaltic forces and ciliary motion leads to quick turnover times in the range of 4–6 h in the rat GIT and approximately 10–20 min in the human nasal tract, respectively.20,59,60 Mucus is not only a barrier for foreign microorganisms but also provides the environment for commensal bacteria, native to the intestinal and nasal flora, which compete with microbes for attachment sites and nutrients.61 The normal commensal flora may exert an anti-infectious and anti-inflammatory influence and protect epithelial cells from toxins.62 Most of the native intestinal bacteria do not penetrate the mucus layer and occupy only the luminal surface (Fig. 2).63
2.2. The structure of airway and intestinal epithelial barriers
In general, epithelial cells are cells forming a protective monolayer on all internal and external surfaces of the human body—the epithelium. An epithelium is a continuous sheet of cells with junctional domains (tight junctions, TJs) between each cell, which forms a barrier as well as a membrane with defined permeability for drugs, nutrients, and pathogens.64 It prevents the uncontrolled passage into the host of partially digested food, bacteria and bacterial products as well as regulates fluid and electrolyte absorption and secretion. All epithelia are internally bounded by a basement membrane. Substances can cross the epithelium by three different processes. The first one is ion and small molecule movement through a cell accomplished by the differential distribution of membrane transporters/carriers on opposite sides of the cell. Second is the transport of larger, macromolecular cargo through cells within membrane-bounded carriers in the process called transcytosis. Paracellular transport (the movement between adjacent cells), regulated by TJs, is the third way to cross the epithelium.65 Because of their size, the particulate transepithelial drug carrier systems pass the barriers by the two latter mechanisms. Epithelial cells are considered the first line of defence against pathogens. They are able to provide immediate response and subsequent initiation of downstream immunological responses. These processes are mediated mostly by interaction of components of microbes with pattern recognition receptors (PRRs) located on epithelial cells. The most prominent ones are the Toll-like receptors that recognize components like bacterial lipopolysaccharides (LPS) of gram-negative bacteria, bacterial flagellin, microbial CpG (cytosine–phosphate–guanine) motifs of DNA, and lipoteichoic acids from gram-positive bacteria.66 An overview of the most important PRR classes is given by De Koker and co-workers.67The lung is composed of more than 40 different cell types, with approximately one-third being epithelial cells.73Their distribution in rat lung has been thoroughly investigated by Souma using electron microscopy techniques.74 The thickness of the lung epithelium decreases in the direction of the alveoli. In terminal bronchioles only a monolayer of cuboidal cells can be found, approximately half of which is ciliated. Squamous pneumocytes (type 1), agranular pneumocytes (type 2), and brush alveolar cells (type 3) constitute the absorptive, alveolar epithelium (Fig. 3).74 Type 1 cells are predominant (over 95%) and display squamous cytoplasmatic extensions. Type 2 cells are cuboidal, display blunt microvilli, and contain many organelles and multivesicular bodies. Type 3 cells are rarely found in humans and may be involved in water absorption, chemoreception, or endocrine function.64,75 Mucus-secreting cells as well as basal cells, which help in the adhesion of columnar cells to the basement membrane, are also present in the airway epithelium.76,77
 | ||
| Fig. 3 Electron microscopy image of a pair of distal brush cells with thick microvilli (0.3 μm in thickness). Magnification × 4,700. Reproduced from ref. 74 with kind permission from Springer Science. | ||
Among these complexes, three major groups can be distinguished: tight junctions (TJs), which firmly close the cleft between adjacent epithelial cells; adherent junctions, which stabilize intercellular contact; and desmosomes—localized spot-like adhesions (Fig. 4).15 The leaky paracellular tight junctions that can be found in jejunum and ileum are considered as the major site of drug absorption.79 The recent advances in research on protein structures and on factors influencing the regulation of the junctional complex permeability have been reviewed elsewhere.80–86 Nevertheless, the gut epithelium is not a perfect barrier, and exogenous pathogens may cross it through breaks between tight junctions, most probably at the villi tips, where the cells are shed. Furthermore, it was recently shown that respiratory, intestinal, and other epithelial cells express the crystallized fragment receptor (FcRn), which, apart from transporting immunoglobulin G (IgG) antibodies, can also be used as a target for vaccine candidates.87 Transport via FcRn is bidirectional and pH-dependent, activated at pH 6, deactivated at pH 7.4.88 Successful vaccination was achieved by coupling the Fc region of IgG2a to the HSV-2gD antigen and intranasal coadministration with the CpG oligonucleotide as an adjuvant. The vaccine completely protected wild-type, but not FcRn deficient mice after intravaginal challenge with virulent HSV-2 186 even after 6 months from inoculation.89 The FcRn-IgG transcellular transport pathway may provide a general delivery route, which avoids MALT for subunit vaccines against many mucosal pathogens.
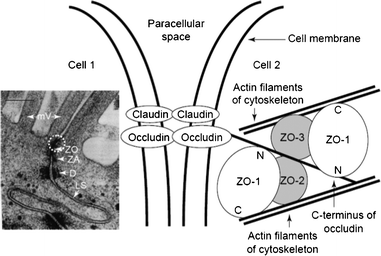 | ||
| Fig. 4 Electron micrograph of the intercellular junctional complex between two Caco-2 cells with circled TJ (left). Schematic representation of interactions among tight junction proteins (right). Abbreviations: D, desmosome; LS, lateral space; mV, microvilli; ZA, zonula adherens (adherens junction); ZO, zonula occludens (tight junction). Reproduced from ref. 79. | ||
2.3. The structure of MALT
Since the mucosal surfaces are the major site of pathogen entry, in particular the GIT and airways, an effective surveillance by the immune system of the mucosa is required. It is achieved by transport of complete antigens, for example, macromolecules and microorganisms through the epithelial barriers to the cells of the mucosal immune system. These cells are concentrated at specialized sites in so called mucosal lymphoid follicles where the cooperation between epithelial cells with antigen-presenting and lymphoid cells takes place.90 The presence of lymphoid follicles is the common feature of mucosa-associated lymphoid tissues (MALT). The structure and localization of MALTs within organisms have been extensively reviewed elsewhere.8,14,16,17,91–95 Mucosal tissues, those of the intestine in particular, contain more macrophages, plasma cells, and T-cells than any other lymphoid tissue of the entire immune system.96 MALT is subdivided according to anatomical regions to nasopharynx-associated lymphoid tissue (NALT), bronchus-associated lymphoid tissue (BALT), gut-associated lymphoid tissue (GALT), conjunctiva-associated lymphoid tissue, lacrimal duct-associated lymphoid tissue, larynx-associated lymphoid tissue, salivary duct-associated lymphoid tissue, skin-associated lymphoid tissue, vulvovaginal-associated lymphoid tissue, and rectal lymphoepithelial tissue.93,97,98 The distribution and composition of MALT varies with species, age, and tissue state (normal state or chronically inflamed). For example, NALT in mice is composed of organized lymphoid aggregates located in the palate at the entrance of nasopharyngeal duct and less organized structures throughout the nasal passages, whereas the human NALT comprises tonsils and adenoids (Waldeyer's ring).99 The development of mucosal and systemic tissues is, to a large extent, dependent on the previous exposure to mucosal microbiota.100 For instance, BALT is not present in all species as well as age groups and can be classified as a tertiary lymphoid organ. A hypothesis is proposed for a two-step vaccination protocol: first, BALT is induced and activated, and second, an antigen (the vaccine) is applied locally.101 The most thoroughly studied MALT is GALT, which comprises Peyer's patches (PPs), the appendix, and isolated lymphoid follicles (ILFs). Human PPs are about 3 mm in diameter, made of 5–200 aggregated lymphoid follicles, and located mainly in the distal ileum.94,102 Mucosal follicles of PPs consist of an assembly of naive B cells, often with a germinal center, supported by a network of follicular dendritic cells (DCs). ILFs, on the other hand, are similar in structure but much smaller (approx. 200 μm). At least 30000 ILFs are distributed throughout the human gut, distally increasing in density.103 PPs as well as ILFs are covered by follicle-associated epithelium (FAE) and separated from it by a subepithelial “dome” region that is rich in T and B lymphocytes and DCs (Fig. 1). Cells from the dome region are responsible for the cooperation between the immune system and mucosa. Furthermore, distinct populations of intraepithelial lymphocytes (IELs), T-cells mostly and DCs, are present at the basal surface of FAE cells. IELs are, most probably, responsible for immunosurveillance of epithelial cell alterations and regulation of epithelial cell viability.96 FAE shows significant differences from the villus epithelium. It contains few or no goblet and enteroendocrine cells, instead 10 to 20% of specialized epithelial M cells are present in the FAE.104
It produces lower amounts of mucus, defensins, and lysozymes, and it is unable to transport protective IgA from the interstitium to the lumen.105 These features facilitate the contact of antigens and pathogens with the FAE surface. It also expresses different glycosylation patterns and receptors, (such as β1-integrin and CD9, which bind to Arg-Gly-Asp (RGD) and Arg-Gly-Glu (RGE)) than the villus epithelium, which can be recognized by microorganisms.106–109 The M cells reside not only in the FAE but also in other parts of the intestinal tract such as the colon and rectum.110,111 They are not limited to GALT and are also present in NALT, BALT, and tonsils.112 M cells can be distinguished by the lack of surface microvili, a fewer number of cytoplasmic lysosomes, a greater number of mitochondria, and much thinner glycocalyx and mucus layer covering their surfaces.113,114 Their apical membrane has a microfold (or membranous) topography, hence the name M cell. Within their basolateral membrane, a deep invagination can be found. Inside this “pocket”, lymphocytes (including DCs) and some phagocytic cells are present.115 M cells transport antigens and microorganisms across the epithelial barrier by endo-, phago-, or pinocytosis and transcytosis to subsequently deliver them directly to the underlying immune system. Due to the presence of the invagination, the distance that the cargo has to cross is much shorter. Some specific PRRs, such as Toll-like receptor-4, platelet-activating factor receptor, α5β1 integrin, and α-L-fucose-specific lectin, are expressed on the surface of M cells.116–118 Since the M cells have high transcytotic activity and thin surface glycocalyx, particles and microorganisms can easily adhere to its apical membrane and be transported through a short distance to the pocket. Therefore, the M cells of the FAE remain the primary vehicles for antigen sampling and their introduction to the immune system; they are very attractive for targeted drug/vaccine delivery.119,120 Apart from M cells, the DCs also exhibit the ability to sample the antigens from the lumen, outside of organized MALT, and present them to the lymphocytes. DCs are distributed along the entire intestinal epithelium as well as in airways.121–124 They belong to the group of antigen presenting cells (APCs), which are able to uptake, transport, and process antigens, and to present them to T-cells.125–127 There are plenty of specialized DC subtypes that differ in the differentiation state, location, migratory pathways, detailed immunological function, and dependence on infections or inflammatory stimuli for their generation.128 CX3CR1-positive lamina propria DCs express TJ proteins, open the TJs between epithelial cells, send dendrites outside the epithelium, and can take up microorganisms directly while preserving the integrity of the epithelial barrier (Fig. 5).129,130 Hence, targeting DC receptors with particulate drug/vaccine formulations may offer an alternative route of crossing the epithelium.131–134 One especially attractive group of these receptors are C-type lectins (CLRs), which bind to specific carbohydrate epitopes.135–137 Within CLRs the mannose receptor (DC-SIGN, CD209), which has the ability to recognize mannose type saccharides, present on viruses, bacteria, and fungi, has attracted most of researcher's attention.138–142 DC-SIGN interacts with HIV envelope glycoprotein gp120, therefore, by blocking this receptor with mannosylated compounds, it is possible to inhibit the binding of the virus to CD4+ T-cells.143–145
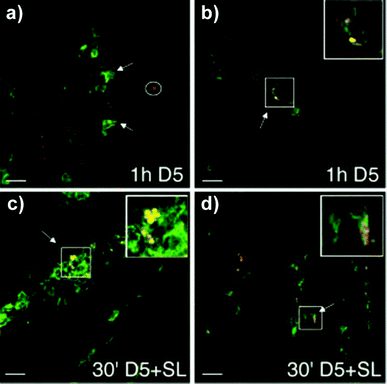 | ||
| Fig. 5 CLSM images of cryosections (7 μm), fixed with 1% PFA and immunostained for DCs (green) and E. coli (red). (a) DCs creeping between epithelial cells (see arrows) in search of E. coli D5 (red, circled) and (b) internalizing bacteria (see arrow and inset, which is magnified ×2), which are stained yellow due to the colocalization. (c–d) Coinfection of E. coli (D5) with S. typhimurium (SL) increases the number of recruited DCs and accelerates the process and magnitude of bacterial internalization. (c) Cell full of bacteria (see arrow indicate and inset, which is magnified ×2). (d) DC dendrite outside the epithelium during the process of internalization. Reproduced from ref. 129 with permission from Macmillan Publishers Ltd. | ||
3. Particulate functional polymers for transepithelial drug delivery
The multitude of materials studied for oral, nasal, and pulmonary drug delivery can be divided into two groups, namely, the ones naturally occurring or derived from natural sources as well as the artificially produced ones.146,147 The nature-originating materials include polysaccharides and the derivatives thereof (chitosan, alginates, dextran, cellulose, starch, pullulan, poly(sialic acid), chondroitin, mannans, and lipopolysaccharides) as well as poly(amino acids), like poly(γ-glutamic) acid and poly(L-lysine), among others.148,149 On the other hand, synthetic polymers employed in the investigations of mucosa-targeted drug delivery systems comprise poly(D/L-lactide), poly(D,L-lactide-co-glycolide), poly(acrylic acid) and its derivatives (carbomers, polycarbophil), polyethers (poly(ethylene oxide), poloxamers), polystyrene (latex beads), poly(meth)acrylate-based copolymers (e.g., Eudragit®), poly(acrylamides) (e.g., N-(2-hydroxypropyl) methacrylamide copolymers), poly(caprolactones), poly(ethylene imines), and a number of others.150 All the materials mentioned above differ in their chemical structure, charges, hydro-\lipophilicity, and biodegradability. Therefore, they form a plethora of particulate systems with varying sizes and zeta potentials, and thus, different biological activities and pharmacokinetics as well as biocompatibility.151,152 Furthermore, in order to obtain targeted delivery of the drug after non-parenteral administration to and through the airway or gastrointestinal mucosal surfaces, not only the physical properties of the particles, that govern their stability/degradation in gastric/intestinal fluid and penetration through mucus, but also the molecular structural features of their surface have to be adjusted for the specific uptake or transport through the epithelium.10,153–156 Since the mucosal surfaces serve as the most common route of pathogen entry, it is logical that by employing structural features, presented on their exterior, precise targeting may be achieved.113,149 M-cells, DCs, and enterocytes possess specific receptors either for recognition of pathogens or nutrient uptake. Therefore, a variety of targeting ligands may be applied to the particle surface to direct the drug formulation to a distinct cell population and enhance its bioavailability. The most commonly targeted receptors are the C-type lectins, carbohydrate-binding proteins that recognize the oligosaccharidic motifs of the glycosylated proteins on the cell surface, and PRRs.138,141,157–159 Moreover, MALT, described in Section 2.3., provides an additional way of entry for particles, and the efficiency of transepithelial vaccinations with particulate systems is well-documented and reviewed.6,8,67 Furthermore, the particulated drug delivery formulations offer some additional advantages. They protect the drug or antigen from destructive pH, enzymatic activities, and offer targeting to a specific cell type. Additionally, more components may be included inside the particle, for example, adjuvant substances that will enhance the response of the organism to the administered drug.160–162Selected examples of functionalized materials, their characterization, administration, and evaluation of bioactivity in in vitro, ex vivo as well as in vivo models are presented herein. The formulations applied in targeted transepithelial drug delivery, from the current literature, are highlighted starting with the most abundantly used polymer class of polyester-derived particulate materials.
3.1 Poly(ε-caprolactone) (PCL), poly(D,L-lactide) (PLA), and poly(D,L-lactide-co-glycolide) (PLGA)
These three polymers belong to the group of FDA-approved “Generally Recognized as Safe” (G.R.A.S.) biodegradable polyesters. As a consequence, they are the most commonly studied materials for controlled drug delivery.163 All can be synthesized by ring opening polymerization (ROP) employing various initiator and solvent systems (Scheme 1). Their biodegradability, crucial for sustained release of drugs, and hydrophilicity are dependent on the degree of crystallinity and molar mass of the (co)polymer as well as on the monomer ratios. Furthermore, for the drug release profiles (burst or sustained) from particulate drug carriers, not only do the polymerization conditions and the resulting structures play a key role, but also the processing parameters, thus, particle size, surface, and inner porosities.164 Methods of PCL synthesis either by polycondensation of 6-hydroxyhexanoic acid or ROP of ε-caprolactone, employing various initiator/catalyst and solvent systems as well as the properties of the resulting material have been thoroughly reviewed.165 Highly significant is the ROP of ε-caprolactone by an activated monomer mechanism using poly(ethylene glycol) (PEG) as macroinitiator.166 Block copolymers obtained by this method offer the possibility to modulate the degradability, biocompatibility, and hydrophilicity of the material. PCL is enzymatically degradable in the environment but non-enzymatically in the body.163 Due to slow degradation within organisms, its particles have found applications in long-term delivery systems, including transdermal and transmucosal, for a multitude of drugs.163,167 The methods for preparation of PCL-based micro- and nanospherical drug carriers include oil/water (o/w) emulsion solvent extraction/evaporation, water/oil/water (w/o/w) emulsion solvent evaporation, spray drying, solution-enhanced dispersion by supercritical fluids, and hot melt techniques.167 Some advantages of PCL include high permeability to small drug molecules, exceptional ability to form blends with other polymers, no generation of an acidic environment during degradation as compared to PLA and PLGA, therefore, sustained drug release from PCL, which can extend even to a period of more than one year.168 | ||
| Scheme 1 Schematic representation of the syntheses of PCL, PLA, and PLGA. A complete list of catalysts/initiators used in ROP of ε-caprolactone can be found in ref. 165. | ||
PLA and its glycolide copolymer PLGA (also known under the commercial name of Resomers®) are the most extensively studied materials for formation of particulate (nano- and microspheres) drug delivery systems.169 The synthesis, self-assembly and some biomedical applications of PLA-based amphiphilic block copolymers have been recently reviewed.170 They are produced by ROP of the respective cyclic diesters of lactic and glycolic acids. Both of these monomers are obtained by esterification and catalytic cyclization of naturally occurring acids, namely lactic acid—from bacterial fermentation of corn starch and sugar cane, as well as glycolic acid—isolated from sugar beets, grapes, pineapples, among other sources. The main advantage of PLA and PLGA is their decomposition by hydrolysis of ester bonds to compounds naturally present in the organism and subsequent resorption with removal of the monomers via metabolic pathways. The polymerization conditions, ratios of comonomers as well as D- to L-lactide and types of initiator influence the biodegradability of these polymers. They also affect the drug release profile and the in vivo performance, as was shown for the oral delivery of estradiol to rats.171 The higher the glycolide content the faster the degradation of PLGA.172 Furthermore, ester-endcapped polymers (more hydrophobic) and those containing higher amounts of pure D- or L-lactide (more crystalline) degrade more slowly.173,174 The drug encapsulation techniques as well as the preparation of PLA/PLGA-based micro- and nanoparticles have been extensively studied and reviewed.149,175–177 Moreover, the processing parameters of PLGA can be adjusted to obtain particles of desired sizes, e.g. for pulmonary delivery.178 Aspiration of PLGA microspheres was found to be safe over 90 days of monitoring and caused no histological abnormalities in internal organs of hamsters.179 In addition, it has been extensively used as an anticancer drug carrier.180 Since the contact with mucus and underlying epithelia is mediated by the vehicle surface, the selected examples of polyester-based particles modified with biomimetic ligands for transepithelial drug delivery are presented within this chapter and summarized in Table 3. Furthermore, for the reader's convenience, the PCL, PLA and PLGA materials have been divided by their application and route of administration.
| Polymer | Surface modifier | Cargo | Cells studieda | Administration method | Studies | Reference |
|---|---|---|---|---|---|---|
| a AM: alveolar macrophage, DC: dendritic cell, MΦ: macrophage, Calu-3: human airway epithelial cell, A549: human alveolar basal epithelial adenocarcinoma cell, J774A.1: mouse monocyte macrophage, Caco-2: human epithelial colorectal adenocarcinoma cell, Raji: B- cell lymphoma line. | ||||||
| Resomer® RG755 | PVA | Rhodamine B (Rho) | AM | In vitro | Model and influence of particle: AM ratio on uptake | 184 |
| Resomers®: RG755, RG503, RG503H | PVA | Rho, BSA, IgY | AM | In vitro | Polymer and PVA influence on encapsulation and uptake | 186 |
| Resomer® RG755 | PVA | Rifampicin (RFP) | AM | In vitro, inhalation to rat lungs, intratracheal to guinea pigs | Size dependence of uptake, eradication of Mycobacterium tuberculosis | 191,195,196 |
| Resomer® RG503H | WGA, GRGDSPK, α-D-Man, BSA, PLL, GRADSPK | 6-Coumarin | AM | In vitro | Influence of targeting ligands on specific uptake | 197 |
| PLGA- (75:2, 90-126 kDa) |
Dipalmitoylphosphatidylcholine, poloxamer 338 | Rho | AM | In vitro | Reduction of uptake by particle coating | 198 |
| PLGA | PLL-g-PEG-RGD, PLL-g-PEG-RDG | 6-Coumarin | AM, DC | In vitro | Ligand specific targeting of MS to MΦs and DCs | 199 |
| PLLA | PVA | Tobramycin | — | Endotracheal to rat lung | Avoided clearance, sustained delivery | 200 |
| PLGA | Mannitol | RFP, indocyanine green | — | Intratracheal to rat lungs | Dispersion and retention of NPs in lungs, higher RFP uptake | 202 |
| PLGA | PVA | BSA, fluorescein Na salt | Calu-3 | In vitro | Development of lung mucosa model | 203 |
| PEG-PLA | stearylamine, Solutol® HS 15 | — | A549 | Endotracheal to mice lung | Influence of charge on toxicity of particles | 204 |
| PLGA | Chitosan | Elcatonin | — | Nebulized to guinea pig lungs via trachea | Adherence of coated particles to mucus, improvement of drug activity | 205 |
| PLGA | PVA | Insulin | — | Nebulized to guinea pig lungs via trachea | Prolongation of hypoglycemic effect | 210 |
| PLGA | Chitosan | RFP | A549 | In vitro | Nebulization efficiency mucoadhesive properties, cytotoxicity | 211 |
| PLGA | PVA, chitosan, PEG | GFP plasmid | A549 | Intranasal to mice | Transfection efficiency and toxicity | 213 |
| PLGA | CS-grafted PLA | — | Calu-3 | In vitro | Internalization within chlamydial inclusions of infected cells | 214 |
| PLA | PEG, CS | Tetanus toxoid | — | Oral and intranasal to mice | Facilitation of transport by PEGylation | 215 |
| PLGA | CS | HBsAg | — | Intranasal to mice | Elucidation of systemic, mucosal, cellular responses | 222 |
| PLGA | PVA | S. equi antigens | — | Intranasal to mice | Full protection against S. equi infection | 223 |
| PLA | PVA, GCS, alginate | S. equi antigens | J774A.1 | Intranasal to mice | Adsorption of antigens, mucosal, humoral and cellular immune responses | 227 |
| PLGA | Bacterial lipopoly-saccharide | West Nile virus envelope protein, OVA | MΦ, DCs | Subcutaneous, intranasal and oral to mice | High uptake by DCs, inflammasome pathway in MΦ, protection from infection | 232 |
| PLGA | PVA, poloxamer, Clostridium perfringens enterotoxin C-terminal peptide | Rho | — | Intranasal and oral to mice | Enhancement of uptake by M-cells, more significant in PPs than in NALT | 239 |
| Resomer® RG503H | WGA, BSA | Fluorescein cadaverine | Caco-2 | In vitro, vertical and horizontal setup | Highest association to cells of WGA-modified particles | 241 |
| PLGA | Biot-PEG-NH2 | FITC-NAv | Caco-2 A549 | In vitro | Strategy for attachment of various functional groups | 243 |
| PEG-PCL | RGD | OVA | Caco-2 Raji B coculture | In vitro model of human FAE, oral to mice | Photografting of the lectin significantly increased transport through the FAE model and number of mice producing IgG | 245,246 |
| PEG-PCL and PLGA | RGD, RGDp, LDVd, LDVp, Man | OVA | J774, Caco-2 Raji B coculture | Intraduodenal, oral to mice | Internalization after 1 h of incubation, uptake three times higher of LDVp and carbohydrate-carrying NPs, compared to non-targeted | 247 |
| PLGA, PDLA, PLLA | — | 6-Coumarin | — | Oral to mice | Evaluation of the particle absorption by Peyer's patches | 248 |
| PLGA | Eudragit® L100-55 CMEC | OVA | — | Oral to mice | Particle stability, drug loading efficiency, and immunogenicity | 251 |
| PLGA | Chitosan-4-thiobutylamine | Curcumin | — | Freshly excised porcine intestine | Residence time on the mucosa, improvement of the release profile | 252 |
| PLGA | Chitosan | Elcatonin | Intragastric to non-fasted rats | Sustained release, reduction of blood Ca2+ levels | 209 | |
| PCL | Eudragit®RS | Aspart-insulin | Oral to diabetic rats | Decrease of fasted glycemia, improvement of the glycemic response to glucose | 253 | |
:25 and a molar mass of 15 kDa as estimated from viscosity measurements. Oil/water emulsion and solvent extraction methods, using poly(vinyl alcohol) (PVA) as the emulsifier, were employed to obtain particles with sizes below 5 μm, 72%, and in the range from 5 to 10 μm, 27% (DLS). Since the particle geometry influences the process of phagocytosis,185 the microspheres were further analyzed by scanning electron microscopy (SEM). The particles were spherical and had a smooth surface (Fig. 6). In order to evaluate the cellular uptake by means of fluorescence microscopy and flow cytometry, rhodamine B (Rho) base was included in the organic phase during the particle preparation. Furthermore, to establish a reliable in vitro model of phagocytic activity, the influence of cryopreservation, interanimal variability as well as culture conditions, i.e. adherent and in suspension, were investigated. It was shown that the intensity of phagocytosis was dependent on the duration of incubation, and a microsphere:AM ratio of 1:1 yields the highest efficiency. The flow cytometric analysis of the fresh and cryopreserved AM showed similar phagocytic activities, however, these results were not in agreement with the microscopic evaluation. This discrepancy might result from different culture treatment for both investigations. For microscopic analysis, AM were adherent cells, whereas for flow cytometry the AMs were maintained in suspension. In this study, the AMs tended to engulf an increasing number of MS during incubation without any discrimination of the size. In addition, the results of flow cytometry analysis yield a correlation between AM population with increased granularity and fluorescent events, therefore, showing that the investigation of phagocytosis with non-fluorescent, antigen-loaded MS is also possible. This method was employed to investigate the influence of the PLGA hydrophilicity, the residual PVA content, and the nature of encapsulated proteins on the MS phagocytosis efficiency.186 To obtain the protein-loaded particles, the w/o/w method was used, and SDS-PAGE of the extracted BSA and IgY confirmed their integrity after the encapsulation procedure. The obtained microspheres varied in sizes from 3.0 to 18.7 μm (DLS), nevertheless, all are potential candidates for phagocytosis, since AM measure up to 30 μm.187 Thus, murine bone marrow-derived macrophages were shown to ingest particles 1.44 times their diameter or 3 times their volume.187,188 Similar hydrodynamic diameters (avg. 6.0 μm) were obtained from RG755-encapsulated Rho using various concentrations (0.01–1%) of PVA. No influence of the encapsulated protein on the particle size (avg. 9.5 μm) was observed for more hydrophobic, ester-terminated polymers, i.e. RG755 and RG503 (50:50, 9 kDa). On the other hand, IgY-loaded MS prepared from hydrophilic RG503H (50:50, 9 kDa), carrying a free carboxylic acid endgroup, were larger (avg. 12.0 μm). The IgY-MS were more porous and less spherical than the rhodamine-loaded MS, and the presence of pores increased with the hydrophilicity of the polymer. RG503H was extensively porous. The BSA-carrying MS were spherical with fewer pores on their surfaces. The entrapment efficiencies for the more hydrophobic polymers were higher for IgY-MS (95–99%) than for BSA-loaded ones (57–63%). When RG503H was used for the encapsulation of the antibody, the efficiency significantly decreased to 35 ± 3%, showing that the hydrophilicity of the polymer represents an important factor in this process. All BSA-MS exhibited high burst release within the first hour of approximately 50% without any further significant release until 350 h. The release of IgY was dependent on the hydrophilicity of the polymer and the porosity of particles, being the highest with the initial burst of 25% for RG503H and slowly progressing for RG755 and RG503 (up to ca. 12% at 350 h). The zeta potentials (ζ) of the obtained MS, measured in the presence of fetal bovine serum, were all negative and varied between −3 mV for rhodamine-loaded RG755 and −12 mV for IgY-RG503. However, no significant influence of ζ on the phagocytosis was found, which was in good correlation with previous results, where similar activities were found for positively (+20 mV) and negatively (−20 mV) charged microspheres.189 The influence of residual PVA on phagocytosis was investigated by flow cytometry using fluorescent RG755 MS. The highest phagocytic activity was obtained for the particles prepared with the lowest surfactant content. Higher phagocytosis intensity was observed for the IgY-loaded particles compared to BSA, independent of the materials used. This fact may be explained by partial adsorption of the antigen on the surface of the MS, therefore, opsonization and promotion of receptor-mediated uptake of the particles.188,190 Furthermore, the phagocytosis of IgY-MS increased with the hydrophobicity of the employed polymer in the order RG755 > RG503 > RG503H. It was concluded that ligand-mediated particle uptake should be able to surpass the negative effect of hydrophilicity, thus, surface functionalization with targeting moieties plays a crucial role.
 | ||
| Fig. 6 Left: SEM image of Rho-loaded PLGA microspheres (scale bar 10 μm); right: optical micrograph obtained after cell sorting of alveolar macrophages, which have phagocytosed numerous PLGA microspheres. Reproduced from ref. 184 with permission from Wiley. | ||
Since phagocytosis may also be affected by the particle size, the anti-tuberculosis drug rifampicin (RFP) was encapsulated in PLGA (75:25, 10 kDa) to yield MS of different sizes (1–10 μm).191 The particles were prepared by the o/w method using 2% (w/v) PVA as surfactant. Microspheres having volume-averaged diameters (DLS) of 1.47 μm, 3.31 μm, 6.06 μm, and 10.03 μm were obtained by the membrane emulsification technique, i.e., by injection of PLGA and RFP, dissolved in dichloromethane, into a PVA-containing aqueous phase through porous glass filters with pore sizes of 0.48, 1.00, 1.95, and 3.63 μm, respectively. The obtained encapsulation efficiencies of 59% and size-dependent drug release rates were similar to the previously reported ones.192 Furthermore, the particle sizes of 1 to 10 μm, obtained in this study, are considered as favorable for phagocytosis by AM, as observed for the poly(L-lactide)-based microparticles.193 Moreover, microspheres with aerodynamic diameters of 1 to 5 μm are respirable and suitable for delivery to the periphery airway surfaces of the lung.194 Subsequently, phagocytosis was determined as a function of the added particle number per AM, and the number of phagocytosed MS was counted by light microscopy after incubation for 4 h. The amount of 1 and 3 μm particles taken up by AM gradually increased up to a MS:AM ratio of 3:1. A significant increase in the uptake at higher ratios was observed with 10 μm particles being less phagocytosed than the smaller ones, and it was concluded that the 3 μm particles are the most suitable for efficient delivery of the drug by phagocytosis. Furthermore, the RFP-loaded PLGA MS were effective in eradication of Mycobacterium tuberculosis (MTB) in AM cells after pulmonary inhalation to rat lungs and intratracheal administration to guinea pig lungs.195,196
In another study, 6-coumarin-loaded, spherical particles were prepared from Resomer® RG503H using the spray drying method.197 The average volume diameter of 2.5 μm and large size distribution of 0.6–15.4 μm were obtained from DLS analysis and confirmed by SEM measurements. The particles were further functionalized with different ligands, namely wheat germ agglutinin (WGA), GRGDSPK (Gly-Arg-Gly-Asp-Ser-Pro-Lys), and α-D-mannose-PEG3-NH2 as well as non-targeting molecules: BSA, poly(L-lysine) (PLL), GRADSPK (Gly-Arg-Ala-Asp-Ser-Pro-Lys) by covalent conjugation to the free carboxyl groups on the MS surface applying a carbodiimide method. The grafting efficiencies were quantified by measuring the residual content of the ligand in the reaction medium. The covalent attachment, not electrostatic or hydrophobic adhesion, of WGA and BSA to the surfaces was confirmed by CLSM imaging, using fluorescent derivatives of the proteins (Fig. 7). The grafting efficiencies were dependent on the molar mass of the ligands and the non-targeting molecules, with the highest (880 pmol mg−1) for the mannosylated ligand, medium for RGD- and RAD-containing peptides (220 and 230 pmol g−1, respectively), and the lowest for proteins (43 pmol mg−1). For PLL, the yields were not estimated but its successful grafting was confirmed by the increase of ζ from −81 mV for RG503H particles to +34 mV. The particles with other functionalities exhibited higher ζ than the unfunctionalized ones, ranging from −26 (mannosylated) to −51 mV (RAD). Furthermore, atomic force microscopy (AFM) was used to analyze the MS. It revealed the presence of a discontinuous monomolecular layer on the surface of the WGA-grafted particles. Due to high polydispersity of the obtained particles, which can influence the results of fluorescence intensity measurements (e.g. by FACS), CLSM was employed for the quantification of the cellular uptake of the MS by porcine AM. Moreover, application of this method allowed discrimination between particles, internalized or attached on the AM surface. Since AMs do not possess receptors for BSA and RAD, the MS/AM ratio-dependent uptake of particles with these moieties did not differ significantly from the unmodified PLGA microparticles and involved only non-specific mechanisms. Much higher internalization was observed for the microspheres grafted with ligands. At an MS:AM ratio of 20, the cellular uptake of the mannose-, mannitol-, and RGD-carrying particles was around four, three, and two times higher than for the ungrafted MS, respectively. The positively charged PLL-functionalized particles, which directly interact with cellular membranes, were taken up to a significantly larger degree than the uncoated ones. The uptake linearly increased with the MS/AM ratio, whereas, for the ligand-carrying particles saturation was observed. These results can be attributed to non-linear, saturable, specific mechanisms of the internalization, e.g. receptor-mediated phagocytosis. In order to further investigate this process, the experiments were carried out under different conditions, namely in the presence of free ligand, at 4 °C, or with cytochalasin D, which inhibits the actin-dependent phagocytosis. The specific and non-specific uptake varied according to the ligands. At higher MS/AM ratios, the non-specific interactions were dominant, with exception of the RGD-grafted spheres. At the lower MS/AM ratio, corresponding to the in vivo situation, the specific uptake contribution dominated. These results demonstrate that the ligand grafting strategy is suitable for improving the cellular uptake of micron-sized particles.
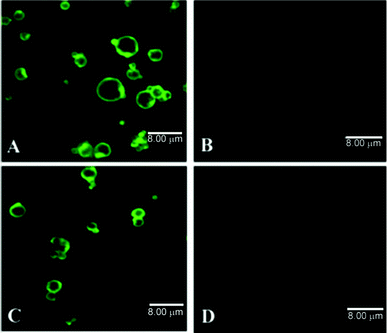 | ||
| Fig. 7 CLSM images of ligand-grafted PLGA microspheres: (A) WGA-FITC and (C) BSA-FITC grafted microspheres (B) and (D) ungrafted microspheres after incubation with proteins without COOH activation. Reproduced from ref. 197. | ||
On the other hand, the particulate drug delivery systems designed for prolonged retention within the peripheral lung must avoid sequestering and loss of bioavailability inside the AM. Therefore, the inhibition of phagocytosis of PLGA- (75:25, 90–126 kDa) based particles (2 to 3 μm in diameter) by coating was investigated.198 The fluorescent, rhodamine B-loaded particles were prepared by an o/w solvent evaporation method and coated with either dipalmitoylphosphatidylcholine (DPPC) or poloxamer 338 (3,300 Da, 80% poly(oxyethylene) content) by simple incubation in aqueous dispersions containing the coating material. The presence of the adsorbates was confirmed by FTIR spectroscopy. FACS was employed to assay the internalization of the particles by primary cultures of rat peritoneal MΦ (PM) and AM as well as by a continuous rat AM culture (NR8383). Coating with DPPC induced strong inhibition of phagocytosis by both AM types, however, less pronounced in the PMs. The poloxamer-coated PLGA particles did not reduce the uptake by NR8383, nevertheless, they reduced the number of MS contained in primary AM but not the percentage of phagocytic cells. As for the PM, the polyether decreased the phagocytic activity up to a MS:AM ratio of 5. In another study, nonspecific phagocytosis of PLL-g-PEG-coated, carboxylated polystyrene (PS) or biodegradable PLGA microspheres was evaluated and compared with ligand specific interactions using PLL-g-PEG-RGD and an inactive ligand conjugate, PLL-g-PEG-RDG.199 The polyether coating significantly reduced the nonspecific phagocytic activity of human blood-derived MΦs towards the microspheres in serum (Fig. 8) and acted as a repellent for these cells. On the other hand, the phagocytosis and the uptake by DCs were significantly enhanced by the presence of RGD which was abolished when using the inactive ligand conjugate—RDG. It was concluded that coatings of PLL-g-PEG-ligand conjugates provide a method for ligand specific targeting of microspheres to these MΦs and DCs while reducing nonspecific phagocytosis.
 | ||
| Fig. 8 Phagocytosis of noncoated (a) and PLL-g-PEG-coated (b) PLGA MS in macrophages as analyzed by CLSM. The outline of the individual cells was visualized by staining the actin cytoskeleton with rhodamine-phalloidin (red). MS were labeled with 6-coumarin (green). Optical sections (xy, left panel) and yz-projections (right panel) allow extracellular and internalized microspheres to be distinguished from each other. Reproduced from ref. 199 with permission from Elsevier. | ||
It has been reported for polystyrene-based materials that nanoparticles, unlike microparticles, are barely taken up by AM.201 Therefore, a system of NPs encapsulated inside MS was developed.202 By using a four-nozzle spray drier, mannitol (MAN) microparticles containing rifampicin (RFP) or indocyanine green (ICG)-loaded PLGA (75:25, 14.4 kDa) nanoparticles were prepared in one step. The mean diameters (DLS) of RFP-PLGA and ICG-PLGA nanoparticles were 213 nm and 200 nm, respectively. These particles were dispersed in MAN, forming MS with mean diameters of 3.2 μm for (RFP-PLGA)MAN and 2.0 μm for (ICG-PLGA)MAN. The rifampicin-carrying microspheres have shown high in vitro aerosol performance and in vivo uptake by AM in rat lungs after intratracheal administration. The uptake of the drug from (RFP-PLGA)MAN particles was larger than for RFP-MAN and RFP-PLGA (2.1 μm) MS. Furthermore, the in vivo dispersion of the micro- and nanoparticles in the rat lungs was investigated using ICG as a fluorescent marker. The 2.0 μm ICG-PLGA tended to localize in the direction of the trachea 1 h after administration, suggesting rapid excretion from the lungs due to mucociliary clearance. On the other hand, the fluorescence of (ICG-PLGA)MAN of similar sizes was widely observed in the lungs for at least 12 h. It was suggested that the mannitol coating dissolved in the lungs and released the PLGA NPs, which were subsequently dispersed and deposited, avoiding the clearance mechanisms (Fig. 9). Thus, the RFP-PLGA nanoparticles were retained in the lungs, phagocytosed by AM, and the uptake of the drug was increased.
 | ||
| Fig. 9 In vivo fluorescent images of lungs of rats after administration of ICG-PLGA and (ICG-PLGA)MAN microspheres. Reproduced from ref. 202 with kind permission from Springer Science. | ||
Highly porous particles, consisting of BSA and the sodium salt of fluorescein coencapsulated in PLGA (64 kDa), were applied in the development of a physiologically-relevant model of the pulmonary epithelial barrier based on Calu-3 human bronchial epithelial cells.203 The microparticles, having 9.9 μm hydrodynamic and 6.4 μm aerodynamic diameters, were aerosolized onto the cell monolayers, which were grown on permeable filter inserts under air-interfaced culture (AIC) or liquid-covered culture (LCC) conditions. The cells exhibited similar morphological features, with microvilli, under SEM analysis and regularly expressed TJs as well as adherens junction proteins along the intercellular interfaces. However, a dense mucus coating was found on AIC cultured Calu-3 but not on LCC grown monolayers, which may protect the cell surface during particle impinging. Moreover, a significant decrease in the integrity of the LCC grown monolayer as compared to the AIC cultured one was observed by trans-epithelial electrical resistance (TEER) measurement upon the removal of the apical medium prior to impingement. Nevertheless, after day 13, TEER in both culture conditions remained similar. The models exhibited significant differences in the permeability towards impinged PLGA microparticles. After the deposition, monolayers grown under LCC conditions showed a considerable increase in particle flux, correlated to a decrease in TEER of approximately 1600 Ω cm2, compared to the control (no impingement) and air-impinged cultures. In contrast, monolayers grown under AIC exhibited no significant change in permeability or TEER values. It was concluded that Calu-3 cells cultured under air-interfaced conditions provide more accurate in vitro data for the transport of aerosolized particles through the tracheo-bronchial region of airway epithelium, since they possess a uniform mucus layer on their surface. However, this model does not include the in vivo mucociliary clearance mechanisms, thus, shares the common limitation of the other cell culture models.
In another study, the safety and tolerability of differently-charged nanoparticles designed for local pulmonary delivery was investigated with particles prepared from PEG-PLA.204 In order to obtain cationic NPs, stearylamine was added to the oil phase during the particle formation process. A typical positively charged formulation consisted of PEG-PLA Mw = 100 kDa (3% w/w), Solutol®HS 15 (1% w/w), stearylamine (0.2% w/w), and Tween 80 (1% w/w) in double-distilled water. The anionic particles were prepared in the same way but without the cationic lipid. The average diameters of the negatively (−31 mV) and positively (32 mV) charged nanoparticles were 129.3 nm and 141.3 nm, respectively. Firstly, the in vitro cytotoxicity towards the human lung adenocarcinoma cell line A549 was evaluated (MTT assay). PEG-PLA NPs irrespective of their surface charge showed no adverse effects on the cell viability. The aerosolized particulate formulations were administered by endotracheal instillation to the lungs of female BALB/c mice. The particle safety was evaluated following 5 daily deliveries. On the 8th (D8) and 9th (D9) day, the animals were sacrificed and analyzed. As a result, local pulmonary response of the immune system to the application of cationic NPs was prolonged and involved AMs and lymphocytes. In contrast, the anionic particles induced moderate and reversible response that involved AMs to a lesser extent. It was established that the positively charged PEG-PLA NPs increased lung side effects, caused transient systemic toxicity, mainly on white blood cells, and are not recommended for repeated pulmonary instillation. On the other hand, the anionic particles can be considered as drug carriers for frequent aspirations. Nevertheless, positively-charged, chitosan (CS)-coated PLGA nanoparticles were evaluated for pulmonary delivery of a peptide drug, which reduces the blood Ca2+ levels, i.e. elcatonin.99,205 It was demonstrated that CS exhibits desirable properties for the improvement of drug absorption through epithelia, like the protection of the drug against enzymatic degradation, enhancement of mucoadhesion, and opening of the intercellular TJs in the GIT and nasal mucosa, which is thoroughly reviewed in several publications.206–208 Therefore, peptide-loaded PLGA (75:25, 20 kDa) nanospheres were prepared by the emulsion solvent diffusion method in oil.209 Subsequently, the particles were incubated in mixed solutions of 1% PVA and 1% CS (150 kDa) for approximately 5 min, centrifuged, washed two times to remove unencapsulated drug, and freeze-dried. This procedure yielded NPs of ca. 650 nm average diameter with chitosan coating, as confirmed by the change of ζ at pH 4.4 from negative (−3.7 mV) for pure PLGA particles to positive (21.2 mV). It was shown that the CS modification had no influence on the particle diameter, dispersibility, and satisfactory inhalation properties as well as drug release behavior of the nanospheres. After initial burst release of approx. 30%, the liberation of elcatonin was sustained for more than 2 weeks in 0.2 M KH2PO4–0.2 M NaOH, pH 6.8, and reached 60% at that time. Suspensions of CS-coated PLGA NPs were administered to guinea pig lungs via the trachea using a bath-type ultrasonic nebulizer, as reported previously for the insulin-loaded PLGA-based particles.210 Subsequently, the nanosphere retention in lungs was evaluated using pyrene-loaded NPs. The CS-modified particles could adhere to mucus and the epithelial cells of lungs and trachea, thus, slowing the elimination mechanisms, as compared to the unmodified PLGA-nanospheres, which were removed about three times faster. Moreover, the pharmacological activity of elcatonin was significantly improved when the drug was administered to the lungs after encapsulation in CS-coated PLGA particles. A considerably prolonged reduction in blood Ca2+ levels appeared for over 24 h in comparison to the free drug solution and the unmodified NPs.205 Another study evaluated rifampicin (RFP) loaded PLGA, chitosan, and CS-coated PLGA microspheres for alveolar delivery.211 The particles were prepared by emulsion or precipitation techniques. The obtained microparticles had mean diameters ranging between 1.47 μm and 2.9 μm with narrow size distributions (0.16–0.57). The ζs varied from −4.8 mV for pure PLGA (75:25, 85.2 kDa) to 37.0 mV for CS (medium molar mass, 87% deacetylated) and PLGA particles containing 0.75% (w/v) CS. The RFP encapsulation efficiencies (EEs) ranged between 15% and 86% of the drug that was initially added to the formulation. For pure CS particles the EE decreased with increasing concentration of the polysaccharide possibly due to higher viscosities of the more concentrated solutions.212 On the other hand, pure PLGA microspheres encapsulated RFP with higher efficiencies of around 60%; for the polyester and polysaccharide mixtures, they were increasing linearly with the amount of added CS, reaching a value of 86% at 0.75% (w/v) of polysaccharide, similarly to the results obtained by others.194 Furthermore, the nebulization efficiency (NE), immediately after particle preparation and removal of the unencapsulated drug as well as the stability during freeze-drying or/and nebulization were evaluated. For all the microparticles no negative influence of the freeze-drying process was observed. The CS-coated PLGA particles (with encapsulated RFP, with 0.50 or 0.75% polysaccharide) exhibited the highest NE and stability as well as good mucoadhesive properties and a comparably low cytotoxicity. Therefore, a chitosan coating represents a considerable advantage for many therapeutic applications. Furthermore, it was used to enhance the gene delivery capability of PLGA particles.213 Since the NPs consisting of pure polyester exhibit a negative charge which limits their transfection efficiencies (DNA complexation) as well as transport through mucosal barriers, the nanospheres were modified with cationic CS. An emulsion-diffusion-evaporation technique using a PVA–chitosan or a PVA–chitosan–PEG blend as stabilizers was employed to obtain particles with hydrodynamic diameters below 200 nm in a reproducible fashion. The PLGA particles were complexed with a reporter plasmid encoding green fluorescent protein (GFP). A549 (lung cancer epithelial cells) were used in an in vitro assay to study the transfection efficiencies. The NPs transferred the EGFP gene, but were less efficient than the lipofectamine control. Nevertheless, both formulations facilitated gene delivery and expression in vivo with increased efficiency and without causing inflammation after intranasal administration to mice. Furthermore, polydisperse (0.5–4.0 μm) microspheres consisting of a CS-grafted PLA shell and a core of PLGA nanocapsules (Fig. 10) exhibited enhanced physical stability, compared to the polyester alone, and excellent aerosol characteristics as determined by inertial impaction studies.214 These particles could be efficiently dispersed in hydrofluoroalkane propellant, taken up and internalized within chlamydial inclusions by Calu-3 (airway epithelial) cells that have been infected with Chlamydia pneumoniae. As a perspective, this drug carrier system may find application in pulmonary delivery using pressurized-metered dose inhalers.
 | ||
| Fig. 10 Fluorescent field microscope image of core-shell particles containing fluorescent green dye loaded PLGA nanocapsules. Inset: TEM image of a core-shell particle. Reproduced from Ref. 214 with permission from Elsevier. | ||
However, the encapsulation process poses some problems, such as low loadings, loss of antigen activity upon entrapment and difficulties in controlling the release of the drug. Therefore, the adsorption of important proteins onto the particle surface has been studied.224 The loading by adsorption represents an interesting alternative to encapsulation since it avoids harsh formulation conditions, like protein contact with organic solvents, high shear agitation and the acidity of the microenvironment resulting from the degradation of PLA and PLGA.225,226 Surface-adsorbed antigens can act as an immune stimulant and as targeting moiety to MALT, thus, increasing the transport across the mucosa. Different surface coatings (PVA (13-23 kDa, 87–89% hydrolyzed), low viscosity alginate (ALG), and glycolchitosan (GCS)) of PLA-based particles were investigated to optimize the adsorption of Streptococcus equi cell wall proteins extract.227 The coatings were obtained by double emulsion (w/o/w) solvent evaporation procedure. The antigen adsorption increased with time over the first hour of incubation in double-distilled water, reaching maximum efficiency values (in % w/w) of 75 for PVA-PLA, 85 for GCS-PLA, and 78 for ALG-PLA. The highest adsorption for the GCS-modified particles can be explained by the preferential binding of the proteins to the positively-charged NPs, as was shown for chitosan-coated PCL nanoparticles.228,229 Protein loading had no significant influence on the particle sizes, which were dependent on the applied coating (PVA (256 nm) being significantly smaller than GCS and ALG (407 nm, and 346 nm, respectively)). Desorption studies revealed a burst release within the first 6 h for all formulations, followed by a sustained release profile. Furthermore, the mucosal, humoral, and cellular immune responses in a mouse model, after intranasal administration of S. equi antigens associated by adsorption or encapsulation to PLA nanospheres, modified by mucoadhesive polymers and absorption enhancers were studied. It was shown that, in contrast to the entrapment process, the adsorption process extensively increases the amount of protein associated to the PLA nanospheres. In addition, the obtained particles were not cytotoxic towards BALB/c J774A.1 cells. It is generally agreed that particles with sizes below 10 μm are taken up by cells, however, only those of between 1 and 1,000 nm are transported by NALT and rapidly enter the bloodstream after intranasal administration.230 It was shown for a peptide entrapped in PLGA particles that the smaller ones of mean sizes below 500 nm were better inducers of cytotoxic T-lymphocytes (immune response) than the larger microparticles (mean sizes of 2 μm and above).231 Therefore, these PLA nanoparticles, with antigens adsorbed on the surface, may have crossed the epithelial barriers through nasal mucosa APCs, i.e., M cells as well as DCs or MΦs, since they induced better immune responses than the free S. equi extract after intranasal administration. Overall, it was concluded that PLA-GCS nanospheres, carrying encapsulated antigen, do not require co-administration of any adjuvants to induce strong systemic and mucosal activity, which is crucial for protection against S. equi infection.
In order to introduce moieties that target APCs into PLGA-based NPs, a bacterial LPS from E. coli strain 0111:B4 was incorporated into particles carrying a West Nile virus envelope protein antigen (rWNVE).232 A modified w/o/w emulsion method was used to simultaneously encapsulate rWNVE and to attach the LPS moieties to the nanosphere surface. By using ovalbumin (OVA) as an antigen model it was demonstrated that the LPS enhanced the EE by approximately 16%, similarly to other amphipathic fatty acids and lipids.233–235 The surface-modified NPs contained 17 μg of rWNVE and 13 μg of LPS per 1 mg of particles. Furthermore, the LPS activity was confirmed by the Limulus Amebocyte Lysate endotoxin assay. In addition to MΦ activation by stimulation of the inflammasome pathway and high uptake by DCs (Fig. 11), yielding high T-cell responses, the LPS-modified NPs proved to be suitable carriers and adjuvants for the rWNVE vaccine. Subcutaneously (s.c.), intranasally and orally vaccinated mice with these particles showed significantly higher antibody titers than the control, i.e. injected s.c. with LPS-PLGA NPs loaded with OVA. The highest anti-rWNVE IgG titers were found after parenteral administration and conferred complete protection to the mice against the viral challenge. Nevertheless, the orally and intranasally administered NPs resulted in 75% and 80% survival, respectively. The results indicate that these particles may be effective carriers for transporting the antigen across epithelial barriers and can provide some protection for the drug in the gut, since oral vaccination with rWNVE alone resulted in lower survival.
| Polymer | Protein loaded | Size (nm)a | ζ potential (mV)a | Encapsulation efficiency (%)a |
|---|---|---|---|---|
| a Results are presented as mean (n = 3) ± standard deviation | ||||
| PLA | Tetanus toxoid | 192 ± 12 | −47.9 ± 1.5 | 36.7 ± 0.3 |
| PEG-PLA | Tetanus toxoid | 196 ± 20 | −23.9 ± 1.2 | 31.1 ± 0.5 |
| CS-PLGA | Tetanus toxoid | 500 ± 29 | + 21.8 ± 1.1 | 90.0 ± 3.8 |
| CS | Tetanus toxoid | 354 ± 27 | + 37.1 ± 5.9 | 55.1 ± 3.4 |
| CS | Insulin | 337 ± 14 | + 36.9 ± 0.3 | 94.7 ± 2.1 |
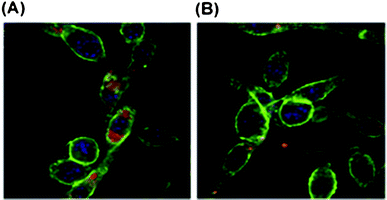 | ||
| Fig. 11 CLSM images of DCs incubated with nanoparticles encapsulating rhodamine B and analyzed by confocal microscopy (green: phalloidin-FITC, blue: DAPI, and red: rhodamine B): (A) NPs modified with LPS taken up by DCs; (B) NPs without LPS do not enter DCs. Reproduced from ref. 232 with permission from Elsevier. | ||
:15, 50–75 kDa) and stabilized with either PVA (30–70 kDa, 87–90% hydrolyzed) or poloxamer 188(Mw = 8.5 kDa). The addition of the polyether to the formulation increased the in vivo uptake of the particles after intranasal administration, even though the hydrophilicity of the NPs was increased by poloxamer. Studies suggest that M-cells revealed an improved uptake of hydrophobic particles.240 Furthermore, the influence of pH and ionic strength on the ζ was investigated. The charge of the particles at physiological conditions (pH 7.4 and 1.7 × 10−1 M) was close to zero and decreased with the impairment of ionic strength till −26 mV at 1.7 × 10−5 M. This fact had a significant influence on the in vivo uptake behavior since at low ionic strength dispersion conditions NPs were taken up in higher numbers, regardless of presence or absence of the targeting peptide or incorporation of poloxamer, and similar to the uptake of bacterial strains. The CPE-carrying particles were also better taken up than non-targeted particles, suggesting that the uptake by NALT is both active and selective, and also it depends on the ionic strength of the buffer. Since the receptor for CPE is also expressed by the M-cells of Peyer's patch, sub-micron PLGA-based particles carrying this ligand were also evaluated for oral administration. The presentation of the protein on the NPs surface resulted in internalization of particles, even though the ligand constituted only 2 wt% of the whole mass. The targeting peptide survived gastric juices and digestive enzymes. CLSM images of excised Peyer's patch regions of the intestine showed that the CPE-modified NPs were transported through the mucosa and were taken into follicles. The uptake was more significant in PPs after oral administration than in NALT possibly due to a longer residence time within the intestine.
A versatile strategy for coupling various targeting moieties and/or dyes to the preformed PLGA particles was established in another study.243 NPs of 210 nm with very narrow dispersities were obtained by using the simple oil-in-water method. Since they possess carboxyl moieties on the surface, biotinylated poly(ethylene glycol) (BPEG) carrying an amino group was coupled via cyanuric chloride to the particles, thus affording an anchor where molecules with avidin (Av) and its homologues streptavidin (SAv) as well as NeutrAvidin (NAv) can be attached. The biotin (Biot)–Av interactions are one of the strongest and most rapid in nature (Kd = 10−13–10−16 M), and therefore suitable for the stable attachment of functional groups.244 The amount of surface-associated biotin was 850 pmol per milligram of polymer, corresponding to roughly 2,650 molecules of biotin per NP. In order to investigate and visualize binding as well as uptake of these particles by monolayers of epithelial cells (Caco-2 and A549), FITC-NAv-labeled NPs were applied in case the cell membranes were stained with Rho-RCA (ricinus communis agglutinin I) or Rho-NAv-labeled-NPs if the cell membranes were stained with FITC-WGA. CLSM imaging (Fig. 12) revealed in case of A549 cells that nanoparticles localized in close association with the cell membranes after 6 h of incubation and showed a clear uptake after 24 h. In the case of the Caco-2 cells, no particle uptake could be observed even after 24 h of incubation. Particles were only attached to the cell membranes. The same results were found for both dyes, therefore, confirming the suitability of this method for rapid and stable attachment of fluorescent markers.
In another study to improve the intestinal uptake of particles, RGD (M-cell targeting motif) was photografted via a bifunctional molecular clip (O-succinimidyl-4-(p-azido-phenyl)butanoate) mainly on the PEG moiety of the PEG-PCL copolymer (Scheme 2).245 The lectin-functionalized block copolymer was then used for the formulation of 3H-labeled OVA-loaded PLGA particles (200 to 220 nm) by a modified w/o/w method.246 The RGD labeling of NPs significantly increased their transport through co-cultured monolayers of Caco-2 and Raji B cells, representing an in vitro model of the human follicle associated epithelium, due to interactions between the lectin and the integrins detected at the apical surface of the model. Furthermore, in vivo studies demonstrated that RGD-carrying NPs are particularly concentrated in M cells of the intestine after oral administration to mice. The use of the RGD ligand at the nanoparticle surface to target M cells provoked a slight increase in the number of mice producing IgG after immunization, thus, confirming the importance of targeting of the carriers. However, no increase of the antibody production in serum was observed with the use of the RGD ligand-modified PEG-PCL, OVA-carrying NPs. This could be due to a partial degradation of the peptide in the gut. Therefore, it was recommended that a non-peptidic β1 integrin ligand could be used and grafted onto the nanoparticles. In a follow up trial, ligands targeting intestinal M cells or APCs other than RGD, i.e., novel non-peptidic and peptidic analogs (RGD peptidomimitic (RGDp), LDV derivative (LDVd), and LDV peptidomimetic (LDVp)) as well as mannose were grafted on the PEG chain of PEG-b-PCL and incorporated in PLGA-based nanoparticles in order to evaluate their efficiencies in oral vaccination, as shown in (Scheme 2).247 Transport enhancement studies on an in vitro model of FAE (coculture of Caco-2 and Raji cell lines) have demonstrated increased transfer of nanoparticles (200 to 260 nm in size) carrying Man, RGD and RGDp moieties. In addition, ligand grafting increased the particle uptake by murine macrophages of the cell line J774. Three times more LDVp and carbohydrate-carrying NPs, compared to non-targeted ones, were internalized after 60 min of incubation. Meanwhile, the uptake of RGD and LDVd doubled in contrast to RGDp-particles where no influence on phagocytosis was observed. Furthermore, upon intraduodenal administration the RGDp-, LDVd-, or mannose-functionalized OVA-loaded NPs elicited a higher production of IgG antibodies than the intramuscular injection of free OVA or intraduodenal administration of either non-targeted or RGD-functionalized nanoparticles.
Microspheres (MS) of 1 to 10 μm, consisting of biodegradable materials (PLGA with various lactide to glycolide ratios, poly(D-lactide) (PDLA), poly(L-lactide) (PLLA) and poly(hydroxybutyrate)) as well as non-biodegradable (polystyrene, poly(methyl methacrylate), cellulose acetate hydrogen phthalate, cellulose triacetate, and ethyl cellulose) were evaluated for their absorption by Peyer's patches after oral administration to mice.248 Mice were administered with a single dose of 20 mg of MS suspended in 0.5 mL of tap water using a blunt-tipped feeding needle inserted into the stomach. It was found, except for cellulose derivatives, that the absorption efficiency correlates well with the relative hydrophobicity of the material, and the particles accumulated predominantly in the dome regions of PPs. Following the oral inoculation of the dose of PLGA (85:15) MS, fluorescence microscopic observations revealed that internalized microspheres were present in the Peyer's patches 24 h post administration, and at all times tested through 35 days. Additionally, the particles that penetrated deep into PPs were almost exclusively ≤5 μm in diameter.
 | ||
| Fig. 12 Xz cross sections from a stack of confocal laser scans (apical up) of Rho-RCA-stained A549 cells, some particles taken up after 24 h (A), Rho-RCA-stained Caco-2 cells, particles attached to the membrane (B), and FITC-WGA-stained Caco-2 cells, fluorescent glycocalyx (C). Cells were imaged after 6 and 24 h incubation with FITC-NAv-labeled BPEG PLGA NP (in the case of Rho-RCA-stained A549 and Caco-2 cells) and Rho-NAv-labeled BPEG PLGA NP (in the case of FITC-WGA-stained Caco-2 cells) (rows A and B: bar = 50 μm × 5 μm. row C: bar = 20 μm × 9 μm). Reproduced from ref. 243 with permission from Elsevier. | ||
 | ||
| Scheme 2 Schematic representation of the PEG-directed photografting of various ligands onto PEG-PCL. | ||
3.2. Polysaccharides
Polysaccharides represent the second most abundantly investigated group of materials in the production of transepithelial drug carriers. It is estimated that each year approximately 4 × 1011 tons of carbohydrates are biosynthesized on earth by plants and photosynthesizing bacteria. The majority of these sugars are produced as polysaccharides.256 They constitute a class of biological macromolecules consisting a large number of monosaccharide units, which exist in an enormous structural diversity due to the variety of sugar units, the differences in their stereochemistry, and the position of linking and further branching. Being G.R.A.S. (Generally Regarded As Safe) materials, sugar compounds with a unique stereochemistry provide a ubiquitous platform to obtain cost effective chemicals and polymers.257 The structures of the most commonly used polysaccharides in transepithelial drug delivery systems are depicted in Scheme 3. Polysaccharides play a major role in diverse areas of protein interactions, drug (vaccine) delivery, cancer treatment as well as cell and tissue engineering. As a consequence, polysaccharides are the subject of glycomics—a newly emerging field, which studies structure–function relationships of these complex carbohydrates.258,259 Polysaccharides are major constituents of the epithelial glycocalyx, mucoproteins, see Section 2.1., (aggregates of central, glycosylated core proteins and polycarbohydrates), virus capsids and bacterial cell walls. Their biochemical function is determined by their nanoscale organization. These biological nanostructures provide the inspiration for developing techniques to tune the assembly of polysaccharides at the nanoscale, which have been thoroughly reviewed.260 Various physicochemical and biological methods have been described to obtain polycarbohydrate-coated drug delivery systems, which offer enhanced mucoadhesion as well as paracellular transport via opening of TJs.261–263 Among all the polysaccharides, chitosan and its trimethylated, thiolated, acetylated as well as glutamate derivatives have been most thoroughly investigated and reviewed as permeation-enhancing, particulate delivery systems.264–266 To obtain more knowledge about chitosan-based drug carriers, the reader is referred to some excellent reviews.267–269 Herein, selected examples of polysaccharidic transmucosal delivery systems, other than CS-based, are presented.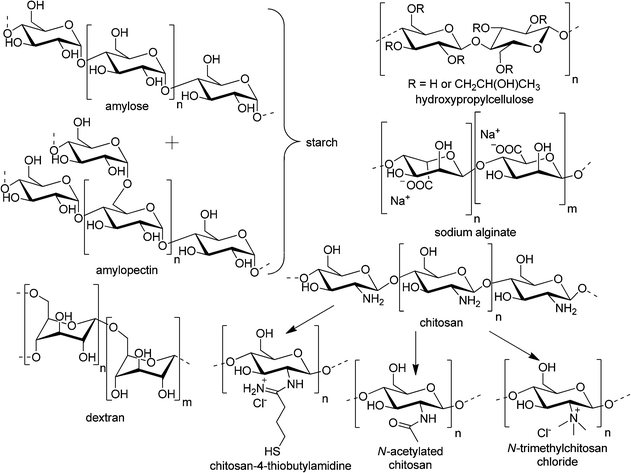 | ||
| Scheme 3 Schematic representation of the most commonly used polysaccharides for transepithelial delivery. | ||
:4 showed rapid absorption in less than one minute and thus achieved 88% bioavailability, a value 1.9 times higher than for free, crystalline fluorescein powder. This formulation was further used for the enhancement of the delivery of beclomethasone (BDP) to lungs.273 In total, 95% of HPC 2.5 to 2.9 μm particles, containing amorphous BDP, were absorbed from the guinea pig lungs within 180 min. In contrast, the same particles loaded with a crystalline form of the drug demonstrated prolonged retention of the drug within the lungs and significantly inhibited eosinophil infiltration into the airways of antigen-induced asthmatic animals up to 24 h. BDP's therapeutic duration was increased by 18 to 23 h, using a reduced dose (1.37 → 0.25 mg kg−1) of the drug encapsulated in HPC microsphere system. Furthermore, it was shown that HPC, among other polysaccharides (chitosan, hyaluronic acid, and alginate), may be employed for encapsulation of FITC-labeled BSA by a spray-drying method without any degradation of the protein.274 This technique yielded particles with a mass median aerodynamic diameter between 2.9 and 4.7 μm and high protein loading. They were blended with mannitol and tested for aerosolization efficiency. The particles consisting of low viscosity HPC exhibited the highest fine particle fraction of 26%, being the best for pulmonary administration to date.
Microparticles composed of HPC, CS, hyaluronic acid (HYA), alginate, gelatin, OVA, and PLGA were individually tested for cytotoxicity towards Calu-3 monolayers. There was no significant difference in viability between the cells impinged with 2 mg mL−1 doses of the carriers and the untreated control, thus, confirming their G.R.A.S. status from the US FDA. HPC and gelatin particles showed the fastest release of BSA, as assayed in a Franz diffusion cell.275 The induction of inflammatory response (release of the cytokine IL-8), changes in the monolayer integrity, i.e. transepithelial permeability (transport of fluorescein sodium salt across the monolayer), and electrical resistance were investigated on cells cultured under AIC and LCC after impingement with particles. HPC-based particles did not cause any decrease in the epithelial integrity or inflammatory reactions. On the other hand, the production of the inflammatory cytokine was significantly increased in the case of PLGA and gelatin microparticles. In addition, the polyester, even though it is the most commonly studied excipient for controlled release in the lungs, had the highest toxicity of the investigated polymers.
Nevertheless, sodium alginate conjugated with spermine successfully encapsulated a rotavirus (RRV) and enhanced virus-specific immunity in mice after oral administration.283 This formulation did not reduce the infectivity of the virus. The formed monodisperse microcapsules of approx. 5 μm were stable in simulated gastric acid, and provided the largest volume of encapsulated material. Furthermore, rhodamine-labeled alginate-spermine particles were detected within cells of PP, mesenteric lymph nodes, and spleen until at least 28 days after oral administration. Three out of three mice orally vaccinated with RRV in these capsules developed virus-specific antibodies, whereas, none out of four inoculated with the same dose of free virus revealed the specific response. Thus, the particles delivered the rotavirus antigen to GALT at greater levels than the free pathogen. Furthermore, the alginate-spermine formulation was found to successfully encapsulate reovirus, deliver the antigen via the oral route, bypass maternal antibody, passively transferred by suckling to neonates, and effectively induce systemic and mucosal responses.284 Alginate was not only able to encapsulate whole viruses or proteins, but also a pneumococcal capsular polysaccharide type 19 (PS19), which was conjugated to the B-subunit of cholera toxin (PS19-CTB) in order to enhance its immunogenicity.285 The microspheres with diameters of less than 5 μm, were prepared by a diffusion-controlled interfacial gelation technique. They encapsulated the polysaccharide with 60% efficiency and showed 80% antigen liberation within one day. Peroral immunization with 25 μg of PS19-CTB entrapped in the alginate microspheres (AMS) evoked both the mucosal IgA and systemic IgM responses to PS19 in small intestine and in sera, respectively. Furthermore, 80% of the mice orally vaccinated with PS19-CTB-loaded AMS were protected from lethal intranasal challenge with S. pneumoniae, whereas more than 60% of the mice in the other control groups died upon infection.286 In a follow up study, recombinant pneumococcal surface adhesin A (rPsaA), a 37-kDa metal-binding lipoprotein, which plays a critical role in bacterial adherence to respiratory mucosa and in virulence, was entrapped in polydisperse (0.5 to 5.0 μm) alginate MS.287 In order to enhance the immunogenicity of the antigen, CTB was coencapsulated or coadministered orally to mice. Encapsulated rPsaA provided higher immunogenicity than the free antigen. Furthermore, the AMS were able to induce cross-protective immunity for the prevention of pneumococcal infection against five other strains of S. pneumoniae. Small alginate microspheres were considered to be an efficient carrier of antigens to the PPs and through the lymphatic system.
Additionally, an alginate-coating process was developed and optimized for CS-based particles, to improve their stability and to prevent the immediate release of the encapsulated drug in the gastrointestinal fluid.288 OVA was adsorbed on the surface of chitosan nanospheres, subsequently coated with alginate, and crosslinked by addition of Ca2+ ions. Successful coating was achieved at alginate/CS-OVA NPs ratios above 2 at pH 7.4 as confirmed by the inversion of ζ (from 41 to −35 mV) as well as differential scanning calorimetry (DSC) and FTIR. The coating decreased the antigen desorption in simulated intestinal fluid (SIF) by less than 40% of OVA released within 10 h, as compared to uncoated particles, which immediately liberated more than 90% of the drug. These particles were further used as a delivery system for oral vaccination of mice with the recombinant hepatitis B surface antigen either alone or in combination with the immunopotentiator, synthetic oligodeoxynucleotide containing an immunostimulatory CpG motif as adjuvant.289 A different approach was applied to obtain insulin-loaded particles having an alginate core with CS electrostatically adsorbed on their surface.290 Nanoparticles exhibited negative charge and had a mean size of 750 nm and, therefore, were suitable for uptake within the gastrointestinal tract. The insulin association efficiency was higher than 70%, and the drug was released in a pH-dependent manner under simulated gastrointestinal conditions. NPs lowered the basal serum glucose levels by more than 40% with 50 and 100 IUkg−1 doses sustaining hypoglycemia for over 18 h after oral administration to rats. The encapsulation of insulin into mucoadhesive nanoparticles was a key factor in the improvement of its absorption and bioactivity after oral administration. Alginate-based delivery systems were applied not only for the encapsulation of viruses, polysaccharides or proteins, but also in the delivery of smaller drug molecules. Isoniazid, a 137.13 g mol−1 compound, was entrapped in sodium alginate crosslinked with Ca2+ ions by a modified emulsification method.291 SEM investigations revealed that heterogeneous particles were obtained with the highest number of an average size of 3.72 μm, a smooth surface, and a discreet spherical shape. The EE, up to 91%, and the extent of drug release increased with the increased concentration of the crosslinking agent, up to 7.5%. Approximately 25% of the drug was released in the simulated gastric fluid (SGF), pH 1.2, in 5 h and 71% in SIF, pH 7.4, in 30 h. This behavior may be explained by the shrinkage of alginate MS due to tightening of the gel meshwork at acidic pH. Under basic conditions the polymer is eroded and the drug is released in a sustained manner caused by slow degradation of the polymeric matrix and diffusion.292
Furthermore, the favorable bioadhesive properties of the microspheres were established in an ex vivo model of albino rat small intestine. The additional confirmation was obtained from the in vivo biodistribution studies of the 99mTc-radiolabeled AMS after oral administration. The mucoadhesive properties of the alginate MS prolonged their retention in the small intestine. They could be observed in the intestinal lumen at 4 h and were detectable even after 24 h. The dissolution and γ-scintigraphy investigations confirmed the applicability of this formulation for enteric drug delivery. Additionally, alginate-based microparticles were designed for pulmonary delivery and loaded with the anti-tumor drug paclitaxel.293 By selecting appropriate parameters for the emulsification and ionic gelation technique, microparticles with a mean volume diameter of 3.0 μm, a mass median aerodynamic diameter of 5.9 μm, a fine particle fraction of 13.9%, and an encapsulation efficiency of 61.0% could be obtained. The microparticles exhibited sustained release under in vitro conditions in PBS. Studies with A549 and Calu-3 (human non-small lung cancer) cells showed effective growth inhibition by paclitaxel-loaded microparticles. Therefore, the possibility of using this formulation as a suitable candidate for inhalation chemotherapy for in vivo experimental models was proven.
 | ||
| Fig. 13 Microscopy images and size distribution (DLS) of alginate particles prepared by inkjet printing (A: drop-on-demand, and B: continuous inkjet). Reproduced from ref. 280. Copyright American Chemical Society. | ||
The adjuvant effect of this polysaccharide was demonstrated in mice using covalently coupled human serum albumin (HSA) as a model antigen.299,300 Out of the parenteral routes of administration tested, intraperitoneal administration gave the strongest humoral immune response, almost as high as with complete Freund's adjuvant (CFA), thus, proving the potential of microparticles containing starch as an adjuvant for human use. Upon oral administration, the HSA-microparticles (2 to 3 μm) induced a good, diversified immune response without any signs of tolerance development. The covalent linkage between HSA and the starch residues assured that the adjuvant and the antigen are taken up by the same APCs, and the size range of the particles allowed for their passage into PPs of the intestine. Furthermore, the immune response profiles depended strongly on the route chosen for administration.301 Moreover, starch microparticles grafted with 3-(triethoxysilyl)-propyl-terminated poly(dimethyl-siloxane) (TS-PDMS), a biocompatible silicone polymer, were used for entrapment of HSA.302 Following oral administration, TS-PDMS-grafted microparticles stimulated significantly stronger serum IgG and IgA responses compared to those elicited by soluble antigen and ungrafted microparticles. The antibody responses of mice orally immunized with a pneumococcal polysaccharide (type 23F) or a pneumococcal polysaccharide conjugated to the outer membrane protein complex of Neisseria meningitides spray-coated onto starch cores and enterocoated with Eudragit® L30 D50 were examined.303 The obtained microcapsules (approx. 0.7 mm in diameter) were insoluble in gastric pH and dissolved at higher (pH > 5) values. This drug carrier system was the first to induce S. pneumoniae polysaccharide-specific serum IgG antibodies following immunization by the oral route with relatively small amounts (a total of 2 μg) of polysaccharide and without the inclusion of cholera toxin or its B subunit as an additional adjuvant. A similar approach was further employed to obtain a vaccine delivery system against the enteral/parenteral nematode parasite Trichinella spiralis.304 Vaccination with loaded microcapsules offered significant protection against the challenging infection. The adjuvant effect of polyacryl starch was further demonstrated in mice following oral vaccination with antigens from Salmonella enterica serovar Enteritidis covalently conjugated to microparticles.305 After bacterial infection, a significant reduction in the colony forming units was found for all immunized animals and correlated with weight loss (10.3% for the non treated mice, 4.0% for vaccinated). In addition, the group that received orally free antigens mixed with microparticles had the highest local immune response in the intestine and showed the highest protection in the challenge. This might be attributed to protein adsorption on the surface of polyacryl starch and protection from the degradation in the gut. Bioadhesive carriers based on starch and poly(acrylic acid) were also employed for an intranasal vaccine delivery system. Liquid nasal PBS vaccines were prepared out of manually milled, freeze dried mixtures of starch and Carbopol® 974P particles (from spray-drying), containing heat-inactivated influenza virus combined with LTR192G adjuvant.306 It was demonstrated that the use of this drug carrier system facilitated the induction of a systemic anti-hemagglutinin antibody response after intranasal vaccination with a whole virus influenza vaccine and the presence of Carbopol® 974P improved the kinetics of the immune responses.
 | ||
| Fig. 14 SEM micrographs: a general view (×200, the bar represents 100 μm) and the surface morphology (×6000, inserts, the bar represents 2 μm) of microspheres. (A) mechanically treated (MT) starch loaded with: (1) insulin, (2) BSA; (B) acid-hydrolyzed (AH) starch loaded with: (1) insulin, (2) BSA; (C) a mixture of MT/AH loaded with (1) insulin, (2) BSA. Reproduced from ref. 298 by permission of The Royal Society of Chemistry. | ||
3.3 Poly(γ-glutamic acid) (γ-PGA)
γ-PGA is of significant interest in drug delivery because of its natural origin (obtained by bacterial fermentation) biodegradability, hydrophilicity, lack of toxicity, biocompatibility, and nonimmunogenicity.307 Its production, as well as wide application as an anticancer drug carrier system, immunestimulatory agent, food additive and for cosmetics has been thoroughly reviewed.308–310γ-PGA is a high-molar mass polypeptide composed of γ-linked glutamic acid units. Its γ-carboxylate side chains can be chemically activated by water soluble carbodiimides, for example, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide. The activated carboxylic groups can be further modified with various primary amine-bearing compounds to yield amide-grafted, functional polymers.311 For example, the carboxyl moieties of γ-PGA (Mn = 300000 Da; D:L ratio of 6:4) were grafted with L-phenylalanine ethyl ester (PAE) to yield an amphiphilic polymer (γ-PGA-graft-L-PAE). The degree of grafting, thus, the hydrophobicity of γ-PGA could be adjusted by varying the molar ratio between the glutamic acid units and the carboxyl-activating reagent. 49, 53 and 61% grafted polymers formed monodispersed nanoparticles by a precipitation and dialysis method.312 Different water miscible solvents (DMSO, DMF, DMAc, NMP) were applied for this procedure and yielded particles of similar mean diameters in the range of 100 to 200 nm and zeta potentials (ζ) of −23 to −24 mV, as analyzed by dynamic light scattering (DLS). TEM images of the particles prepared from DMSO showed structures of smooth spherical morphology with a PAE-core and a PGA shell. Subsequently, ovalbumin (OVA) as a model antigen was either covalently immobilized on the nanoparticle surface, by the water soluble carbodiimide method, or entrapped inside the particles by nanoprecipitation of DMSO solutions of the amphiphilic polymer into phosphate-buffered saline (PBS) solutions of OVA. Entrapment efficiencies up to 50 wt% at 0.5 mg mL−1 of OVA were reached. On the other hand, the efficiency of the covalent protein attachment was about 5%. In addition, sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis confirmed the successful OVA incorporation. Upon entrapment of OVA, the size of the NPs was greatly increased up to ca. 400 nm, without any significant change of ζ. Furthermore, the in vitro release studies in PBS (pH = 7.4) at 37 °C showed that no OVA was released (less than 5%) over 10 days and no degradation of the amphiphilic polymer at neutral pH was observed. However, the degradation of γ-PGA NPs by γ-glutamyl transpeptidase (γ-GTP), which is widely distributed among living organism and catalyzes the hydrolysis of γ-glutamyl compounds, was confirmed (Fig. 15).313 As a consequence, the encapsulated OVA should be released upon the enzymatic degradation of the particles. Furthermore, the NPs with the entrapped protein were suitable for lyophilization in presence of 2.5% glucose as stabilizer and showed no cytotoxic effects toward leukemic HL-60 cells.
This initial research was followed by investigations of the γ-PGA-graft-L-PAE potential as a carrier for a series of proteins. Their incorporation into particulated formulations was achieved by adsorption, surface immobilization, and encapsulation methods.314 Among the proteins thyroglobulin, catalase, Con A, OVA, BSA, a peroxidase, β-lactoglobulin, myoglobin, lysozyme, α-lactalbumin, and cytochrome c were examined. The highest catalase activity was obtained when it was encapsulated inside the γ-PGA-graft-L-PAE nanoparticles, thus, the protein flexibility was not significantly impaired. Other conjugation methods, like adsorption by electrostatic interactions and covalent binding to the particle surface, markedly reduced catalase activity. The conformational stability of the protein inside the carrier system is of crucial importance for the induction of adequate immune response after vaccination.315 In further studies, the γ-PGA-graft-L-PAE NPs were employed to encapsulate the CD8(+) T-cell epitope peptide of Listeria monocytogenes in order to analyze their in vitro interaction with B cells, macrophages (MΦ), and immature dendritic cells (iDCs) as well as their vaccine activity in vivo after intravenous (i.v.) administration to mice.316 For this purpose the amphiphilic polymer was conjugated with a fluorescent label (5-aminoacetamido fluorescein isothiocyanate, FITC) in the presence of water soluble carbodiimide. The uptake of the FITC-NPs by the above-mentioned cells was investigated by flow cytometry (FC) and their intracellular localization was visualized by confocal laser scanning microscopy (CLSM). The nanoparticles were more efficiently taken up in a dose- and time-dependent manner by iDCs than by MΦ or B cells, and they preferentially localized in lysosomal compartments. The FITC-NP-associated form of OVA was taken up much more efficiently than free FITC-OVA. The exposure of iDCs to γ-PGA-graft-L-PAE NPs caused their maturation, production of the innate inflammatory cytokines IL-12p40 and TNF-α as well as CD40 expression and stimulation ofallogenic T-cells in a dose-dependent fashion. In fact, 4 h after i.v. administration, B cells, spleen DCs (the highest number), and MΦ but not in T-cells were FITC-positive. The vaccination with OVA-NPs strongly induced OVA-specific IFN-γ producing cells, cytotoxic T-lymphocyte (CTL) activity, and antibody (Ab) production. Moreover, after immunization with NPs carrying a CD8(+) T-cell epitope peptide of Listeria monocytogenes the infected mice were significantly protected from death. To further demonstrate the potential of γ-PGA-based mucosal vaccines, the γ-PGA-graft-L-PAE nanoparticles encapsulating the recombinant HIV-1 (IIIB strain) gp120 protein (gp120-PGA-NPs) were intranasally administered to mice, and immunological responses were examined.317 The animals were immunized once with various concentrations of gp120-PGA-NPs with a mean hydrodynamic diameter of 450 ± 124 nm (DLS) in a total volume of 20 μL to one nostril. The spleen cells obtained from the mice vaccinated with protein-encapsulated nanoparticles showed significant antigen-specific proliferation in a dose-dependent fashion compared to the spleen cells obtained from mice immunized with a mixture of gp120 and the B-subunit of cholera toxin (gp120 + CTB). Long-lived memory CD8(+) T-cells were also upregulated. Even though a substantial decay in the effector memory T-cells was observed over time in the immunized mice, the central memory T-cells remained relatively constant from day 30 to day 238 after immunization. Moreover, it was shown that biodegradable gp120-loaded PGA NPs induced much stronger CD8(+) T-cell responses than nonbiodegradable polystyrene NPs.
 | ||
| Fig. 15 SEM images of γ-PGA NPs enzymatic degradation by γ-GTP. Reproduced from ref. 313 with permission from Wiley. | ||
In further research, different hydrophobic amino acids were grafted onto a γ-PGA backbone, using the water soluble carbodiimide methodology (Scheme 4).318 The L-tyrosine ethyl ester-grafted γ-PGA was well-soluble in water and did not form any nanoparticles. It could be demonstrated that grafting efficiencies of 40 to 50% with L-phenylalanine ethyl ester or L-tryptophan methyl ester introduced a sufficient amount of hydrophobic moieties for the successful preparation of stable nanoparticles by precipitation and dialysis method. The NPs were monodisperse, smooth, and spherical, with a size of around 200 nm and strongly negative ζ (−20 to −30 mV). Interestingly, higher degrees of grafting with the hydrophobic amino acids yielded smaller particles, thus, the hydrodynamic diameters could be tuned. The NPs produced from 53% L-phenylalanine ethyl ester-modified γ-PGA revealed the highest OVA loading and enhanced the antigen delivery to iDCs, causing their maturation. Intranasal vaccination of mice with gp120-loaded γ-PGA NPs significantly upregulated cellular immunity, thus, confirming the previous results.317 Furthermore, the γ-PGA-graft-L-PAE nanoparticles were employed to encapsulate recombinant HIV-1 p24 and to immunize mice via subcutaneous administration.319 Antigen-specific IFN-γ-producing T-cells were activated in spleen and p24-specific serum antibodies were induced by this vaccination. It was demonstrated that the nanoparticles play a critical role in generating cellular immune response through lack of T-cell activation after administration of a p24 and nanoparticle mixture. The γ-PGA NPs showed similar adjuvenating properties to complete Freund's adjuvant (CFA), the standard immunopotentiating agent, in inducing p24-specific antibodies in serum, however, unlike CFA they predominantly activated p24-specific IFN-γ-producing T-cells. Therefore, γ-PGA NPs encapsulating various antigens were proven as efficient protein-based vaccines against infectious diseases, including HIV-1 infection.
Thus, more advanced studies were carried out on rhesus macaques intranasally or subcutaneously vaccinated with recombinant gp120 envelope protein of HIV-1, which was encapsulated in γ-PGA NPs prepared by the above-mentioned methods.320 The immune response and protective efficacy against a challenge inoculation of simian and human immunodeficiency chimeric virus (SHIV) was examined. In contrast to mice, all macaques had to be subcutaneously immunized twice with additional vaccinations, because their immune responses were insufficient after three intranasal immunizations. The nanoparticulate formulation of gp120 induced significantly higher cellular and humoral immune responses than free antigen, however, NPs were ineffective in the generation of protection but, on the contrary, enhanced the SHIV infection. The intravenously challenged virus had replicated more in the vaccinated groups than in the PBS control group. Furthermore, this tendency was more intense in the gp120-NP group than in the free gp120 group. The nanoparticle-generated immune responses were in good correlation with the promotion of viral growth. One explanation of this phenomenon lies in the choice of the encapsulated antigen used for the immunization. Thus, gp120 was employed as the antigen based on the result that the immune response against env (the protein of the viral envelope) was better than that against gag (the protein of the nucleocapsid shell around the RNA of a retrovirus) in the mouse studies.317,319 On the other hand, the strength of the specific immune responses against the env in the crust protein and accessory proteins are correlated to the increase in the quantity of blood virus in HIV patients.321 These results stress the requirement for proper choice of the antigen and the further accumulation of basic information about the relationships within the immune system for protective immunity in HIV vaccine development
 | ||
| Scheme 4 Schematic representation of the synthesis of γ-PGA amphiphilic polymers. | ||
3.4. Other polymers
Apart from macromolecules originating from natural sources (polysaccharides, γ-PGA), also numerous synthetic polymers, other than PLGA, have found application in the formation of particulated transmucosal drug delivery systems. The development of polymerization techniques of well-defined polymeric bioconjugates opened an avenue to obtain tailor-made functional materials for a wide range of biological applications.322,323 Furthermore, new methods for incorporation of bioactive (targeting) moieties allow precise control over the amounts and the distribution of various functional groups on the polymeric backbone.324,325 However, since most of the synthetic polymers are not biodegradable, they have to be administered with great care and monitored for accumulation in different organs. Nevertheless, U.S. FDA-approved, PEG-functionalized drugs have shown improved pharmacokinetic properties, resulting in better patient quality of life.326 Even though the first approved PEGylated products have been on the market for 20 years already, there is still an extensive quantity of research concerning the possible side effects, such as: hypersensitivity, unexpected changes in pharmacokinetic behavior, toxic side products, and an antagonism arising from the easy degradation of the polymer under mechanical stress resulting from its structure and/or non-biodegradability.327 On the other hand, synthetic polymers offer a wide range of tunable properties such as pH and thermal responsiveness, self-assembly for improved drug encapsulation/release as well as mucoadhesiveness. Herein, selected examples of artificial polymers are presented, which improved transmucosal transport of drugs. An overview of the polymeric structures, functional groups and applications can be found in Table 5.| Schematic representation of the polymer structures | Surface modifier | Dye, drug, antigen | Application | Reference | ||
|---|---|---|---|---|---|---|
| Polystyrene: | — | Fluorescent | Markers for antigen tracking to lymph nodes | 330 | ||

|
— | Fluorescent | Influence of size, ζ, CS coating on particle clearance from airways | 331 | ||
| — | 46Sc-labeled | Kinetics of microparticle distribution and elimination after intranasal application to mice | 332 | |||
| Sulfate, carboxyl, amine | Fluorescent | HSMPT in mucus | 18,21,333 | |||
| Neutravidin | Biotinylated lectins | Association with FAE glycolipids | 334 | |||
| NH2-lectins | HIV particles | Capture of inactivated HIV-1, intranasal immunization of macaques | 335–337 | |||
| Gantrez ®: | Sambucus Nigra agglutinin, BSA | Fluorescent | Association to Caco-2 monolayers | 338 | ||
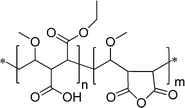
|
Thiamine | OVA | Passage to ileum tissue after oral administration to rats, Th1 stimulation, high immune response after oral administration to mice | 343,344 | ||
| Vitamin B12, dextrans | OVA | Oral administration to rats, delivery to distal portions of GIT, improved bioadhesion, induction of strong immune response | 345,346 | |||
| Salmonella entiriditis flagellin, Man | — | Induced strong and long-lasting immune responses after oral vaccination | 347 | |||
| Con A | BSA | MΦ targeting, increase of phagocytosis efficiency | 348 | |||
| HPMA: | For more information on HPMA copolymers, their synthesis, evaluation of biological properties, and explorations of their potential as carriers of biologically active compounds to colon, the reader is referred to an excellent review from Kopecek and coworkers.348 | |||||

|
||||||
| Polyethers: | — | Fluorescein | Influence on uptake through intestinal mucosa after oral administration | 255 | ||
|
||||||
| — | — | AFM investigation of poloxamer adhesion to mucus | 352 | |||
| — | PLGA | Improvement of transport through mucus | 353 | |||
| Mixture of polycarbophil, divinyl glycol-crosslinked poly(acrylic acid) | HBsAg, plasmid DNA | Thermoresponsive gels for intranasal antibody and gene delivery | 354 | |||
| DSPE | Rifampicin | Aerosolizable pulmonary drug delivery system | 356 | |||
| Poly(meth)(acrylic acids): | — | Tat protein of HIV-1 | Long-lasting cellular and humoral responses after intranasal, intramuscular and subcutaneous immunization | 360 | ||

|
||||||
| Spermine and magnesium | Calcitonin | Reversibly enhanced permeability of Caco-2 monolayers, prolonged hypocalcemic efficacy after oral administration | 361 | |||
| Cysteine, glutathione | Human growth hormone | Nasal microparticulate delivery system | 290 | |||
| Thiolated | — | Mucoadhesive and enzyme-inhibitory permeation enhancers | 362–364 | |||
| P(68)-10: | — | siRNA | Aerosolizable pulmonary drug delivery system, does not cause proinflammatory responses, 16-fold lower toxicity than poly(ethylene imine), comparable to Lipofectamine 2000 | 366 | ||
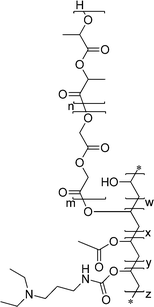
|
||||||
| lung surfactant | pDNA | Potent pulmonary gene delivery vector | 369 | |||
| Poly(propylene sulfide): | Pluronic® OH | Thiolated antigens | DC targeting in lymph nodes after interstitial injection | 377 | ||

|
||||||
| Pluronic® OH | Thiolated antigens | Cross-presentation of disulfide conjugated antigens | 378 | |||
| Pluronic® OH | Thiolated OVA, cysteine derivative of bacterial flagellin | Intranasally administered vaccination system | 379 | |||
 | ||
| Fig. 16 CLSM images of a rat small intestine showing: (left) 2 μm particles (arrows) at the mucosal surface of enterocytes (ms), within enterocytes (e), and within the villous lamina propria (lp), a goblet cell (g), bar = 2 μm; (middle) a 6 μm particle near the interface of the villous epithelium (ve) and lamina propria (lp), bar = 6 μm; (right) a 20 μm particle close to the villous tip and a 10 μm particle (insert) associated with the mucosal epithelium (me), bar = 20 μm. Reproduced from ref. 330 with kind permission from Springer Science. | ||
Another study investigated the effect of particle size, zeta-potential and mucoadhesive particle properties on mucociliary particle clearance from the airways in a trachea-based in vitro model.331 Using 50 nm up to 6000 nm-sized polystyrene particles, it was demonstrated that the diameters of the inhaled particulate matter showed no significant differences on the speed of their clearance. On the other hand, mucoadhesive chitosan-PLGA particles were transported at the slowest rate (0.7 ± 0.3 mm min−1) of all particles tested. In another study, scandium-46 labeled styrene-divinyl benzene (7 μm in diameter) microspheres (MS) were applied to investigate the kinetics of microparticle distribution and elimination following intranasal application to mice.332 The animals were administered with the same doses (0.250 mg) of MS suspended in either 50 μL or 10 μL of PBS. The smaller dose resulted in restriction of the material within the nasal cavity and an appreciable uptake into NALT. On the other hand, approximately 50% of the administered dose was deposited in the bronchopulmonary region when a larger instillation volume was used. This, however, resulted in higher radioactivity translocation into systemic tissues and implies that the epithelial barriers in the lungs are less exclusive for the entry of microparticulates into systemic compartments than those of mucosae in the nose or intestine. Furthermore, the application of fluorescent NPs, 100, 200, and 500 nm in size, allowed the use of high-speed multiple-particle tracking (HSMPT) to quantify transport rates of individual polymeric particles of various sizes and in samples of fresh human cervicovaginal mucus.333 For carboxyl-functionalized PS nanospheres it was demonstrated that much larger fractions of 100 nm particles were immobilized or otherwise hindered by mucus than the large 200 to 500 nm particles. NPs coated with PEG showed lower adsorption of avidin and diffused through mucus much faster in a size-dependent manner with the largest particles having the highest effective diffusion (Deff) coefficient, only 4- and 6-fold lower than that for the same particles in water. The significant differences in Deff within each group of particles suggested that different mechanisms exist of particle transport through mucus. This hypothesis is supported by visual observations of both immobile and rapidly moving particles in the same movie from the HSMPT. The NPs larger than the reported mesh-pore size range (10 to 200 nm) in mucus are able to undergo rapid diffusional transport through mucus barriers as long as they are properly coated, i.e. PEGylated.21 HSMPT was further employed to evaluate the dependence of transmucosal transport rates on the zeta potential of 200 nm PS nanospheres.18 Fluorescent NPs carrying sulfate (ζ = –55 mV), carboxyl (ζ = −37 mV), and amine groups (ζ = 7 mV) were examined in native mucus and in a purified mucin model. The negatively charged particles were 20 to 30 times faster than the cationic ones. Additionally, it was shown that purified mucin preparation does not provide an accurate model system for native mucus.
The accessibility of glycolipid and oligosaccharide epitopes on rabbit villus and FAE was investigated using fluorescent PS particles of virus-like size (100 nm) and bacterium-sized (1 μm), functionalized with different lectins.334 The NP were coated with neutravidin resulting in 10 nmol biotin binding sites per milligram of polystyrene. Subsequently, biotinylated lectins (CTB, binding to glycolipid GM1; RCA-I, specific for terminal β(1,4)-linked galactose; and Maackia amurensis lectin II (Mal II), which recognizes a hierarchy of oligosaccharides containing α(2,3)-linked sialic acid or biocytin) were conjugated to the NP's surface. The micron-sized particles were prepared analogously. The adhesion of PS lectin carriers was examined in an in vitro model (Caco-2BBe cell line) as well as on freshly excised live or fixed rabbit Peyer's patch mucosa. The CTB-carrying NPs adhered to aldehyde fixed tissues but not to live PP mucosa due to filamentous brush border glycocalyx present on villus and FAE enterocytes as well as a thin glycoprotein coat on M cells, which prevented lectin access to GM1. The crosslinking of proteins during the fixation process may have unmasked certain receptors that are not available in vivo. The RCA-I functionalized particles did not show specific binding to any epithelial tissue. Both lectin-carrying and control particles exhibited non-specific sparse adhesion to intestinal epithelium. Nevertheless, the RCA-I coated NPs adhered to live or fixed Caco-2 cells 39 times better than the control. This result underscores the fact that the accessibility of protein-linked oligosaccharide epitopes on epithelial cells grown in vitro may not be the same as on enterocytes in vivo. In contrast to other lectin-modified NPs, the Mal II-carrying particles (120 nm) adhered to both live and fixed tissues (Fig. 17). They exhibited preference towards FAE (adhered 4.5 times better than control particles), possibly reflecting an abundance of lectin-binding sites in the thick brush border glycocalyx. The 1 μm RCA-I-coated particles failed to adhere to villus enterocytes, FAE or M cells, consistent with the results obtained using 120 nm particles. Correspondingly, the 1 μm Mal II-coated particles exhibited unspecific binding to any cell surface, indicating that the α(2,3)-linked sialic acid-containing determinants present on the FAE that were accessible to virus-sized particles were not accessible to particles as large as bacteria (Fig. 17).
Another method for functionalization of PS nanospheres was applied for conjugation of concanavalin A (Con A) and subsequent capture of HIV-1 virions.335 For this reason, poly(methacrylic acid)-covered polystyrene nanospheres (mean diameter 360 nm) were prepared by copolymerization of styrene with the poly(tert-butyl methacrylate) macromonomer followed by acid hydrolysis. Afterwards, NH2-lectins (ConA, lens culinaris agglutinin, galanthus nivalis lectin, and hippeastrum hybrid lectin as well as the galactose-specific ricinus communis agglutinin (RCA120)) were immobilized onto the NP's surface carboxyl groups by water soluble carbodiimide condensation (Fig. 18). The resulting conjugates were tested for HIV virion capturing. Out of the mannose-specific lectins tested, nanospheres with immobilized concanavalin A achieved up to 95% reduction of gp120 level and viral infectivity at a concentration of 0.5 mg mL−1. The PS NPs functionalized with ConA and HIV gp120 protein (HIV-NPs) were further evaluated as a vaccine delivery system. Different routes of administration to mice as well as size dependence for the capture of virions were examined. The amount of immobilized Con A per NP was dependent on the surface area of the particle. Moreover, these lectin-functionalized nanospheres with different sizes could equally capture inactivated HIV-1.336 Intranasal administration with HIV-NPs elicited the highest vaginal IgA responses and the vaginal washings neutralized the virus. In addition, macaques which were intranasally immunized with nanospheres, capturing simian/human immunodeficiency virus KU-2, exhibited partial protection when vaginally and systemically challenged with pathogenic viruses.337
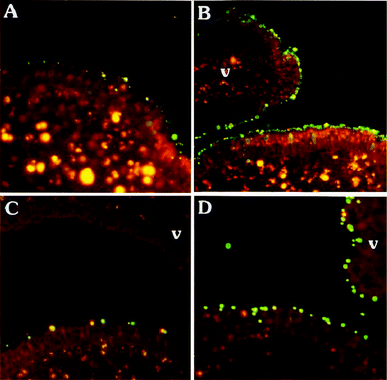 | ||
| Fig. 17 Adherence of Mal II-coated 120 nm and 1 μm particles to FAE and villus (v) epithelium. Live or aldehyde fixed Peyer's patch explants were exposed to a 1:1 mixture of Mal II-coated particles (green) and biocytin-quenched particles (red) for 40 min. A: On live tissue, Mal II-coated 120 nm particles bound preferentially to FAE, including enterocytes and M cells. B: On fixed tissue, Mal II-coated 120 nm particles bound to both FAE and villus epithelium. C: On live tissue, Mal II-coated 1 μm particles bound to FAE, but this binding was nonspecific since control particles were present in equal numbers. D: On fixed tissue, Mal II-coated particles (1 μm) bound to both FAE and villus epithelium. (scale bar = 100 μm). Reproduced from ref. 334 with permission from The American Physiological Society. | ||
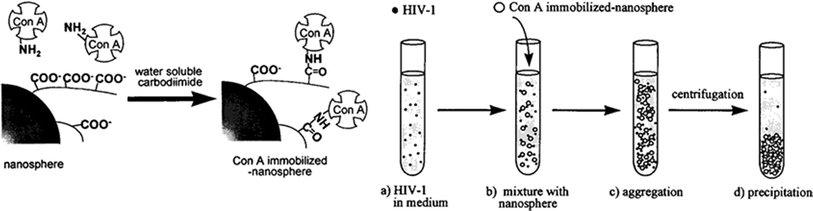 | ||
| Fig. 18 Immobilization of ConA onto the surface of polystyrene nanospheres and capture of HIV-1 virions. Reproduced from ref. 335. Copyright American Chemical Society. | ||
 | ||
| Fig. 19 Visualization of the nanoparticles in normal mucosa (M) of the ileum and in follicle-associated epithelium of Peyer's patches (PP) by fluorescence microscopy. (a) Thiamine-coated nanoparticles in normal mucosa; (b) control nanoparticles in normal mucosa; (c) thiamine-modified NPs in Peyer's patches; (d) unmodified NPs in Peyer's patches. Reproduced from ref. 343 with permission from Elsevier. | ||
In another study, vitamin B12 was either attached to the surface of the PVM/MA nanoparticles or linked to the copolymer chains in dimethylformamide prior to NP formation.345 The vitamin B12 functionalized particles were monodisperse, 200 nm in size and negatively charged. Higher association efficiency and surface stability was achieved when the coupling of the bioadhesive ligand was performed before nanoprecipitation. Also, these particles exhibited important tropism to the distal portions of the gut after oral administration to rats, which was about two and 3.5 times higher than the tropism observed for the exclusively surface functionalized and the control NPs, respectively. A further improvement of the bioadhesion was achieved by grafting different dextrans onto the polyanhydride backbone.346 Oral administration of particles pre-functionalized with dextrane induced stronger and more balanced serum anti-OVA titers of IgG2a (Th1) and IgG1 (Th2) compared to control OVA-NPs. In addition, oral immunization induced a higher mucosal IgA response than subcutaneous administration. Similar results were obtained for bioadhesive PVM/MA nanoparticles coated with flagellin, derived from Salmonella enteritidis, or mannose after oral vaccination.347 The mannosylated and flagellin-functionalized NPs demonstrated stronger, long lasting systemic and mucosal immune responses than the respective non-conjugated antigen carriers. PVM/MA was also used to improve the association of ConA to PLGA-based antigen carriers designed to target MΦs.348 The lectin-carrying, BSA-loaded microspheres, 5 to 7 μm in size, obtained by spray-drying, were four times more efficiently grafted with the ligand than plain PLGA MS. In addition, the attachment of Con A to microspheres induced oxygen consumption, increased phagocytosis efficiency and even the production of NO by MΦs. These results suggest that Con A, and possibly also other lectins, grafted onto PLGA-PVM/MA microparticles can serve as potential adjuvants by modulating protein delivery and MΦ activation
PEGylated particles have shown their mucopenetration ability as well as prolongation of their blood circulation half-life by several orders of magnitude due to formation of a hydrophilic protective layer around the nanoparticles that is able to repel the absorption of proteins.355 Therefore, block copolymers of mPEG (2000 and 5000 Da) and distearoyl phosphatidyl-ethanolamine (DSPE) were used to prepare rifampicin-loaded nanoparticles for pulmonary delivery by aerosolization.356 Lyophilized drug-loaded micelles were resuspended in hexane to give NPs and their size distribution was examined by DLS. It was observed that the size of nanoparticles decreased with an increase of PEG content in the copolymer composition. The mPEG2000-DSPE NPs varied from 230 to 400 nm, while those of mPEG5000-DSPE were from 160 to 230 nm in size depending on the formulation composition. Nevertheless, the aerodynamic characteristics of the particles as well as the entrapment efficiencies (EE) were not influenced by the molar mass of the copolymers. The EE of 100% was achieved with a drug to polymer ratio of 1:5. The particles exhibited stable in vitro drug release over the period of 3 days. Both mPEG-DSPE copolymers are therefore promising candidates for drug delivery to lungs.
 | ||
| Fig. 20 SEM image of SPM–PAA NPs (left) and CLSM microphotographs of Caco-2 cell monolayers after 2 h incubation with FITC-labeled PAA solution (0.06% w/v) (A), magnesium-PAA NPs (0.04:0.06% w/v) (B), and SPM-PAA NPs (0.05:0.06% w/v) (C). Reproduced from ref. 361 with permission from Elsevier. | ||
3.5. Particles of other synthetic polymers
In the recent years (2005–2011) a few newly developed particles composed of non-commercially available polymers have been investigated for transmucosal drug delivery.:1 with high loading efficiency (93%). The NPs did not show any sign of agglomeration over 12 h at 25 °C and were 166 nm in size and with a positive ζ value. They exhibited a maximum siRNA release after 14 h incubation in PBS pH 7.4. Moreover, P(68)-10 nanoparticles displayed a 16-fold lower toxicity than poly(ethylene imine) with 25 kDa, used as positive control in the MTT cytotoxicity assay, and did not cause proinflammatory responses after intratracheal administration to the lungs of mice, as compared to PS nanoparticles.369In vitro knockdown of firefly luciferase reporter gene was then used to demonstrate the potential of the nanoparticles as siRNA carriers in a human lung epithelial cell line (H1299 luc).368 At the optimized N:P (nitrogen to phosphorus) ratio the NPs were comparable to Lipofectamine 2000, a commercial transfection reagent. Additionally, a formulation containing pDNA-loaded P(68)-10 NPs coated with lung surfactant proved to be a potent pulmonary gene delivery vector due to its high stability during aerosolization with a vibrating mesh nebulizer and favorable biological activity.370
 | ||
| Fig. 21 Schematic representation of the synthesis of functionalized PPS-Pluronic® NPs. Reproduced from ref. 375. Copyright American Chemical Society. | ||
PPS nanoparticles of 20 to 45 nm, but not 100 nm, prepared with hydroxyl-endcapped Pluronic® were shown to target DCs in lymph nodes without the use of a targeting ligand and cause an immune response to conjugated antigen after interstitial injection.377 The Pluronic® F-127-stabilized PPS NPs with disulfide-conjugated antigenic peptides were delivered by to the APCs. They could be cross-presented on major histo-compatibility complex class I and II molecules, leading to receptor transgenic T-cell activation both in vitro and in vivo.378 The above-mentioned studies led to the development of an intranasally-administered vaccine consisting of PPS nanoparticles conjugated with thiolated OVA, as a model antigen, and a cysteine derivative of bacterial flagellin (a toll-like receptor 5 ligand), as an adjuvant.379 These particles were able to penetrate the nasal mucosa, transit via M cells and be taken up by the APCs in NALT (Fig. 22). The vaccination induced cellular and humoral responses at mucosal and systemic levels. Furthermore, the co-conjugation of the adjuvant significantly enhanced mucosal antibody levels even at distant locations, including vagina and rectum, from the site of administration, and induced a Th1 bias in cellular responses.
 | ||
| Fig. 22 The distribution of Alexa647-labeled PPS NPs (red) in nasal tissue sections shows mucosal uptake: (A) ×20 and (B) ×63 magnification at 12 h post-administration; (C) ×20 magnification at 72 h post-administration; (D) ×20 magnification of untreated nasal tissue section. Reproduced from ref. 379 with permission from Elsevier. | ||
Conclusions
The development of polymeric particles for transmucosal drug/antigen delivery is a truly interdisciplinary field of research. It requires combined knowledge of organic and polymer chemistry, particle and colloid physics, material science, biochemistry, cell and molecular biology as well as immunology and physiology to produce efficient carrier systems. To the best of our knowledge there is no universal, all-purpose transmucosal drug carrier system. Since different types of anatomorphological barriers are encountered by different methods of administration, the physicochemical properties of each and every particulate have to be evaluated and adjusted for the chosen route of delivery. In addition, the processing parameters (formulation, freeze-drying, etc.) have a significant influence on the charge distributions and polydispersity indices of the obtained particles. As a consequence, only the application of well-defined particulates, with narrow zeta potential and size variations, is able to provide accurate knowledge on the mechanisms involved in transepithelial transport as well as immunological responses. Not only the addition of various surfactants, but also the targeting moieties and their type of attachment have a strong influence on the transepithelial transport properties and have to be taken into account while designing new drug/antigen carriers. Prior to any in vitro or in vivo studies, the preservation of the biological activity of a drug, upon its release from the particles, has to be confirmed. The advantages of every polymeric material have to be carefully considered for each of the reviewed classes. Its choice depends mainly on the planned administration route since the degradation rate and porosity of the particles play the key role in the temporal as well as spacial release of the drug. Moreover, the adjuvant properties of the polymer significantly enhance the efficiency of transmucosal vaccines. However, most pharmaceutical research is concentrated on commercially available materials, which are very often inadequately characterized, and does not take advantage of novel self-assembling polymeric systems or of high-throughput particle production methods and systematic polymer libraries. The application of well-defined, temperature, pH and oxidation responsive macromolecular structures together with introduction of bioactive moieties via efficient coupling methods, e.g. “click chemistry”, will result in advanced drug/antigen carriers of tunable sizes and charges by proper formulation parameters. In addition, custom-made materials of defined molar masses yielding particles in a narrow size range can be applied to further advance the knowledge of the key aspects of mucosal immunity and provide transepithelial vaccines against mucosally transmitted diseases.LIST OF ABBREVIATIONS
| AFM | atomic force microscopy |
| AIC | air-interfaced culture |
| AM | alveolar macrophage |
| AMS | alginate microsheres |
| APC | antigen-presenting cell |
| BSA | bovine serum albumin |
| CFA | complete Freund's adjuvant |
| CLR | C-type lectin |
| CLSM | confocal laser scanning microscopy |
| Con A | concanavalin A |
| CS | chitosan |
| CTB | B-subunit of cholera toxin |
| CTL | cytotoxic T lymphocyte |
| DC | dendritic cell |
| DLS | dynamic light scattering |
| EE | encapsulation efficiency |
| FACS | fluorescence-activated cell sorting |
| FAE | follicle-associated epithelium |
| FITC | fluorescein isothiocyanate |
| GALT | gastrointestinal-associated lymphoid tissue |
| GIT | gastrointestinal tract |
| γ-PGA | poly(γ-glutamic acid) |
| HPC | hydroxypropylcellulose |
| HSMPT | high-speed multiple-particle tracking |
| IgG | immunoglobulin G |
| LCC | liquid-covered culture |
| LPS | lipopolysaccharides |
| MALT | mucosa-associated lymphoid tissue |
| MΦ | macrophage |
| MS | microspheres |
| NALT | nasopharynx-associated lymphoid tissue |
| NP | nanoparticle |
| OVA | ovalbumin |
| PAE | L-phenylalanine ethyl ester |
| PBS | phosphate buffered saline |
| PCL | poly(ε-caprolactone) |
| PEG | poly(ethylene glycol) |
| PLA | poly(D,L-lactide) |
| PLGA | poly(D,L-lactide-co-glycolide) |
| PLL | poly(L-lysine) |
| PLLA | poly(L-lactic acid) |
| PP | Peyer's patch |
| PPS | poly(propylene sulfide) |
| PRR | pattern recognition receptors |
| PS | polystyrene |
| PVA | poly(vinyl alcohol) |
| PVM/MA | poly(methyl vinyl ether-co-maleic anhydride), Gantrez® |
| RCA | ricinus communis agglutinin |
| RFP | rifampicin |
| ROP | ring opening polymerization |
| SDS-PAGE | sodium dodecyl sulphate-polyacrylamide gel electrophoresis |
| SEM | scanning electron microscopy |
| SPM | spermine |
| TEER | trans-epithelial electrical resistance |
| TJ | tight junction |
| WGA | wheat germ agglutinin |
Acknowledgements
The authors are grateful to the Thuringian Ministry for Education, Science, and Culture (grant #B514-09051, NanoConSens) and to Carl-Zeiss-Foundation for financial support, They would also like to thank Dr Stephanie Schubert for helpful comments.References
- D. T. O'Hagan, A. Wack and A. Podda, Clin. Pharmacol. Ther., 2007, 82, 740–744 CrossRef PubMed.
- M. J. Pearse and D. Drane, Adv. Drug Delivery Rev., 2005, 57, 465–474 CrossRef CAS PubMed.
- B. Heurtault, B. Frisch and F. Pons, Expert Opin. Drug Delivery, 2010, 7, 829–844 CrossRef CAS PubMed.
- J. Wilschut, Immunol. Lett., 2009, 122, 118–121 CrossRef CAS PubMed.
- W. J. Stark, Angew. Chem., Int. Ed., 2011, 50, 1242–1258 CrossRef CAS PubMed.
- A. C. Rice-Ficht, A. M. Arenas-Gamboa, M. M. Kahl-McDonagh and T. A. Ficht, Curr. Opin. Microbiol., 2010, 13, 106–112 CrossRef CAS PubMed.
- D. Mohanan, B. Slutter, M. Henriksen-Lacey, W. Jiskoot, J. A. Bouwstra, Y. Perrie, T. M. Kundig, B. Gander and P. Johansen, J. Controlled Release, 2010, 147, 342–349 CrossRef CAS PubMed.
- K. Chen and A. Cerutti, Immunity, 2010, 33, 479–491 CrossRef CAS PubMed.
- F. A. Sharp, D. Ruane, B. Claass, E. Creagh, J. Harris, P. Malyala, M. Singh, D. T. O'Hagan, V. Pétrilli, J. Tschopp, L. A. J. O'Neill and E. C. Lavelle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 870–875 CrossRef CAS PubMed.
- S. D. Xiang, A. Scholzen, G. Minigo, C. David, V. Apostolopoulos, P. L. Mottram and M. Plebanski, Methods, 2006, 40, 1–9 CrossRef CAS PubMed.
- P. Heegaard, L. Dedieu, N. Johnson, M.-F. Le Potier, M. Mockey, F. Mutinelli, T. Vahlenkamp, M. Vascellari and N. Sørensen, Arch. Virol., 2011, 156, 183–202 CrossRef CAS PubMed.
- R. A. Goldsby, T. J. Kindt, B. A. Osborne and J. Kuby, Immunology, 5th edn, W.H. Freeman, New York, 2003 Search PubMed.
- C. Nagler-Anderson, Nat. Rev. Immunol., 2001, 1, 59–67 CrossRef CAS PubMed.
- O. Borges, F. Lebre, D. Bento, G. Borchard and H. E. Junginger, Pharm. Res., 2010, 27, 211–223 CrossRef CAS PubMed.
- G. Roda, A. Sartini, E. Zambon, A. Calafiore, M. Marocchi, A. Caponi, A. Belluzzi and E. Roda, World J. Gastroenterol., 2010, 16, 4264–4271 CrossRef PubMed.
- D. A. Bland, C. A. Barrera and V. E. Reyesin Mucosal Immunology and Virology, ed. S. K. Tyring, Springer-Verlag, London, 2006, pp. 23–52 Search PubMed.
- E. Lavelle, Cell. Mol. Life Sci., 2005, 62, 2750–2770 CrossRef CAS PubMed.
- J. S. Crater and R. L. Carrier, Macromol. Biosci., 2010, 10, 1473–1483 CrossRef CAS PubMed.
- R. A. Cone, Adv. Drug Delivery Rev., 2009, 61, 75–85 CrossRef CAS PubMed.
- S. K. Lai, Y. Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171 CrossRef CAS PubMed.
- Y. Cu and W. M. Saltzman, Nat. Mater., 2009, 8, 11–13 CrossRef CAS PubMed.
- S. K. Lai, Y. Y. Wang, D. Wirtz and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 86–100 CrossRef CAS PubMed.
- R. Bansil and B. S. Turner, Curr. Opin. Colloid Interface Sci., 2006, 11, 164–170 CrossRef CAS.
- S. Kirkham, J. K. Sheehan, D. Knight, P. S. Richardson and D. J. Thornton, Biochem. J., 2002, 361, 537–546 CrossRef CAS PubMed.
- O. Svensson and T. Arnebrant, Curr. Opin. Colloid Interface Sci., 2010, 15, 395–405 CrossRef CAS.
- D. A. Norris and P. J. Sinko, J. Appl. Polym. Sci., 1997, 63, 1481–1492 CrossRef CAS.
- J. Dekker, J. W. A. Rossen, H. A. Büller and A. W. C. Einerhand, Trends Biochem. Sci., 2002, 27, 126–131 CrossRef CAS PubMed.
- J.-L. Desseyn, J.-P. Aubert, N. Porchet and A. Laine, Mol. Biol. Evol., 2000, 17, 1175–1184 CrossRef CAS PubMed.
- S. J. Gendler and A. P. Spicer, Annu. Rev. Physiol., 1995, 57, 607–634 CrossRef CAS PubMed.
- J. Perez-Vilar and R. Mabolo, Histol. Histopathol., 2007, 22, 455–464 CAS.
- D. J. Thornton, I. Carlstedt, M. Howard, P. L. Devine, M. R. Price and J. K. Sheehan, Biochem. J., 1996, 316, 967–975 CrossRef CAS PubMed.
- B. van Klinken, J. Dekker, H. A. Buller and A. W. Einerhand, Am. J. Physiol., 1995, 269, G613–G627 CAS.
- D. J. Thornton, K. Rousseau and M. A. McGuckin, Annu. Rev. Physiol., 2008, 70, 459–486 CrossRef CAS PubMed.
- M. R. Knowles and R. C. Boucher, J. Clin. Invest., 2002, 109, 571–577 CrossRef CAS PubMed.
- K. R. Bhaskar, P. Garik, B. S. Turner, J. D. Bradley, R. Bansil, H. E. Stanley and J. T. Lamont, Nature, 1992, 360, 458–461 CrossRef CAS PubMed.
- J. P. Celli, B. S. Turner, N. H. Afdhal, R. H. Ewoldt, G. H. McKinley, R. Bansil and S. Erramilli, Biomacromolecules, 2007, 8, 1580–1586 CrossRef CAS PubMed.
- S. Schreiber and P. Scheid, Am. J. Physiol.-Gastroint. Liver Physiol., 1997, 272, G63–G70 CAS.
- S. S. Hehar, J. D. T. Mason, A. B. Stephen, N. Washington, N. S. Jones, S. J. Jackson and D. Bush, Clin. Otolaryngol., 1999, 24, 24–25 CrossRef CAS PubMed.
- L. A. J. M. Creuwels, L. M. G. Van Golde and H. P. Haagsman, Lung, 1997, 175, 1–39 CrossRef CAS PubMed.
- R. Veldhuizen, K. Nag, S. Orgeig and F. Possmayer, Biochim. Biophys. Acta, Mol. Basis Dis., 1998, 1408, 90–108 CrossRef CAS.
- N. Mygind and R. Dahl, Adv. Drug Delivery Rev., 1998, 29, 3–12 CrossRef CAS.
- H. Rosen and T. Abribat, Nat. Rev. Drug Discovery, 2005, 4, 381–385 CrossRef CAS PubMed.
- J. Widdicombeed., Airway surface liquid: concepts and measurements., Birkhaüser, Basel, 1997 Search PubMed.
- M. A. Sleigh, J. R. Blake and N. Liron, Am. Rev. Respir. Dis., 1988, 137, 726–741 CrossRef CAS PubMed.
- J. Ally, A. Amirfazli and W. Roa, J. Aerosol Med.-Depos. Clear. Eff. Lung, 2006, 19, 491–509 CrossRef CAS PubMed.
- D. J. Smith, E. A. Gaffney and J. R. Blake, Respir. Physiol. Neurobiol., 2008, 163, 178–188 CrossRef CAS PubMed.
- I. Behrens, P. Stenberg, P. Artursson and T. Kissel, Pharm. Res., 2001, 18, 1138–1145 CrossRef CAS.
- M. Copeman, J. Matuz, A. J. Leonard, J. P. Pearson, P. W. Dettmar and A. Allen, J. Gastroenterol. Hepatol., 1994, 9, S55–S59 CrossRef PubMed.
- R. D. Pullan, G. A. O. Thomas, M. Rhodes, R. G. Newcombe, G. T. Williams, A. Allen and J. Rhodes, Gut, 1994, 35, 353–359 CrossRef CAS PubMed.
- K. Matsuo, H. Ota, T. Akamatsu, A. Sugiyama and T. Katsuyama, Gut, 1997, 40, 782–789 CrossRef CAS PubMed.
- L. A. van der Waaij, H. J. M. Harmsen, M. Madjipour, F. G. M. Kroese, M. Zwiers, H. M. van Dullemen, N. K. de Boer, G. W. Welling and P. L. M. Jansen, Inflamm. Bowel Dis., 2005, 11, 865–871 CrossRef PubMed.
- M. Dawson, D. Wirtz and J. Hanes, J. Biol. Chem., 2003, 278, 50393–50401 CrossRef CAS PubMed.
- D. P. Wolf, L. Blasco, M. A. Khan and M. Litt, Fertil. Steril., 1977, 28, 41–46 CAS.
- G. Forstner, Annu. Rev. Physiol., 1995, 57, 585–605 CrossRef CAS PubMed.
- R. A. Conein Mucosal Immunol., ed. M. Jiri, E. L. Michael, R. M. Jerry, B. John, M. Lloyd and S. Warren, Academic Press, Burlington, 3rd edn, 2005, pp. 49–72 Search PubMed.
- B. Amsden, Macromolecules, 1999, 32, 874–879 CrossRef CAS.
- M. Davidovich-Pinhas and H. Bianco-Peled, Expert Opin. Drug Delivery, 2010, 7, 259–271 CrossRef CAS PubMed.
- D. W. Powell, in Physiology and Membrane Dysfunctions, ed. T. E. Andreoli and S. G. Schultz, Plenum, New York, 2nd edn, 1987, pp. 559–596 Search PubMed.
- M. S. Ali and J. P. Pearson, Laryngoscope, 2007, 117, 932–938 CrossRef CAS PubMed.
- J. F. Schuhl, J. Investig. Allergol. Clin. Immunol., 1995, 5, 333–336 CAS.
- I. von Ossowski, J. Reunanen, R. Satokari, S. Vesterlund, M. Kankainen, H. Huhtinen, S. Tynkkynen, S. Salminen, W. M. de Vos and A. Palva, Appl. Environ. Microbiol., 2010, 76, 2049–2057 CrossRef CAS PubMed.
- T. T. MacDonald and G. Monteleone, Science, 2005, 307, 1920–1925 CrossRef CAS PubMed.
- P. Sherman, N. Fleming, J. Forstner, N. Roomi and G. Forstner, Am. J. Pathol., 1987, 126, 527–534 CAS.
- M. Mackay, I. Williamson and J. Hastewell, Adv. Drug Delivery Rev., 1991, 7, 313–338 CrossRef.
- P. L. Tuma and A. L. Hubbard, Physiol. Rev., 2003, 83, 871–932 CrossRef CAS PubMed.
- S. Akira, Adv. Immunol., 2001, 78, 1–56 CAS.
- S. De Koker, B. N. Lambrecht, M. A. Willart, Y. van Kooyk, J. Grooten, C. Vervaet, J. P. Remon and B. G. De Geest, Chem. Soc. Rev., 2011, 40, 320–339 RSC.
- A. Busuttil, I. A. R. More and D. McSeveney, J. Anat., 1977, 124, 445–458 CAS.
- H. Matsui, S. H. Randell, S. W. Peretti, C. W. Davis and R. C. Boucher, J. Clin. Invest., 1998, 102, 1125–1131 CrossRef CAS PubMed.
- N. Csaba, M. Garcia-Fuentes and M. J. Alonso, Adv. Drug Delivery Rev., 2009, 61, 140–157 CrossRef CAS PubMed.
- A. R. Halama, S. Decreton, J. M. Bijloos and P. A. Clement, Rhinology, 1990, 28, 25–32 CAS.
- D. P. Penney, Int. Rev. Cytol., 1988, 111, 231–269 CAS.
- M. Beck-Broichsitter, T. Schmehl, W. Seeger and T. Gessler, J. Nanomater., 2011 Search PubMed.
- T. Souma, Arch. Histol. Cytol., 1987, 50, 419–436 CrossRef CAS.
- R. J. Mason and L. G. Dobbs, J. Biol. Chem., 1980, 255, 5101–5107 CAS.
- M. J. Evans and C. G. Plopper, Am. J. Respir. Crit. Care Med., 1988, 138, 481–483 CAS.
- P. K. Jeffery, Am. J. Respir. Crit. Care Med., 1998, 157, S174–180 CrossRef PubMed.
- S. Sadahiro, T. Ohmura, Y. Yamada, T. Saito and Y. Taki, Surg. Radiol. Anat., 1992, 14, 251–257 CrossRef CAS PubMed.
- P. D. Ward, T. K. Tippin and D. R. Thakker, Pharm. Sci. Technol. Today, 2000, 3, 346–358 CrossRef CAS PubMed.
- M. A. Deli, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 892–910 CrossRef CAS PubMed.
- L. Gonzalez-Mariscal and P. Nava, Adv. Drug Delivery Rev., 2005, 57, 811–814 CrossRef CAS PubMed.
- H. Chiba, M. Osanai, M. Murata, T. Kojima and N. Sawada, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 588–600 CrossRef CAS PubMed.
- G. Krause, L. Winkler, S. L. Mueller, R. F. Haseloff, J. Piontek and I. E. Blasig, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 631–645 CrossRef CAS PubMed.
- L. Paris, L. Tonutti, C. Vannini and G. Bazzoni, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 646–659 CrossRef CAS PubMed.
- J. A. Guttman and B. B. Finlay, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 832–841 CrossRef CAS PubMed.
- M. Kondoh, T. Yoshida, H. Kakutani and K. Yagi, Drug Discovery Today, 2008, 13, 180–186 CrossRef CAS PubMed.
- J. R. McGhee, Nat. Biotechnol., 2011, 29, 136–138 CrossRef CAS PubMed.
- B. L. Dickinson, K. Badizadegan, Z. Wu, J. C. Ahouse, X. P. Zhu, N. E. Simister, R. S. Blumberg and W. I. Lencer, J. Clin. Invest., 1999, 104, 903–911 CrossRef CAS PubMed.
- L. Ye, R. Zeng, Y. Bai, D. C. Roopenian and X. Zhu, Nat. Biotechnol., 2011, 29, 158–163 CrossRef CAS PubMed.
- M. R. Neutra, N. J. Mantis and J.-P. Kraehenbuhl, Nat. Immunol., 2001, 2, 1004–1009 CrossRef CAS PubMed.
- H. Kiyono and S. Fukuyama, Nat. Rev. Immunol., 2004, 4, 699–710 CrossRef CAS PubMed.
- S. Chadwick, C. Kriegel and M. Amiji, Adv. Drug Delivery Rev., 2010, 62, 394–407 CrossRef CAS PubMed.
- P. Brandtzaeg, H. Kiyono, R. Pabst and M. W. Russell, Mucosal Immunol., 2008, 1, 31–37 CrossRef CAS PubMed.
- J. Kunisawa, S. Fukuyama and H. Kiyono, Curr. Mol. Med., 2005, 5, 557–572 CrossRef CAS PubMed.
- P. Brandtzaeg, Immunol. Invest., 2010, 39, 303–355 CrossRef CAS PubMed.
- J. Mestecky, R. S. Blumberg, H. Kiyono and J. R. Mc Ghee, ed., The Mucosal Immune System, 5th edn, Lippincott Williams and Wilkins, Philadelphia, USA, 2003 Search PubMed.
- P. L. Ogra, H. Faden and R. C. Welliver, Clin. Microbiol. Rev., 2001, 14, 430–445 CrossRef CAS PubMed.
- M. F. Cesta, Toxicol. Pathol., 2006, 34, 599–608 CrossRef PubMed.
- H. O. Alpar, S. Somavarapu, K. N. Atuah and V. W. Bramwell, Adv. Drug Delivery Rev., 2005, 57, 411–430 CrossRef CAS PubMed.
- C. L. Ohland and W. K. MacNaughton, Am. J. Physiol.: Gastrointest. Liver Physiol., 2010, 298, G807–G819 CrossRef CAS PubMed.
- R. Pabst and T. Tschernig, Am. J. Respir. Cell Mol. Biol., 2010, 43, 137–141 CrossRef CAS PubMed.
- J. S. Cornes, Gut, 1965, 6, 225–229 CrossRef CAS PubMed.
- F. Trepel, Klin. Wochenschr., 1974, 52, 511–515 CrossRef CAS PubMed.
- L. Owen and A. L. Jones, Gastroenterology, 1974, 66, 189–203 Search PubMed.
- J. Pappo and R. L. Owen, Gastroenterology, 1988, 95, 1173–1177 CAS.
- P. J. Giannasca, K. T. Giannasca, A. M. Leichtner and M. R. Neutra, Infect. Immun., 1999, 67, 946–953 CAS.
- A. Gebert and G. Hach, Gastroenterology, 1993, 105, 1350–1361 CrossRef CAS.
- R. Sharma, E. J. M. vanDamme, W. J. Peumans, P. Sarsfield and U. Schumacher, Histochem. Cell Biol., 1996, 105, 459–465 CrossRef CAS PubMed.
- E. Gullberg, A. V. Keita, S. Y. Salim, M. Andersson, K. D. Caldwell, J. D. Soderholm and P. Artursson, J. Pharmacol. Exp. Ther., 2006, 319, 632–639 CrossRef CAS PubMed.
- L. J. Hathaway and J. P. Kraehenbuhl, Cell. Mol. Life Sci., 2000, 57, 323–332 CrossRef CAS PubMed.
- A. Gebert, M. Goke, H. J. Rothkotter and C. F. Dietrich, Z. Gastroenterol., 2000, 38, 855–872 CrossRef CAS PubMed.
- A. Gebert and R. Pabst, Semin. Immunol., 1999, 11, 165–170 CrossRef CAS PubMed.
- A. Azizi, A. Kumar, F. Diaz-Mitoma and J. Mestecky, PLoS Pathog., 2010, 6 CAS , article no. e1001147.
- A. Frey, K. T. Giannasca, R. Weltzin, P. J. Giannasca, H. Reggio, W. I. Lencer and M. R. Neutra, J. Exp. Med., 1996, 184, 1045–1059 CrossRef CAS PubMed.
- M. R. Neutra, in Defense of Mucosal Surfaces: Pathogenesis, Immunity and Vaccines, Springer-Verlag Berlin, Berlin, 1999, vol. 236, pp. 17–32 Search PubMed.
- M. A. Jepson, M. A. Clark and B. H. Hirst, Adv. Drug Delivery Rev., 2004, 56, 511–525 CrossRef CAS PubMed.
- M. A. Clark, B. H. Hirst and M. A. Jepson, Infect. Immun., 1998, 66, 1237–1243 CAS.
- M. Y. Chou, K. Hartvigsen, L. F. Hansen, L. Fogelstrand, P. X. Shaw, A. Boullier, C. J. Binder and J. L. Witztum, J. Intern. Med., 2008, 263, 479–488 CrossRef CAS PubMed.
- R. KuoLee and W. X. Chen, Expert Opin. Drug Delivery, 2008, 5, 693–702 CrossRef CAS PubMed.
- A. L. Man, M. E. Prieto-Garcia and C. Nicoletti, Immunology, 2004, 113, 15–22 CrossRef CAS PubMed.
- I. Maric, P. G. Holt, M. H. Perdue and J. Bienenstock, J. Immunol., 1996, 156, 1408–1414 CAS.
- A. Porgador, H. F. Staats, Y. Itoh and B. L. Kelsall, Infect. Immun., 1998, 66, 5876–5881 CAS.
- F. L. Jahnsen, E. Gran, R. Haye and P. Brandtzaeg, Am. J. Respir. Cell Mol. Biol., 2004, 30, 31–37 CrossRef CAS PubMed.
- P. G. Holt, M. A. Schon-Hegrad, J. Oliver, B. J. Holt and P. G. McMenamin, Int. Arch. Allergy Immunol., 1990, 91, 155–159 CrossRef CAS.
- J. Banchereau and R. M. Steinman, Nature, 1998, 392, 245–252 CrossRef CAS PubMed.
- K. Shortman and Y.-J. Liu, Nat. Rev. Immunol., 2002, 2, 151–161 CrossRef CAS PubMed.
- J. Tel, A. J. A. Lambeck, L. J. Cruz, P. J. Tacken, I. J. M. de Vries and C. G. Figdor, J. Immunol., 2010, 184, 4276–4283 CrossRef CAS PubMed.
- K. Shortman and S. H. Naik, Nat. Rev. Immunol., 2007, 7, 19–30 CrossRef CAS PubMed.
- M. Rescigno, M. Urbano, B. Valzasina, M. Francolini, G. Rotta, R. Bonasio, F. Granucci, J. P. Kraehenbuhl and P. Ricciardi-Castagnoli, Nat. Immunol., 2001, 2, 361–367 CrossRef CAS PubMed.
- J. H. Niess, S. Brand, X. Gu, L. Landsman, S. Jung, B. A. McCormick, J. M. Vyas, M. Boes, H. L. Ploegh, J. G. Fox, D. R. Littman and H.-C. Reinecker, Science, 2005, 307, 254–258 CrossRef CAS PubMed.
- P. Dubsky, H. Ueno, B. Piqueras, J. Connolly, J. Banchereau and A. Palucka, J. Clin. Invest., 2005, 25, 551–572 Search PubMed.
- B. Pulendran, Immunol. Rev., 2004, 199, 227–250 CrossRef CAS PubMed.
- K. Palucka, J. Banchereau and I. Mellman, Immunity, 2010, 33, 464–478 CrossRef CAS PubMed.
- P. J. Tacken, I. J. M. de Vries, R. Torensma and C. G. Figdor, Nat. Rev. Immunol., 2007, 7, 790–802 CrossRef CAS PubMed.
- W. I. Weis, M. E. Taylor and K. Drickamer, Immunol. Rev., 1998, 163, 19–34 CrossRef CAS PubMed.
- C. G. Figdor, Y. van Kooyk and G. J. Adema, Nat. Rev. Immunol., 2002, 2, 77–84 CrossRef CAS PubMed.
- C. K. Tang, K. C. Sheng, V. Apostolopoulos and G. A. Pietersz, Expert Rev. Vaccines, 2008, 7, 1005–1018 CrossRef CAS PubMed.
- L. Z. He, A. Crocker, J. Lee, J. Mendoza-Ramirez, X. T. Wang, L. A. Vitale, T. O'Neill, C. Petromilli, H. F. Zhang, J. Lopez, D. Rohrer, T. Keler and R. Clynes, J. Immunol., 2007, 178, 6259–6267 CrossRef CAS.
- S. Espuelas, C. Thumann, B. Heurtault, F. Schuber and B. Frisch, Bioconjugate Chem., 2008, 19, 2385–2393 CrossRef CAS PubMed.
- J. B. Torrelles, A. K. Azad, L. N. Henning, T. K. Carlson and L. S. Schlesinger, Curr. Drug Targets, 2008, 9, 102–112 CrossRef CAS PubMed.
- A. Gupta, R. K. Gupta and G. S. Gupta, J. Sci. Ind. Res., 2009, 68, 465–483 CAS.
- J. M. Irache, H. H. Salman, C. Gamazo and S. Espuelas, Expert Opin. Drug Delivery, 2008, 5, 703–724 CrossRef CAS PubMed.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS PubMed.
- O. Martinez-Avila, L. M. Bedoya, M. Marradi, C. Clavel, J. Alcami and S. Penades, ChemBioChem, 2009, 10, 1806–1809 CrossRef CAS PubMed.
- S. Sattin, A. Daghetti, M. Thepaut, A. Berzi, M. Sanchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi, M. Clerici and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed.
- S. Chadwick, C. Kriegel and M. Amiji, Adv. Drug Delivery Rev., 2010, 62, 394–407 CrossRef CAS PubMed.
- A. K. Goyal, K. Khatri, N. Mishra and S. P. Vyas, Expert Opin. Ther. Pat., 2008, 18, 1271–1288 CrossRef CAS.
- M. K. Chourasia and S. K. Jain, Drug Delivery, 2004, 11, 129–148 CrossRef CAS PubMed.
- G. Ratzinger, C. Fillafer, V. Kerleta, M. Wirth and F. Gabor, Crit. Rev. Ther. Drug Carrier Syst., 2010, 27, 1–83 CrossRef CAS.
- A. Sosnik, Á. M. Carcaboso and D. A. Chiappetta, Recent Pat. Biomed. Eng., 2008, 1, 43–59 CrossRef CAS.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787–796 CrossRef CAS PubMed.
- M. G. Lu, K. T. Al-Jamal, K. Kostarelos and J. Reineke, ACS Nano, 2010, 4, 6303–6317 CrossRef PubMed.
- M. S. Duthie, H. P. Windish, C. B. Fox and S. G. Reed, Immunol. Rev., 2011, 239, 178–196 CrossRef CAS PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier and J. P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS PubMed.
- N. K. Garg, S. Mangal, H. Khambete, P. K. Sharma and R. K. Tyagi, Recent Pat. Drug Delivery Formulation, 2010, 4, 114–128 CrossRef CAS.
- N. Mishra, A. K. Goyal, S. Tiwari, R. Paliwal, S. R. Paliwal, B. Vaidya, S. Mangal, M. Gupta, D. Dube, A. Mehta and S. P. Vyas, Expert Opin. Ther. Pat., 2010, 20, 661–679 CrossRef CAS PubMed.
- D. Cooper, J. M. Ilarregui, S. A. Pesoa, D. O. Croci, M. Perretti and G. A. Rabinovich, in Methods in Enzymology, ed. F. Minoru, Academic Press, Amsterdam, 2010, vol. 480, pp. 199–244 Search PubMed.
- F. Gabor, E. Bogner, A. Weissenboeck and M. Wirth, Adv. Drug Delivery Rev., 2004, 56, 459–480 CrossRef CAS PubMed.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- A. M. Harandi, D. Medaglini and R. J. Shattock, Vaccine, 2010, 28, 2363–2366 CrossRef PubMed.
- R. L. Coffman, A. Sher and R. A. Seder, Immunity, 2010, 33, 492–503 CrossRef CAS PubMed.
- V. A. Ferro and O. Perez, Methods, 2009, 49, 299–300 CrossRef CAS PubMed.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- F. Mohamed and C. F. van der Walle, J. Pharm. Sci., 2008, 97, 71–87 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- M. S. Kim, K. S. Seo, G. Khang and H. B. Lee, Macromol. Rapid Commun., 2005, 26, 643–648 CrossRef CAS.
- V. R. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1–23 CrossRef CAS PubMed.
- J. V. Koleske, in Polymer Blends, ed. D. R. Paul and S. Newman, Academic Press, New York, 1978, vol. 2, pp. 369–389 Search PubMed.
- X. Song, X. Zhao, Y. Zhou, S. Li and Q. Ma, Curr. Drug Metab., 2010, 11, 859–869 CrossRef CAS PubMed.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- G. Mittal, D. K. Sahana, V. Bhardwaj and M. Kumar, J. Controlled Release, 2007, 119, 77–85 CrossRef CAS PubMed.
- M. Dorta, O. Munguía and M. Llabres, Int. J. Pharm., 1993, 100, 9–14 CrossRef CAS.
- L. Brandon-Peppas and M. Vert, in Handbook of pharmaceutical controlled release technology, ed. D. L. Wise, Marcel Dekker, New York, 2000 Search PubMed.
- D. A. Perrin and J. P. English, in Handbook of biodegradable polymers, ed. A. J. Domb and D. M. Wiseman, Harwood Academic Publishers, Amsterdam, 1997 Search PubMed.
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS PubMed.
- V. Lassalle and M. L. Ferreira, J. Chem. Technol. Biotechnol., 2010, 85, 1588–1596 CrossRef CAS.
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS PubMed.
- J. Emami, H. Hamishehkar, A. R. Najafabadi, K. Gilani, M. Minaiyan, H. Mahdavi, H. Mirzadeh, A. Fakhari and A. Nokhodchi, J. Microencapsulation, 2009, 26, 1–8 CrossRef CAS PubMed.
- C. Springer, S. Benita, Y. Sherman, N. Gursoy, D. Gilhar and A. Avital, Pediatr. Res., 2005, 58, 537–541 CrossRef CAS PubMed.
- R. Dinarvand, N. Sepehri, S. Manoochehri, H. Rouhani and F. Atyabi, Int. J. Nanomed., 2011, 6, 877–895 CrossRef CAS PubMed.
- K. Lane, B. Egan, S. Vick, R. Abdolrasulnia and V. Shepherd, J. Leukocyte Biol., 1998, 64, 345–350 CAS.
- J. L. Stafford, N. F. Neumann and M. Belosevic, Crit. Rev. Microbiol., 2002, 28, 187–248 CrossRef PubMed.
- F. O. Martinez, L. Helming and S. Gordon, Annu. Rev. Immunol., 2009, 27, 451–483 CrossRef CAS PubMed.
- A. M. Torché, E. Albina, P. Le Corre, A. Jestin and R. Le Verge, J. Controlled Release, 1999, 58, 289–301 CrossRef.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- A. M. Torché, P. Le Corre, E. Albina, A. Jestin and R. Le Verge, J. Drug Targeting, 2000, 7, 343–354 CrossRef.
- B. Charley, Comp. Immunol., Microbiol. Infect. Dis., 1985, 8, 99–108 CrossRef CAS.
- G. J. Cannon and J. A. Swanson, J. Cell Sci., 1992, 101, 907–913 Search PubMed.
- Y. Tabata and Y. Ikada, Biomaterials, 1988, 9, 356–362 CrossRef CAS PubMed.
- M. Koval, K. Preiter, C. Adles, P. D. Stahl and T. H. Steinberg, Exp. Cell Res., 1998, 242, 265–273 CrossRef CAS PubMed.
- K. Hirota, T. Hasegawa, H. Hinata, F. Ito, H. Inagawa, C. Kochi, G. I. Soma, K. Makino and H. Terada, J. Controlled Release, 2007, 119, 69–76 CrossRef CAS PubMed.
- F. Ito and K. Makino, Colloids Surf., B, 2004, 39, 17–21 CrossRef CAS PubMed.
- D. L. Biggs, C. S. Lengsfeld, B. M. Hybertson, K.-y. Ng, M. C. Manning and T. W. Randolph, J. Controlled Release, 2003, 92, 147–161 CrossRef CAS PubMed.
- P. O'Hara and A. J. Hickey, Pharm. Res., 2000, 17, 955–961 CrossRef.
- A. Yoshida, M. Matumoto, H. Hshizume, Y. Oba, T. Tomishige, H. Inagawa, C. Kohchi, M. Hino, F. Ito, K. Tomoda, T. Nakajima, K. Makino, H. Terada, H. Hori and G.-I. Soma, Microbes Infect., 2006, 8, 2484–2491 CrossRef CAS PubMed.
- S. Suarez, P. O'Hara, M. Kazantseva, C. E. Newcomer, R. Hopfer, D. N. McMurray and A. J. Hickey, J. Antimicrob. Chemother., 2001, 48, 431–434 CrossRef CAS PubMed.
- N. Brandhonneur, F. Chevanne, V. Vie, B. Frisch, R. Primault, M. F. Le Potier and P. Le Corre, Eur. J. Pharm. Sci., 2009, 36, 474–485 CrossRef CAS PubMed.
- B. G. Jones, P. A. Dickinson, M. Gumbleton and I. W. Kellaway, Int. J. Pharm., 2002, 236, 65–79 CrossRef CAS PubMed.
- S. Faraasen, J. Voros, G. Csucs, M. Textor, H. P. Merkle and E. Walter, Pharm. Res., 2003, 20, 237–246 CrossRef CAS.
- E. A. Poyner, H. O. Alpar, A. J. Almeida, M. D. Gamble and M. R. W. Brown, J. Controlled Release, 1995, 35, 41–48 CrossRef CAS.
- K. Makino, N. Yamamoto, K. Higuchi, N. Harada, H. Ohshima and H. Terada, Colloids Surf., B, 2003, 27, 33–39 CrossRef CAS.
- K. Ohashi, T. Kabasawa, T. Ozeki and H. Okada, J. Controlled Release, 2009, 135, 19–24 CrossRef CAS PubMed.
- J. Fiegel, C. Ehrhardt, U. F. Schaefer, C.-M. Lehr and J. Hanes, Pharm. Res., 2003, 20, 788–796 CrossRef CAS.
- O. Harush-Frenkel, M. Bivas-Benita, T. Nassar, C. Springer, Y. Sherman, A. Avital, Y. Altschuler, J. Borlak and S. Benita, Toxicol. Appl. Pharmacol., 2010, 246, 83–90 CrossRef CAS PubMed.
- H. Yamamoto, Y. Kuno, S. Sugimoto, H. Takeuchi and Y. Kawashima, J. Controlled Release, 2005, 102, 373–381 CrossRef CAS PubMed.
- H. Takeuchi, Y. Shimotori, H. Yamamoto, T. Hino and Y. Kawashima, Drug Delivery Syst., 1997, 12, 347–352 CrossRef CAS.
- N. G. M. Schipper, S. Olsson, J. A. Hoogstraate, A. G. deBoer, K. M. Vårum and P. Artursson, Pharm. Res., 1997, 14, 923–929 CrossRef CAS.
- V. Dodane, M. Amin Khan and J. R. Merwin, Int. J. Pharm., 1999, 182, 21–32 CrossRef CAS PubMed.
- Y. Kawashima, H. Yamamoto, H. Takeuchi and Y. Kuno, Pharm. Dev. Technol., 2000, 5, 77–85 CrossRef CAS PubMed.
- Y. Kawashima, H. Yamamoto, H. Takeuchi, S. Fujioka and T. Hino, J. Controlled Release, 1999, 62, 279–287 CrossRef CAS PubMed.
- M. L. Manca, S. Mourtas, V. Dracopoulos, A. M. Fadda and S. G. Antimisiaris, Colloids Surf., B, 2008, 62, 220–231 CrossRef CAS PubMed.
- A. Berthold, K. Cremer and J. Kreuter, J. Controlled Release, 1996, 39, 17–25 CrossRef CAS.
- M. Kumar, S. S. Mohapatra, X. Kong, P. K. Jena, U. Bakowsky and C. M. Lehr, J. Nanosci. Nanotechnol., 2004, 4, 990–994 CrossRef CAS PubMed.
- B. Bharatwaj, L. B. Wu, J. A. Whittum-Hudson and S. R. P. da Rocha, Biomaterials, 2010, 31, 7376–7385 CrossRef CAS PubMed.
- A. Vila, A. Sanchez, M. Tobio, P. Calvo and M. J. Alonso, J. Controlled Release, 2002, 78, 15–24 CrossRef CAS PubMed.
- M. Tobío, A. Sánchez, A. Vila, I. Soriano, C. Evora, J. L. Vila-Jato and M. J. Alonso, Colloids Surf., B, 2000, 18, 315–323 CrossRef.
- M. Tobío, R. Gref, A. Sánchez, R. Langer and M. J. Alonso, Pharm. Res., 1998, 15, 270–275 CrossRef.
- R. Fernández-Urrusuno, P. Calvo, C. Remuñán-López, J. L. Vila-Jato and M. José Alonso, Pharm. Res., 1999, 16, 1576–1581 CrossRef.
- P. Calvo, C. Remuñan-López, J. L. Vila-Jato and M. J. Alonso, Pharm. Res., 1997, 14, 1431–1436 CrossRef CAS.
- P. Calvo, J. L. Vila-Jato and M. J. Alonso, Biomaterials, 1997, 18, 1305–1310 CrossRef CAS PubMed.
- T. Jung, W. Kamm, A. Breitenbach, E. Kaiserling, J. X. Xiao and T. Kissel, Eur. J. Pharm. Biopharm., 2000, 50, 147–160 CrossRef CAS PubMed.
- K. S. Jaganathan and S. P. Vyas, Vaccine, 2006, 24, 4201–4211 CrossRef CAS PubMed.
- A. F. Azevedo, J. Galhardas, A. Cunha, P. Cruz, L. M. D. Gonçalves and A. J. Almeida, Eur. J. Pharm. Biopharm., 2006, 64, 131–137 CrossRef CAS PubMed.
- M. A. Benoit, B. Baras and J. Gillard, Int. J. Pharm., 1999, 184, 73–84 CrossRef CAS PubMed.
- V. W. Bramwell and Y. Perrie, J. Pharm. Pharmacol., 2006, 58, 717–728 CrossRef CAS PubMed.
- A. Caputo, K. Sparnacci, B. Ensoli and L. Tondelli, Curr. Drug Delivery, 2008, 5, 230–242 CrossRef CAS.
- H. F. Florindo, S. Pandit, L. M. D. Goncalves, H. O. Alpar and A. J. Almeida, Int. J. Pharm., 2010, 390, 25–31 CrossRef CAS PubMed.
- H. F. Florindo, S. Pandit, L. Lacerda, L. M. D. Gonçalves, H. O. Alpar and A. J. Almeida, Biomaterials, 2009, 30, 879–891 CrossRef CAS PubMed.
- H. F. Florindo, S. Pandit, L. M. D. Goncalves, H. O. Alpar and A. J. Almeida, Vaccine, 2009, 27, 1230–1241 CrossRef CAS PubMed.
- M. D. Donovan and Y. Huang, Adv. Drug Delivery Rev., 1998, 29, 147–155 CrossRef CAS.
- D. F. Nixon, C. Hioe, P. D. Chen, Z. N. Bian, P. Kuebler, M. L. Li, H. Qiu, X. M. Li, M. Singh, J. Richardson, P. McGee, T. Zamb, W. Koff, C. Y. Wang and D. Ohagan, Vaccine, 1996, 14, 1523–1530 CrossRef CAS PubMed.
- S. L. Demento, S. C. Eisenbarth, H. G. Foellmer, C. Platt, M. J. Caplan, W. M. Saltzman, I. Mellman, M. Ledizet, E. Fikrig, R. A. Flavell and T. M. Fahmy, Vaccine, 2009, 27, 3013–3021 CrossRef CAS PubMed.
- K. D. Newman, P. Elamanchili, G. S. Kwon and J. Samuel, J. Biomed. Mater. Res., 2002, 60, 480–486 CrossRef CAS PubMed.
- C. Thomasin, N. T. Ho, H. P. Merkle and B. Gander, J. Pharm. Sci., 1998, 87, 259–268 CrossRef CAS PubMed.
- A. R. Patel, S. Kulkarni, T. D. Nandekar and P. R. Vavia, J. Microencapsulation, 2008, 25, 531–540 CrossRef CAS PubMed.
- T. E. Rajapaksa and D. D. Lo, Curr. Immunol. Rev., 2010, 6, 29–37 CrossRef CAS.
- M. Kondoh, A. Takahashi, M. Fujii, K. Yagi and Y. Watanabe, Biol. Pharm. Bull., 2006, 29, 1783–1789 CAS.
- J. Ling, H. Liao, R. Clark, M. S. M. Wong and D. D. Lo, J. Biol. Chem., 2008, 283, 30585–30595 CrossRef CAS PubMed.
- T. E. Rajapaksa, K. M. Bennett, M. Hamer, C. Lytle, V. G. J. Rodgers and D. D. Lo, J. Biol. Chem., 2010, 285, 23739–23746 CrossRef CAS PubMed.
- A. M. Hillery and A. T. Florence, Int. J. Pharm., 1996, 132, 123–130 CrossRef CAS.
- A. Weissenboeck, E. Bogner, M. Wirth and F. Gabor, Pharm. Res., 2004, 21, 1917–1923 CrossRef CAS.
- G. J. Russell-Jones, H. Veitch and L. Arthur, Int. J. Pharm., 1999, 190, 165–174 CrossRef CAS PubMed.
- B. Weiss, M. Schneider, L. Muys, S. Taetz, D. Neumann, U. F. Schaefer and C. M. Lehr, Bioconjugate Chem., 2007, 18, 1087–1094 CrossRef CAS PubMed.
- N. M. Green, in Advances in Protein Chemistry, ed. C.B. Anfinsen, J. J. T. Edsall and F. M. Richards, Academic Press, London, 1975, vol. 29, pp. 85–133 Search PubMed.
- V. Pourcelle, H. Freichels, F. Stoffelbach, R. Auzély-Velty, C. Jérôme and J. Marchand-Brynaert, Biomacromolecules, 2009, 10, 966–974 CrossRef CAS PubMed.
- M. Garinot, V. Fievez, V. Pourcelle, F. Stoffelbach, A. des Rieux, L. Plapied, I. Theate, H. Freichels, C. Jerome, J. Marchand-Brynaert, Y. J. Schneider and V. Preat, J. Controlled Release, 2007, 120, 195–204 CrossRef CAS PubMed.
- V. Fievez, L. Plapied, A. des Rieux, V. Pourcelle, H. Freichels, V. Wascotte, M.-L. Vanderhaeghen, C. Jerôme, A. Vanderplasschen, J. Marchand-Brynaert, Y.-J. Schneider and V. Préat, Eur. J. Pharm. Biopharm., 2009, 73, 16–24 CrossRef CAS PubMed.
- J. H. Eldridge, C. J. Hammond, J. A. Meulbroek, J. K. Staas, R. M. Gilley and T. R. Tice, J. Controlled Release, 1990, 11, 205–214 CrossRef CAS.
- S. J. Challacombe, D. Rahman and D. T. Ohagan, Vaccine, 1997, 15, 169–175 CrossRef CAS PubMed.
- S. J. Challacombe, D. Rahman, H. Jeffery, S. S. Davis and D. T. Ohagan, Immunology, 1992, 76, 164–168 CAS.
- A. Delgado, E. C. Lavelle, M. Hartshorne and S. S. Davis, Vaccine, 1999, 17, 2927–2938 CrossRef CAS PubMed.
- V. Grabovac and A. Bernkop-Schnurch, Drug Dev. Ind. Pharm., 2007, 33, 767–774 CrossRef CAS PubMed.
- C. Damge, M. Socha, N. Ubrich and P. Maincent, J. Pharm. Sci., 2010, 99, 879–889 CAS.
- J. Wendorf, M. Singh, J. Chesko, J. Kazzaz, E. Soewanan, M. Ugozzoli and D. O'Hagan, J. Pharm. Sci., 2006, 95, 2738–2750 CrossRef CAS PubMed.
- G. A. Brazeau, B. Cooper, K. A. Svetic, C. L. Smith and P. Gupta, J. Pharm. Sci., 1998, 87, 667–677 CrossRef CAS PubMed.
- T. K. Lindhorst, Essentials of Carbohydrate Chemistry and Biochemistry, WILEY-VCH Verlag GmbH, Weinheim, 2000 Search PubMed.
- X. Feng, J. East Anthony, W. Hammond and M. Jaffe, in Contemporary Science of Polymeric Materials, American Chemical Society, 2010, vol. 1061, pp. 3–27 Search PubMed.
- R. Raman, S. Raguram, G. Venkataraman, J. C. Paulson and R. Sasisekharan, Nat. Methods, 2005, 2, 817–824 CrossRef CAS PubMed.
- R. Sasisekharan, R. Raman and V. Prabhakar, Annu. Rev. Biomed. Eng., 2006, 8, 181–231 CrossRef CAS PubMed.
- S. Boddohi and M. J. Kipper, Adv. Mater., 2010, 22, 2998–3016 CrossRef CAS PubMed.
- C. Lemarchand, R. Gref and P. Couvreur, Eur. J. Pharm. Biopharm., 2004, 58, 327–341 CrossRef CAS PubMed.
- S. Maher, D. J. Brayden, L. Feighery and S. McClean, Crit. Rev. Ther. Drug Carrier Syst., 2008, 25, 117–168 CrossRef CAS PubMed.
- G. Di Colo, Y. Zambito and C. Zaino, J. Pharm. Sci., 2008, 97, 1652–1680 CrossRef CAS PubMed.
- F. Esmaeili, S. Heuking, H. E. Junginger and G. Borchard, J. Drug Deliv. Sci. Technol., 2010, 20, 53–61 CrossRef CAS.
- M. Werle and A. Bernkop-Schnürch, J. Pharm. Pharmacol., 2008, 60, 273–281 CrossRef CAS PubMed.
- S. M. van der Merwe, J. C. Verhoef, J. H. M. Verheijden, A. F. Kotzé and H. E. Junginger, Eur. J. Pharm. Biopharm., 2004, 58, 225–235 CrossRef CAS PubMed.
- M. Amidi, E. Mastrobattista, W. Jiskoot and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62, 59–82 CrossRef CAS PubMed.
- N. Duceppe and M. Tabrizian, Expert Opin. Drug Delivery, 2010, 7, 1191–1207 CrossRef CAS PubMed.
- C. Caramella, F. Ferrari, M. C. Bonferoni, S. Rossi and G. Sandri, J. Drug Deliv. Sci. Technol., 2010, 20, 5–13 CrossRef CAS.
- V. Kumar and G. S. Banker, Drug Dev. Ind. Pharm., 1993, 19, 1–31 CrossRef CAS.
- J. W. Lee, J. H. Park and J. R. Robinson, J. Pharm. Sci., 2000, 89, 850–866 CrossRef CAS PubMed.
- M. Sakagami, K. Sakon, W. Kinoshita and Y. Makino, J. Controlled Release, 2001, 77, 117–129 CrossRef CAS PubMed.
- M. Sakagami, W. Kinoshita, K. Sakon, J.-I. Sato and Y. Makino, J. Controlled Release, 2002, 80, 207–218 CrossRef CAS PubMed.
- N. Sivadas, D. O'Rourke, A. Tobin, V. Buckley, Z. Ramtoola, J. G. Kelly, A. J. Hickey and S. A. Cryan, Int. J. Pharm., 2008, 358, 159–167 CrossRef CAS PubMed.
- V. M. Leitner, D. Guggi, A. H. Krauland and A. Bernkop-Schnurch, J. Controlled Release, 2004, 100, 87–95 CrossRef CAS PubMed.
- W. R. Gombotz and S. Wee, Adv. Drug Delivery Rev., 1998, 31, 267–285 CrossRef CAS.
- M. George and T. E. Abraham, J. Controlled Release, 2006, 114, 1–14 CrossRef CAS PubMed.
- L. W. Chan, H. Y. Lee and P. W. S. Heng, Int. J. Pharm., 2002, 242, 259–262 CrossRef CAS PubMed.
- L. W. Chan and P. W. S. Heng, Biomaterials, 2002, 23, 1319–1326 CrossRef CAS PubMed.
- J. T. Delaney, A. R. Liberski, J. Perelaer and U. S. Schubert, Soft Matter, 2010, 6, 866–869 RSC.
- N. Mofidi, M. Aghai-Moghadam and M. N. Sarbolouki, Process Biochem., 2000, 35, 885–888 CrossRef CAS.
- D. Lemoine, F. Wauters, S. Bouchend'homme and V. Préat, Int. J. Pharm., 1998, 176, 9–19 CrossRef CAS.
- P. A. Offit, C. A. Khoury, C. A. Moser, H. F. Clark, J. E. Kim and T. J. Speaker, Virology, 1994, 203, 134–143 CrossRef CAS PubMed.
- S. Periwal, T. Speaker and J. Cebra, J. Virol., 1997, 71, 2844–2850 CAS.
- N.-H. Cho, S.-Y. Seong, K.-H. Chun, Y.-H. Kim, I. Chan Kwon, B.-Y. Ahn and S. Y. Jeong, J. Controlled Release, 1998, 53, 215–224 CrossRef CAS PubMed.
- S.-Y. Seong, N.-H. Cho, I. C. Kwon and S. Y. Jeong, Infect. Immun., 1999, 67, 3587–3592 CAS.
- J. Y. Seo, S. Y. Seong, B. Y. Ahn, I. C. Kwon, H. Chung and S. Y. Jeong, Infect. Immun., 2002, 70, 1143–1149 CrossRef CAS PubMed.
- O. Borges, G. Borchard, J. C. Verhoef, A. de Sousa and H. E. Junginger, Int. J. Pharm., 2005, 299, 155–166 CrossRef CAS PubMed.
- O. Borges, J. Tavares, A. de Sousa, G. Borchard, H. E. Junginger and A. Cordeiro-Da-Silva, Eur. J. Pharm. Sci., 2007, 32, 278–290 CrossRef CAS PubMed.
- B. Sarmento, A. Ribeiro, F. Veiga, P. Sampaio, R. Neufeld and D. Ferreira, Pharm. Res., 2007, 24, 2198–2206 CrossRef CAS PubMed.
- R. Rastogi, Y. Sultana, M. Aqil, A. Ali, S. Kumar, K. Chuttani and A. K. Mishra, Int. J. Pharm., 2007, 334, 71–77 CrossRef CAS PubMed.
- C.-K. Kim and E.-J. Lee, Int. J. Pharm., 1992, 79, 11–19 CrossRef CAS.
- S. Alipour, H. Montaseri and M. Tafaghodi, Colloids Surf., B, 2010, 81, 521–529 CrossRef CAS PubMed.
- http://www.starch.dk/isi/market/index.asp. .
- http://www.nnfcc.co.uk/publications/nnfcc-renewable-chemicals-factsheet-starch. .
- A. M. Smith, Biomacromolecules, 2001, 2, 335–341 CrossRef CAS PubMed.
- P. Artursson, P. Edman, T. Laakso and I. Sjöholm, J. Pharm. Sci., 1984, 73, 1507–1513 CrossRef CAS PubMed.
- L. Elfstrand, A. C. Eliasson and M. Wahlgren, J. Pharm. Sci., 2009, 98, 3802–3815 CrossRef CAS PubMed.
- L. Degling and P. Stjärnkvist, Vaccine, 1995, 13, 629–636 CrossRef CAS PubMed.
- L. D. Wikingsson and I. Sjöholm, Vaccine, 2002, 20, 3355–3363 CrossRef CAS PubMed.
- L. Stertman, L. Strindelius and I. Sjöholm, Vaccine, 2004, 22, 2863–2872 CrossRef CAS PubMed.
- P. L. Heritage, L. M. Loomes, J. Jianxiong, M. A. Brook, B. J. Underdown and M. R. McDermott, Immunology, 1996, 88, 162–168 CrossRef CAS PubMed.
- M. P. Flanagan and J. G. Michael, Vaccine, 1999, 17, 72–81 CrossRef CAS PubMed.
- M. A. Dea-Ayuela, S. Rama-Iniguez, S. Torrado-Santiago and F. Bolas-Fernandez, J. Drug Targeting, 2006, 14, 567–575 CrossRef CAS PubMed.
- L. Strindelius, L. D. Wikingsson and I. Sjoholm, Infect. Immun., 2002, 70, 1434–1442 CrossRef CAS PubMed.
- D. Coucke, M. Schotsaert, C. Libert, E. Pringels, C. Vervaet, P. Foreman, X. Saelens and J. P. Remon, Vaccine, 2009, 27, 1279–1286 CrossRef CAS PubMed.
- J. M. Buescher and A. Margaritis, Crit. Rev. Biotechnol., 2007, 27, 1–19 CrossRef CAS PubMed.
- B. Manocha and A. Margaritis, Crit. Rev. Biotechnol., 2008, 28, 83–99 CrossRef CAS PubMed.
- H. Poo, C. Park, M.-S. Kwak, D.-Y. Choi, S.-P. Hong, I.-H. Lee, Y. T. Lim, Y. K. Choi, S.-R. Bae, H. Uyama, C.-J. Kim and M.-H. Sung, Chem. Biodiversity, 2010, 7, 1555–1562 CAS.
- M.-H. Sung, C. Park, C.-J. Kim, H. Poo, K. Soda and M. Ashiuchi, Chem. Rec., 2005, 5, 352–366 CrossRef CAS PubMed.
- M. Matsusaki, K.-i. Hiwatari, M. Higashi, T. Kaneko and M. Akashi, Chem. Lett., 2004, 33, 398–399 CrossRef CAS.
- T. Akagi, T. Kaneko, T. Kida and M. Akashi, J. Controlled Release, 2005, 108, 226–236 CrossRef CAS PubMed.
- T. Akagi, M. Higashi, T. Kaneko, T. Kida and M. Akashi, Macromol. Biosci., 2005, 5, 598–602 CrossRef CAS PubMed.
- T. Akagi, T. Kaneko, T. Kida and M. Akashi, J. Biomater. Sci., Polym. Ed., 2006, 17, 875–892 CrossRef CAS PubMed.
- E. C. Lavelle and D. T. O'Hagan, Expert Opin. Drug Delivery, 2006, 3, 747–762 CrossRef CAS PubMed.
- T. Uto, X. Wang, K. Sato, M. Haraguchi, T. Akagi, M. Akashi and M. Baba, J. Immunol., 2007, 178, 2979–2986 CrossRef CAS.
- X. Wang, T. Uto, T. Akagi, M. Akashi and M. Baba, J. Virol., 2007, 81, 10009–10016 CrossRef CAS PubMed.
- T. Akagi, X. Wang, T. Uto, M. Baba and M. Akashi, Biomaterials, 2007, 28, 3427–3436 CrossRef CAS PubMed.
- X. Wang, T. Uto, T. Akagi, M. Akashi and M. Baba, J. Med. Virol., 2008, 80, 11–19 CrossRef CAS PubMed.
- A. Himeno, T. Akagi, T. Uto, X. Wang, M. Baba, K. Ibuki, M. Matsuyama, M. Horiike, T. Igarashi, T. Miura and M. Akashi, Vaccine, 2010, 28, 5377–5385 CrossRef CAS PubMed.
- P. Kiepiela, K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker and P. Goulder, Nat. Med., 2007, 13, 46–53 CrossRef CAS PubMed.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- J. P. Magnusson, A. O. Saeed, F. Fernandez-Trillo and C. Alexander, Polym. Chem., 2011, 2, 48–59 RSC.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011 Search PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- http://www.polysciences.com/Catalog/Department/81/categoryId__372/. .
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Eur. J. Immunol., 2008, 38, 1404–1413 CrossRef CAS PubMed.
- K. E. Carr, R. A. Hazzard, S. Reid and G. M. Hodges, Pharm. Res., 1996, 13, 1205–1209 CrossRef CAS.
- A. Henning, M. Schneider, N. Nafee, L. Muijs, E. Rytting, X. Y. Wang, T. Kissel, D. Grafahrend, D. Klee and C. M. Lehr, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, 233–241 CrossRef CAS PubMed.
- J. E. Eyles, I. D. Spiers, E. D. Williamson and H. O. Alpar, J. Pharm. Pharmacol., 2001, 53, 601–607 CrossRef CAS PubMed.
- S. K. Lai, D. E. O'Hanlon, S. Harrold, S. T. Man, Y. Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1482–1487 CrossRef CAS PubMed.
- N. J. Mantis, A. Frey and M. R. Neutra, Am. J. Physiol. Gastrointest. Liver Physiol., 2000, 278, G915–923 CAS.
- M. Akashi, T. Niikawa, T. Serizawa, T. Hayakawa and M. Baba, Bioconjugate Chem., 1998, 9, 50–53 CrossRef CAS PubMed.
- T. Akagi, M. Ueno, K. Hiraishi, M. Baba and M. Akashi, J. Controlled Release, 2005, 109, 49–61 CrossRef CAS PubMed.
- A. Miyake, T. Akagi, Y. Enose, M. Ueno, M. Kawamura, R. Horiuchi, K. Hiraishi, M. Adachi, T. Serizawa, O. Narayan, M. Akashi, M. Baba and M. Hayami, J. Med. Virol., 2004, 73, 368–377 CrossRef CAS PubMed.
- http://online1.ispcorp.com/en-US/Pages/ProductDetail.aspx?BU=Performance%20Chemicals&l1=Trade%20Name&l2=Gantrez%C2%AE&prodName=Gantrez%C2%AE%20AN-119&prdId=74000. .
- L. A. Scalf and J. F. Fowler, J. Am. Acad. Dermatol., 2000, 42, 355–356 CrossRef CAS PubMed.
- P. Arbós, M. Wirth, M. A. Arangoa, F. Gabor and J. M. Irache, J. Controlled Release, 2002, 83, 321–330 CrossRef.
- N. Shibuya, I. J. Goldstein, W. F. Broekaert, M. Nsimba-Lubaki, B. Peeters and W. J. Peumans, J. Biol. Chem., 1987, 262, 1596–1601 CAS.
- W. F. Broekaert, M. Nsimba-Lubaki, B. Peeters and W. J. Peumans, Biochem. J., 1984, 221, 163–169 CrossRef CAS PubMed.
- H. H. Salman, C. Gamazo, M. Agüeros and J. M. Irache, Vaccine, 2007, 25, 8123–8132 CrossRef CAS PubMed.
- I. Tamayo, J. M. Irache, C. Mansilla, J. Ochoa-Reparaz, J. J. Lasarte and C. Gamazo, Clin. Vaccine Immunol., 2010, 17, 1356–1362 CrossRef CAS PubMed.
- H. Salman, C. Gamazo, P. de Smidt, G. Russell-Jones and J. Irache, Pharm. Res., 2008, 25, 2859–2868 CrossRef CAS PubMed.
- A. S. Porfire, V. Zabaleta, C. Gamazo, S. E. Leucuta and J. M. Irache, Int. J. Pharm., 2010, 390, 37–44 CrossRef CAS PubMed.
- H. H. Salman, J. M. Irache and C. Gamazo, Vaccine, 2009, 27, 4784–4790 CrossRef CAS PubMed.
- L. León-Rodriguez, J. Leiro-Vidal, J. Blanco-Méndez and A. Luzardo-Álvarez, Int. J. Pharm., 2010, 402, 165–174 CrossRef PubMed.
- J. Kopecek and P. Kopeckova, Adv. Drug Delivery Rev., 2010, 62, 122–149 CrossRef CAS PubMed.
- I. Schmolka, J. Am. Oil Chem. Soc., 1977, 54, 110–116 CrossRef CAS.
- R. Strickley, Pharm. Res., 2004, 21, 201–230 CrossRef CAS.
- J. Cleary, L. Bromberg and E. Magner, Langmuir, 2004, 20, 9755–9762 CrossRef CAS PubMed.
- M. Yang, S. K. Lai, Y. Y. Wang, W. X. Zhong, C. Happe, M. Zhang, J. Fu and J. Hanes, Angew. Chem., Int. Ed., 2011, 50, 2597–2600 CrossRef CAS PubMed.
- I. K. Han, Y. B. Kim, H. S. Kang, D. Sul, W. W. Jung, H. J. Cho and Y. K. Oh, Methods, 2006, 38, 106–111 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- J. M. A. Abdulla, Y. T. F. Tan and Y. Darwis, AAPS PharmSciTech, 2010, 11, 663–671 CrossRef PubMed.
- http://eudragit.evonik.com/product/eudragit/en/Pages/default.aspx. .
- K. Sparnacci, M. Laus, L. Tondelli, C. Bernardi, L. Magnani, F. Corticelli, M. Marchisio, B. Ensoli, A. Castaldello and A. Caputo, J. Biomater. Sci., Polym. Ed., 2005, 15, 1557–1574 CrossRef.
- R. Voltan, A. Castaldello, E. Brocca-Cofano, G. Altavilla, A. Caputo, M. Laus, K. Sparnacci, B. Ensoli, S. Spaccasassi, M. Ballestri and L. Tondelli, Pharm. Res., 2007, 24, 1870–1882 CrossRef CAS PubMed.
- A. Caputo, A. Castaldello, E. Brocca-Cofano, R. Voltan, F. Bortolazzi, G. Altavilla, K. Sparnacci, M. Laus, L. Tondelli, R. Gavioli and B. Ensoli, Vaccine, 2009, 27, 3605–3615 CrossRef CAS PubMed.
- A. Makhlof, M. Werle, Y. Tozuka and H. Takeuchi, J. Controlled Release, 2011, 149, 81–88 CrossRef CAS PubMed.
- A. E. Clausen and A. Bernkop-Schnürch, J. Pharm. Sci., 2000, 89, 1253–1261 CrossRef CAS PubMed.
- A. Bernkop-Schnürch and S. Steininger, Int. J. Pharm., 2000, 194, 239–247 CrossRef.
- A. Bernkop-Schnürch, H. Zarti and G. F. Walker, J. Pharm. Sci., 2001, 90, 1907–1914 CrossRef PubMed.
- L. A. Dailey, M. Wittmar and T. Kissel, J. Controlled Release, 2005, 101, 137–149 CrossRef CAS PubMed.
- M. Beck-Broichsitter, J. Gauss, C. B. Packhaeuser, K. Lahnstein, T. Schmehl, W. Seeger, T. Kissel and T. Gessler, Int. J. Pharm., 2009, 367, 169–178 CrossRef CAS PubMed.
- M. Wittmar, F. Unger and T. Kissel, Macromolecules, 2006, 39, 1417–1424 CrossRef CAS.
- J. Nguyen, T. W. J. Steele, O. Merkel, R. Reul and T. Kissel, J. Controlled Release, 2008, 132, 243–251 CrossRef CAS PubMed.
- L. A. Dailey, N. Jekel, L. Fink, T. Gessler, T. Schmehl, M. Wittmar, T. Kissel and W. Seeger, Toxicol. Appl. Pharmacol., 2006, 215, 100–108 CrossRef CAS PubMed.
- J. Nguyen, R. Reul, T. Betz, E. Dayyoub, T. Schmehl, T. Gessler, U. Bakowsky, W. Seeger and T. Kissel, J. Controlled Release, 2009, 140, 47–54 CrossRef CAS PubMed.
- A. Rehor, J. A. Hubbell and N. Tirelli, Langmuir, 2005, 21, 411–417 CrossRef CAS PubMed.
- B. L. Allen, J. D. Johnson and J. P. Walker, ACS Nano, 2011, 5, 5263–5272 CrossRef CAS PubMed.
- A. Rehor, N. Tirelli and J. A. Hubbell, Macromolecules, 2002, 35, 8688–8693 CrossRef CAS.
- S. T. Reddy, A. J. van der Vlies, E. Simeoni, V. Angeli, G. J. Randolph, C. P. O'Neil, L. K. Lee, M. A. Swartz and J. A. Hubbell, Nat. Biotechnol., 2007, 25, 1159–1164 CrossRef CAS PubMed.
- A. J. van der Vlies, C. P. O'Neil, U. Hasegawa, N. Hammond and J. A. Hubbell, Bioconjugate Chem., 2010, 21, 653–662 CrossRef CAS PubMed.
- S. N. Thomas, A. J. van der Vlies, C. P. O'Neil, S. T. Reddy, S. S. Yu, T. D. Giorgio, M. A. Swartz and J. A. Hubbell, Biomaterials, 2011, 32, 2194–2203 CrossRef CAS PubMed.
- S. T. Reddy, A. Rehor, H. G. Schmoekel, J. A. Hubbell and M. A. Swartz, J. Controlled Release, 2006, 112, 26–34 CrossRef CAS PubMed.
- S. Hirosue, I. C. Kourtis, A. J. van der Vlies, J. A. Hubbell and M. A. Swartz, Vaccine, 2010, 28, 7897–7906 CrossRef CAS PubMed.
- A. Stano, A. J. van der Vlies, M. M. Martino, M. A. Swartz, J. A. Hubbell and E. Simeoni, Vaccine, 2011, 29, 804–812 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2012 |